
Comparative Analysis of Primers Used for 16S rRNA Gene Sequencing in Oral Microbiome Studies


Abstract
1. Introduction
2. Material and Methods
2.1. In Silico Evaluation of Primers and Phylogenetic Tree Construction
2.2. Study Population and Plaque Sample Collection
2.3. Extraction of Genomic DNA and Next-Generation Sequencing
2.4. Bioinformatic Analysis, Statistical Analysis, and Visualization
3. Results
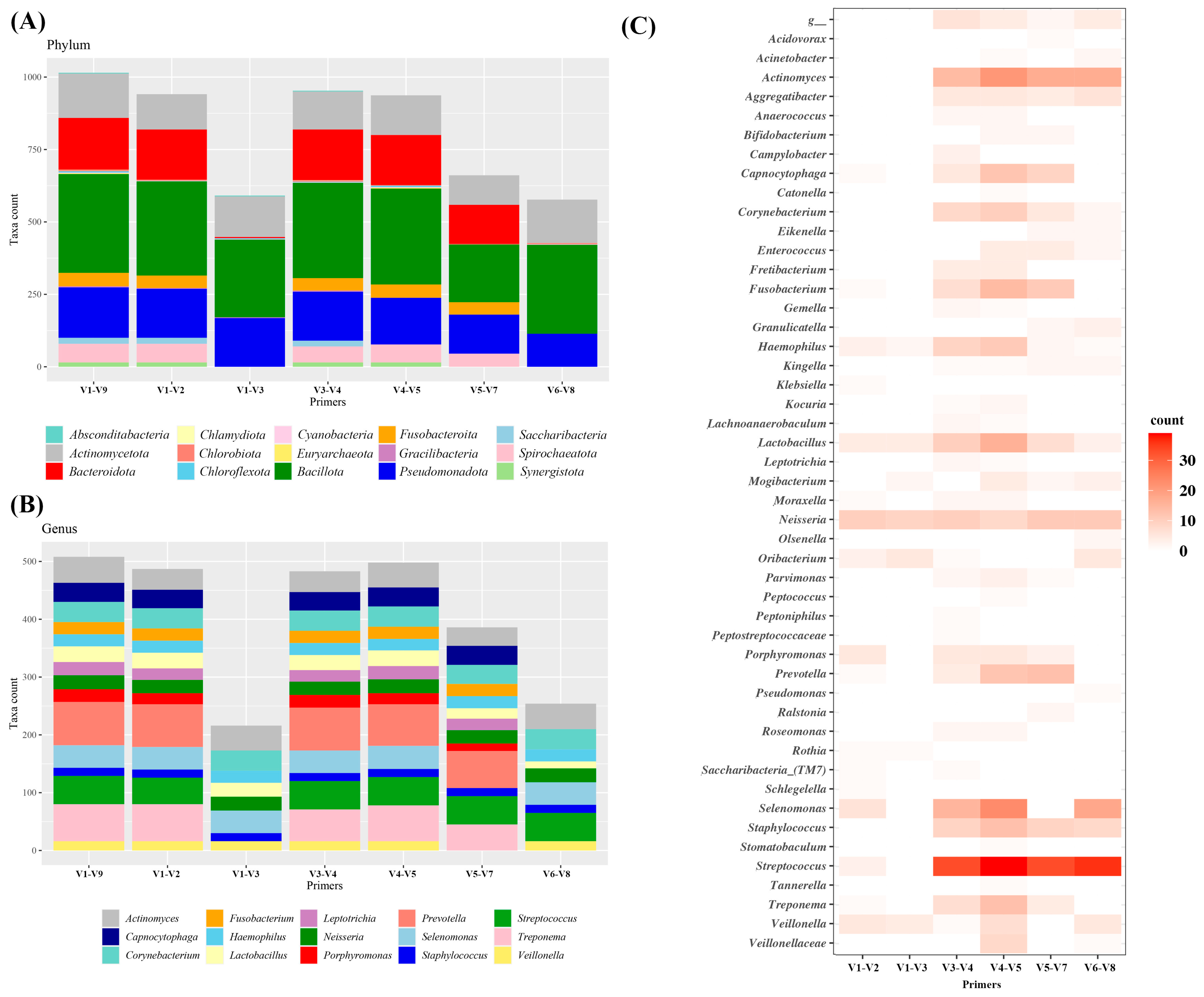
3.1. In Silico PCR Amplicon Detection and Classification
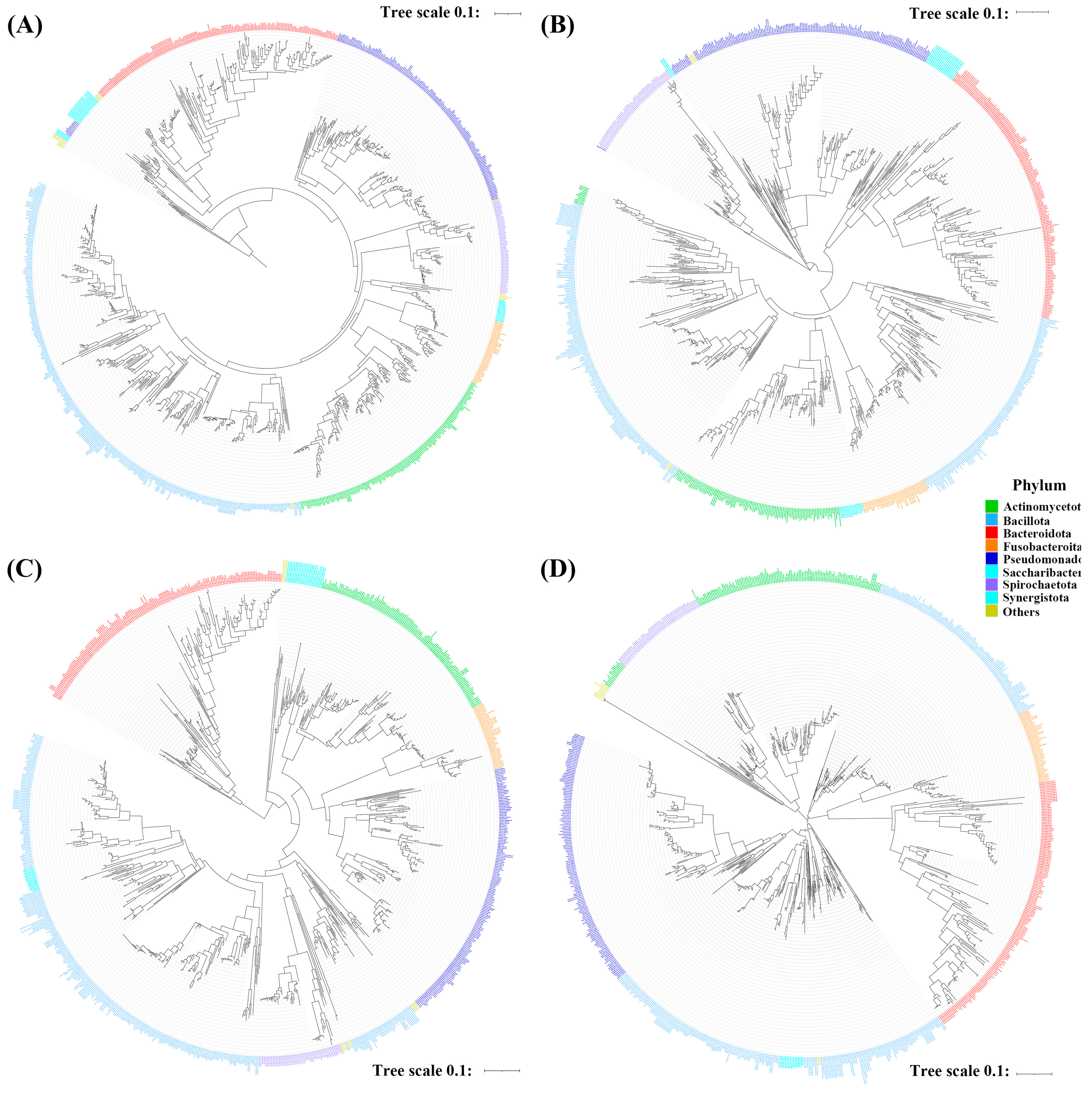
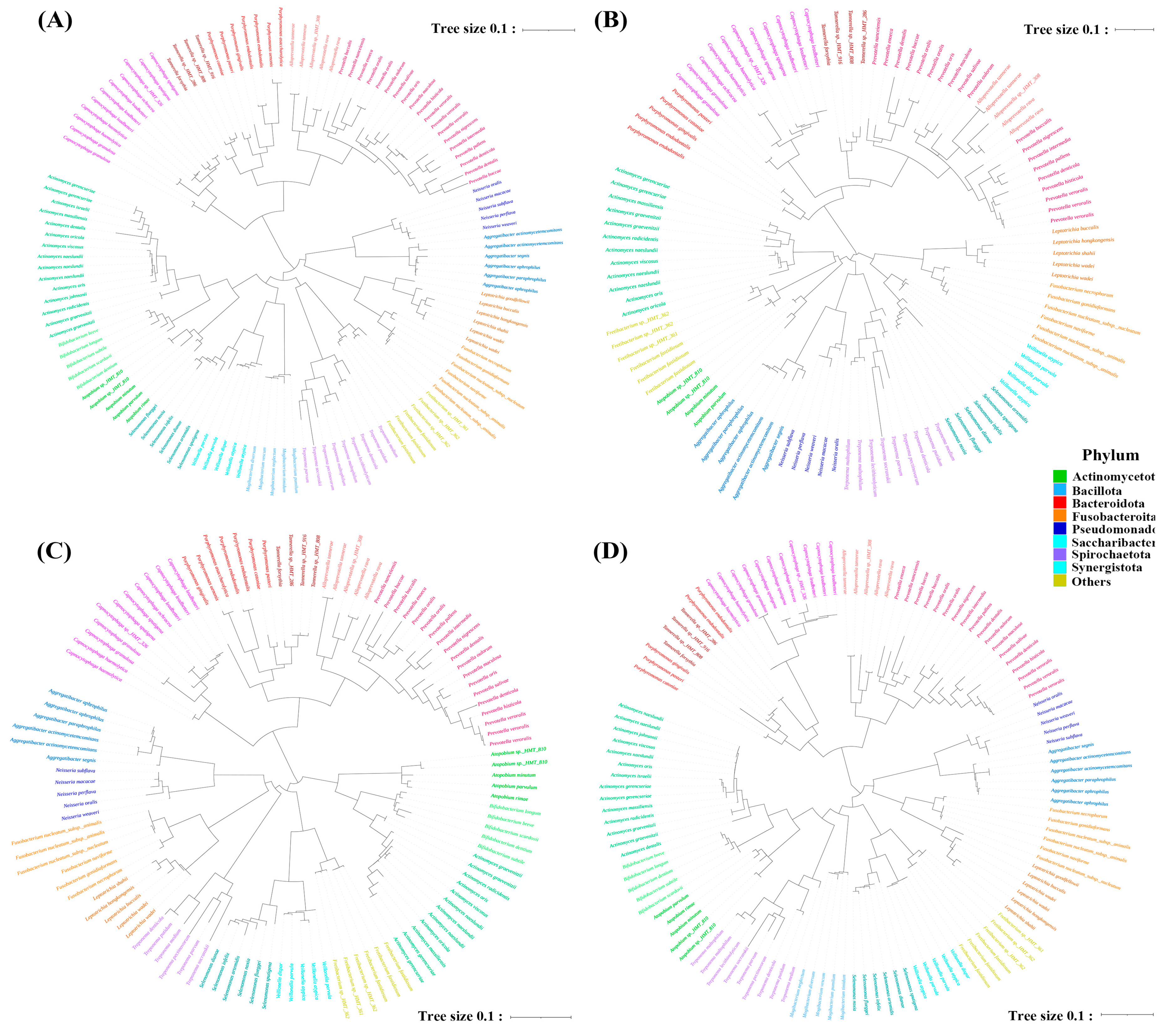
3.2. Phylogenetic Tree Construction
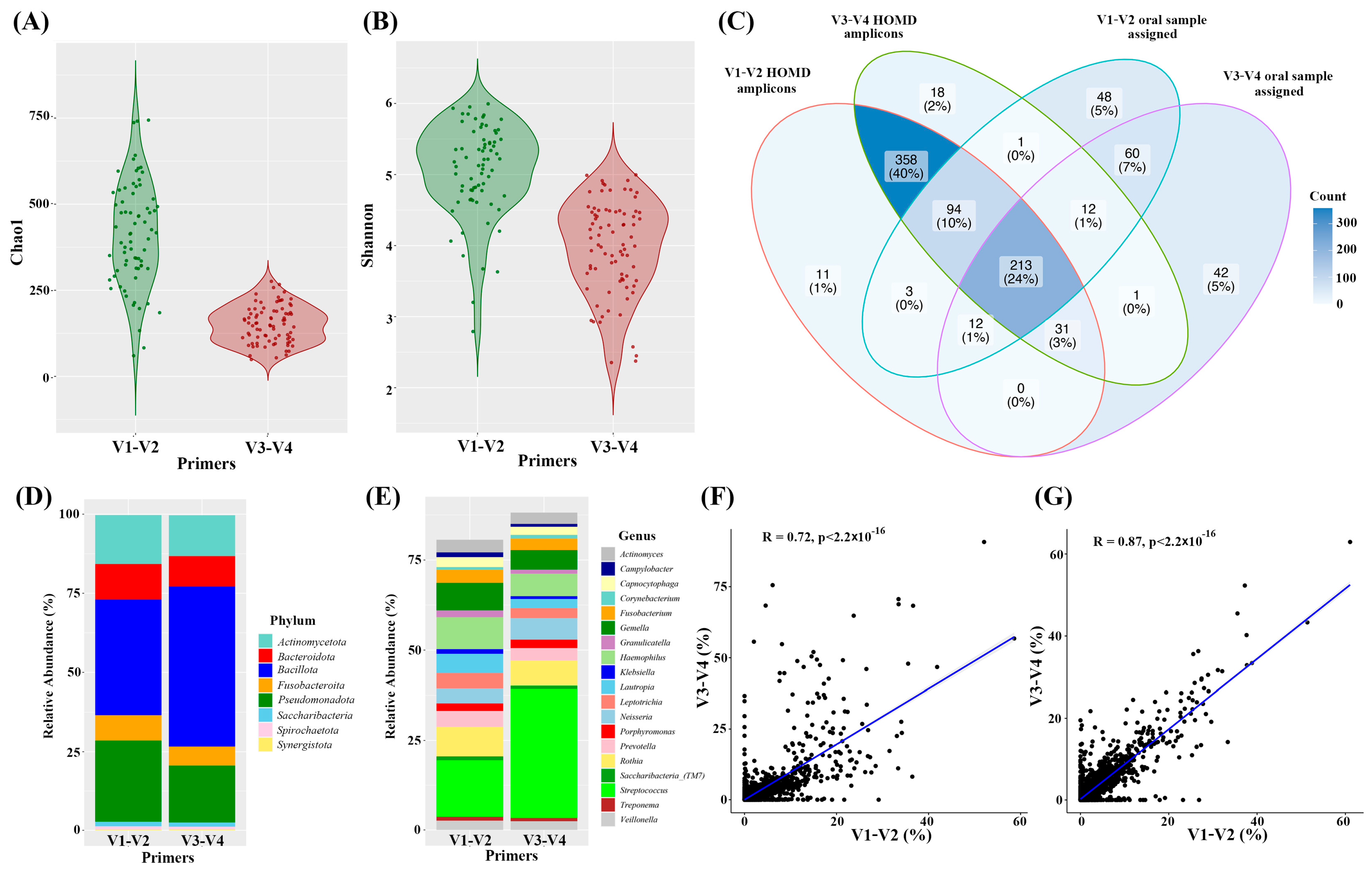
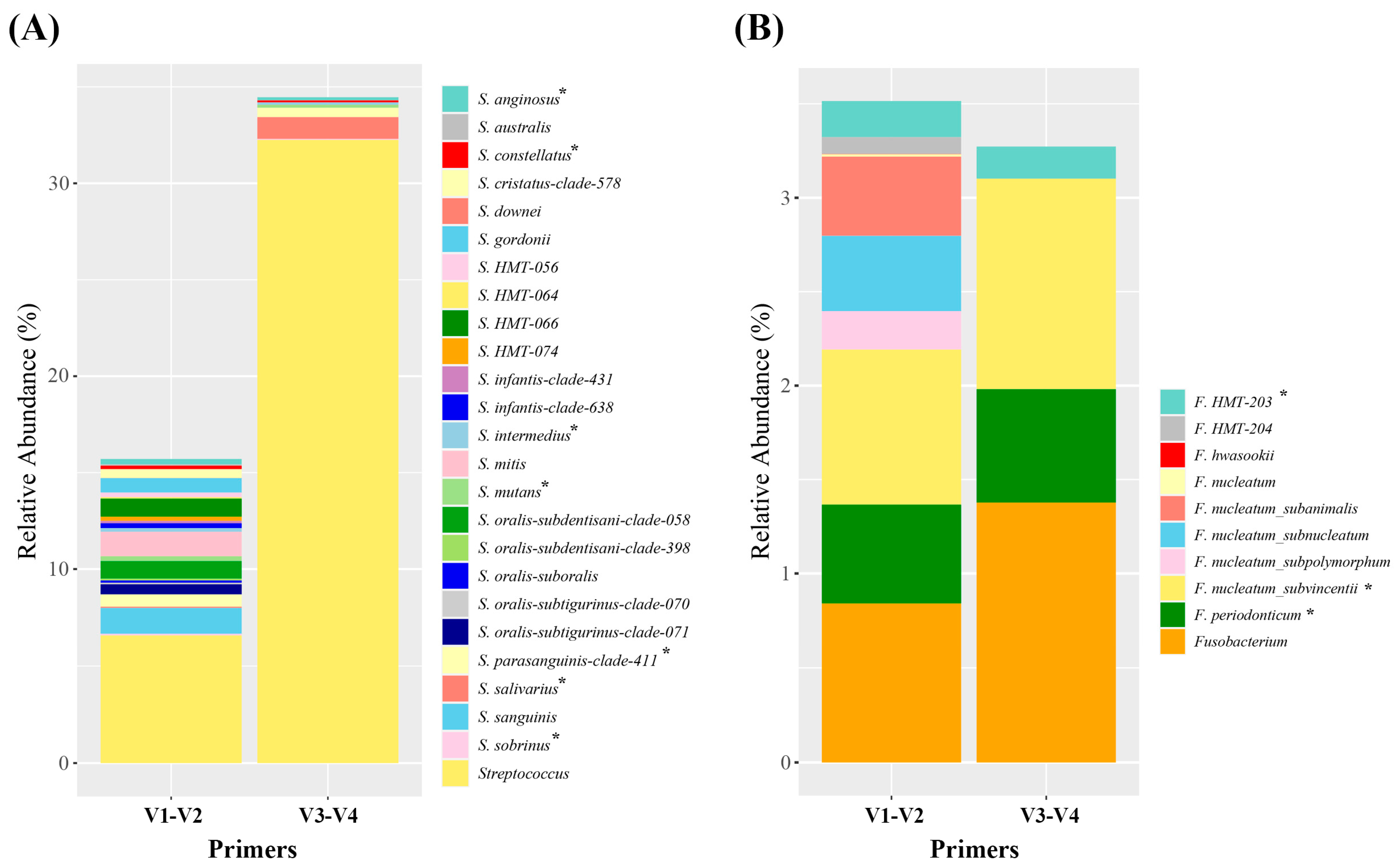
3.3. Oral Microbiome Detected by V1–V2 or V3–V4 Primers
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ramakodi, M.P. Influence of 16S rRNA reference databases in amplicon-based environmental microbiome research. Biotechnol. Lett. 2022, 44, 523–533. [Google Scholar] [CrossRef] [PubMed]
- Fadeev, E.; Cardozo-Mino, M.G.; Rapp, J.Z.; Bienhold, C.; Salter, I.; Salman-Carvalho, V.; Molari, M.; Tegetmeyer, H.E.; Buttigig, P.L.; Boetius, A. Comparison of Two 16S rRNA Primers (V3–V4 and V4–V5) for Studies of Arctic Microbial Communities. Front. Microbiol. 2021, 16, 637526. [Google Scholar] [CrossRef]
- Dewhirst, F.E.; Chen, T.; Izard, J.; Paster, B.J.; Tanner, A.C.; Yu, W.H.; Lakshmanan, A.; Wade, W.G. The Human Oral Microbiome. J. Bacteriol. 2010, 192, 5002–5017. [Google Scholar] [CrossRef] [PubMed]
- Human Microbiome Project Consortium. Structure, function and diversity of the healthy human microbiome. Nature 2012, 486, 207–214. [Google Scholar] [CrossRef] [PubMed]
- Wade, W.G. The oral microbiome in health and disease. Pharmacol. Res. 2013, 69, 137–143. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.S.; Spakowics, D.J.; Hong, B.Y.; Petersen, L.M.; Demkowicz, P.; Chen, L.; Leopold, S.R.; Hanson, B.M.; Agresta, H.O.; Gerstein, M.; et al. Evaluation of 16S rRNA gene sequencing for species and strain-level microbiome analysis. Nat. Commu. 2019, 10, 5029. [Google Scholar] [CrossRef] [PubMed]
- Peiffer, J.A.; Spor, A.; Koren, O.; Jin, Z.; Tringe, S.G.; Dangl, J.L.; Bucker, E.S.; Ley, R.E. Diversity and heritability of the maize rhizosphere microbiome under field conditions. Proc. Natl. Acad. Sci. USA 2013, 110, 6548–6553. [Google Scholar] [CrossRef] [PubMed]
- Cabral, D.J.; Wurster, J.I.; Flokas, M.E.; Alevizakos, W.; Zabat, M.; Korry, B.J.; Rowan, A.D.; Sano, W.H.; Andreatos, N.; Ducharme, R.B.; et al. The salivary microbiome is consistent between subjects and resistant to impacts of short-term hospitalization. Sci. Rep. 2017, 7, 11040. [Google Scholar] [CrossRef]
- Zhang, C.; Zhang, M.; Pang, X.; Zhao, Y.; Wang, L.; Zhao, L. Structural resilience of the gut microbiota in adult mice under high-fat dietary perturbations. ISME J. 2012, 6, 1848–1857. [Google Scholar] [CrossRef]
- Frid, P.; Baraniya, D.; Halbig, J.; Rypdal, V.; Songstad, N.T.; Rosèn, A.; Berstad, J.R.; Flatø, B.; Alakwaa, F.; Gil, E.G.; et al. Salivary Oral Microbiome of Children with Juvenile Idiopathic Arthritis: A Norwegian Cross-Sectional Study. Front. Cell. Infect. Microbiol. 2020, 4, 602239. [Google Scholar] [CrossRef]
- Yu, G.; Phillips, S.; Gail, M.H.; Goedert, J.J.; Humphrys, M.S.; Ravel, J.; Ren, Y.; Caporaso, N.E. The effect of cigarette smoking on the oral and nasal microbiota. Microbiome 2017, 5, 3. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; He, P.; Zhou, M.; Li, S.; Zhang, X.; Tao, X.; Wang, A.; Wu, X. Variations in oral microbiome and its predictive functions between tumorous and healthy individuals. J. Med. Microbiol. 2022, 71, 001568. [Google Scholar] [CrossRef]
- Dashper, S.G.; Mitchell, H.L.; Cao, K.A.L.; Carpenter, L.; Gussy, M.G.; Calache, H.; Gladman, S.L.; Bulah, D.M.; Hoffmann, B.; Catmull, D.V.; et al. Temporal development of the oral microbiome and prediction of early childhood caries. Sci. Rep. 2019, 9, 19732. [Google Scholar] [CrossRef]
- Bell, T.H.; Yergeau, E.; Maynard, C.M.; Juck, D.; Whyte, L.G.; Geer, C.W. Predictable bacterial composition and hydrocarbon degradation in Arctic soils following diesel and nutrient disturbance. ISME J. 2013, 7, 1200–1210. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Lu, G.; Luo, E.; Wu, B.; Li, Z.; Guo, J.; Xia, Z.; Zheng, C.; Su, Q.; Zheg, Y.; et al. Oral, Nasal, and Gut Microbiota in Parkinson’s Disease. Neuroscience 2022, 1, 65–78. [Google Scholar] [CrossRef]
- Kalanzi, D.; Mayanja-Kizza, H.; Nakanjako, D.; Semitala, F.; Mboowa, G.; Mbabali, M.; Kigozi, E.; Katabazi, F.A.; Sserwadda, I.; Kateeta, D.P.; et al. Microbial characteristics of dental caries in HIV positive individuals. Front. Oral Health 2022, 3, 1004930. [Google Scholar] [CrossRef]
- Sousa, V.; Nibali, L.; Spartt, D.; Dopico, J.; Mardas, N.; Petrie, A.; Donos, N. Peri-implant and periodontal microbiome diversity in aggressive periodontitis patients: A pilot study. Clin. Oral Implants Res. 2017, 28, 558–570. [Google Scholar] [CrossRef]
- Liu, Z.; DeSanti, T.Z.; Andersen, G.L.; Knight, R. Accurate taxonomy assignments from 16S rRNA sequences produced by highly parallel pyrosequencers. Nucleic Acids Res. 2008, 36, e120. [Google Scholar] [CrossRef] [PubMed]
- Shen, W.; Le, S.; Li, Y.; Hu, F. SeqKit: A Cross-Platform and Ultrafast Toolkit for FASTA/Q File Manipulation. PLoS ONE 2016, 11, e0163962. [Google Scholar] [CrossRef]
- Hall, M.; Beiko, R.G. 16S rRNA Gene Analysis with QIIME2. Methods Mol. Biol. 2018, 1849, 113–129. [Google Scholar]
- Letunic, I.; Bork, P. Interactive Tree of Life (iTOL) v5: An online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 2021, 49, W293–W296. [Google Scholar] [CrossRef]
- Klidworth, A.; Pruesse, E.; Schweer, T.; Peplies, J.; Quast, C.; Horn, M.; Glöckner, F.O. Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Res. 2013, 41, e1. [Google Scholar] [CrossRef] [PubMed]
- Thijs, S.; Beeck, M.O.D.; Beckers, B.; Truyens, S.; Stevens, V.; Hamme, J.D.V.; Weyens, N.; Vangronsveld, J. Comparative Evaluation of Four Bacteria-Specific Primer Pairs for 16S rRNA Gene Surveys. Front. Microbiol. 2017, 8, 494. [Google Scholar] [CrossRef]
- Cabral, D.J.; Wurster, J.I.; Korry, B.; Penumutchu, S.; Belenky, P. Consumption of a Western-Style Diet Modulates the Response of the Murine Gut Microbiome to Ciprofloxacin. mSystems 2020, 5, e00317–e00320. [Google Scholar] [CrossRef] [PubMed]
- Na, H.S.; Kim, S.Y.; Han, H.; Kim, H.J.; Lee, J.Y.; Lee, J.H.; Chung, J. Identification of Potential Oral Microbial Biomarkers for the Diagnosis of Periodontitis. J. Clin. Med. 2020, 9, 1549. [Google Scholar] [CrossRef] [PubMed]
- Shi, M.; Wei, Y.; Hu, W.; Nie, Y.; Wu, X.; Lu, R. The Subgingival Microbiome of Periodontal Pockets With Different Probing Depths in Chronic and Aggressive Periodontitis: A Pilot Study. Front. Cell. Infect. Microbiol. 2018, 8, 124. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Hemme, C.; Beleno, J.; Shi, Z.J.; Ning, D.; Qin, Y.; Tu, Q.; Jorgensen, M.; He, Z.; Wu, L.; et al. Oral microbiota of periodontal health and disease and their changes after nonsurgical periodontal therapy. ISME J. 2018, 12, 1210–1224. [Google Scholar] [CrossRef]
- Liu, G.; Chen, F.; Cai, Y.; Chen, Z.; Luan, Q.; Yu, X. Measuring the subgingival microbiota in periodontitis patients: Comparison of the surface layer and the underlying layers. Microbiol. Immunol. 2020, 64, 99–112. [Google Scholar] [CrossRef]
- Walker, A.W.; Martin, J.C.; Scott, P.; Parkhill, J.; Flint, H.J.; Scott, K.P. 16S rRNA gene-based profiling of the human infant gut microbiota is strongly influenced by sample processing and PCR primer choice. Microbiome 2015, 3, 32015. [Google Scholar] [CrossRef]
- Chakravorty, S.; Helb, D.; Burday, M.; Connell, N.; Alland, D. A detailed analysis of 16S ribosomal RNA gene segments for the diagnosis of pathogenic bacteria. J. Microbiol. Methods 2007, 69, 330–339. [Google Scholar] [CrossRef]
- Abellan-Schneyder, I.; Matchado, M.S.; Reitmeier, S.; Sommer, A.; Sewald, Z.; Baumbach, J.; List, M.; Neuhaus, K. Primer, Pipelines, Parameters: Issues in 16S rRNA Gene Sequencing. mSphere 2021, 6, e01202–e01220. [Google Scholar] [CrossRef]
- Turnbaugh, P.J.; Ley, R.E.; Hamady, M.; Fraser-Liggett, C.M.; Knight, R.; Gordon, J.I. The human microbiome project. Nature 2007, 449, 804–810. [Google Scholar] [CrossRef] [PubMed]
- Wade, W.G.; Prosdocimi, E.M. Profiling of Oral Bacterial Communities. J. Dent. Res. 2020, 99, 621–629. [Google Scholar] [CrossRef] [PubMed]
- Na, H.S.; Yu, Y.; Kim, S.Y.; Lee, J.H.; Chung, J. Comparison of the Performance of MiSeq and HiSeq 2500 in a Microbiome Study. Microbiol. Biotechnol. Lett. 2020, 48, 574–581. [Google Scholar] [CrossRef]
- Navas-Molina, J.A.; Peralta-Sanchez, J.M.; Gonzalez, A.; McMurdie, P.J.; Vazquez-Baeza, Y.; Xu, Z.; Ursell, L.K.; Lauber, C.; Zhou, H.; Song, S.J.; et al. Advancing our understanding of the human microbiome using QIIME. Methods Enzymol. 2013, 531, 371–444. [Google Scholar] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Region | Forward | Reverse | Size (bp) |
|---|---|---|---|
| V1–V2 | AGAGTTTGATYMTGGCTCAG | TGCTGCCTCCCGTAGRAGT | 311 |
| V1–V3 | TNANACATGCAAGTCGRRCG | WTTACCGCGGCTGCTGG | 450 |
| V3–V4 | CCTACGGGNGGCWGCAG | GACTACHVGGGTATCTAATCC | 444 |
| V4–V5 | GTGYCAGCMGCCGCGGTAA | CCGYCAATTYMTTTRAGTTT | 411 |
| V5–V7 | AACMGGATTAGATACCCKG | ACGTCATCCCCACCTTCC | 394 |
| V6–V8 | CAACGCGAAGAACCTTACC | GACGGGCGGTGWGTRCA | 424 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Na, H.S.; Song, Y.; Yu, Y.; Chung, J. Comparative Analysis of Primers Used for 16S rRNA Gene Sequencing in Oral Microbiome Studies. Methods Protoc. 2023, 6, 71. https://doi.org/10.3390/mps6040071
Na HS, Song Y, Yu Y, Chung J. Comparative Analysis of Primers Used for 16S rRNA Gene Sequencing in Oral Microbiome Studies. Methods and Protocols. 2023; 6(4):71. https://doi.org/10.3390/mps6040071
Chicago/Turabian StyleNa, Hee Sam, Yuri Song, Yeuni Yu, and Jin Chung. 2023. "Comparative Analysis of Primers Used for 16S rRNA Gene Sequencing in Oral Microbiome Studies" Methods and Protocols 6, no. 4: 71. https://doi.org/10.3390/mps6040071
APA StyleNa, H. S., Song, Y., Yu, Y., & Chung, J. (2023). Comparative Analysis of Primers Used for 16S rRNA Gene Sequencing in Oral Microbiome Studies. Methods and Protocols, 6(4), 71. https://doi.org/10.3390/mps6040071