1. Introduction
Different aspects of (001) surface as well as (001) interface phenomena in ABO
3 perovskites are topics of paramount importance in modern solid state physics [
1,
2,
3,
4,
5,
6,
7,
8,
9,
10,
11,
12,
13,
14,
15,
16]. All of our hybrid-DFT computed SrZrO
3, PbZrO
3, PbTiO
3, BaTiO
3 and SrTiO
3 solids belong to the group of so-called ABO
3 perovskite compounds [
17,
18,
19]. Strontium zirconate (SrZrO
3) is an extremely interesting material due to its high-temperature electronic properties [
20]. The SrZrO
3 phase transitions have been very carefully examined by Kennedy et al. [
21] by means of powder neutron diffraction, as well as the Rietveld method [
21]. Maniwa et al. [
22] and Shirane et al. [
23] described PbZrO
3 as antiferroelectric due to its dielectric behaviour. Since then, materials that revealed a structural phase transition between two non-polar phases with a large dielectric anomaly at the high-temperature region of the transition were called antiferroelectrics [
22,
23]. Antiferroelectric oxides, for example PbZrO
3, become highly promising candidate materials for dielectric energy storage capacitors [
24,
25]. PbTiO
3 is a crucial component in a wide range of industrially important applications, including sensors, ultrasonic transducers and actuators [
26]. Polymer ceramic hybrid composites (PVA/PbTiO
3) are highly promising functional materials in a wide variety of research disciplines, demonstrating useful electrical, optical, mechanical, thermal and antibacterial properties [
27]. Ferroelectric materials, for example BaTiO
3, have been mentioned in connection to bacterial degradation [
28]. It is demonstrated that the remnant polarization can induce antibacterial activities [
29] in ferroelectric materials like BaTiO
3. Lead zirconate titanate Pb(Zr,Ti)O
3 is the most widely used worldwide piezoceramic material for electromechanical device applications [
30]. Strontium titanate exhibits photocatalytic activity [
31,
32]. The identification of highly promising thermoelectric materials, like SrTiO
3, is accelerated by theoretical predictions from ab initio calculations [
33,
34,
35,
36]. In general, there is a lot of interest in the science and technology of transition metal oxides, like ABO
3 perovskites, due to their large variety of specific electronic, optical and magnetic properties [
37,
38,
39,
40,
41,
42,
43,
44,
45,
46,
47,
48]. For that reason, during the last 25 years, a lot of theoretical and experimental studies have been performed for PbTiO
3, BaTiO
3, SrTiO
3, SrZrO
3 and PbZrO
3 perovskite (001) surfaces [
49,
50,
51,
52,
53,
54,
55,
56,
57,
58,
59,
60,
61,
62,
63,
64,
65,
66,
67,
68,
69,
70,
71,
72]. At the same time, it is very difficult to understand why a smaller number of first-principles computations, as well as experimental studies, were devoted to BTO/STO and PTO/STO, as well as SZO/PZO (001) heterostructures [
73,
74,
75,
76,
77,
78,
79,
80,
81,
82,
83,
84,
85,
86,
87,
88].
SrTiO
3 (STO) perovskite is paraelectric and maintains its cubic centrosymmetric structure at all observed temperatures [
89]. BaTiO
3 (BTO), as a function of temperature, undergoes three phase transitions. Namely, the BTO crystal structure changes at 183 K from rhombohedral (
R3m) to orthorhombic (
Amm2), then at 278 K to tetragonal (
P4mm), and finally at 403 K to a high-temperature cubic structure (
) [
90]. PbTiO
3 (PTO) undergoes only one phase transition at 766 K from a tetragonal ferroelectric phase, stable at room temperature, with the space group
P4mm to the high-symmetry cubic structure with the space group
[
91]. It is well-known that the classical PbZrO
3 (PZO) perovskite exhibits three different phases. Namely, at low temperatures, PZO has an orthorhombic antiferroelectric phase stable at the temperature region up to 230 °C. The PZO rhombohedral ferroelectric phase is stable at the temperature region from 230 °C to 233 °C. Finally, PZO perovskite possesses a cubic paraelectric phase at temperatures above 233 °C [
69]. Lastly, for the SrZrO
3 (SZO) perovskite, Ligny et al. [
92] discovered the following three phase transitions from enthalphy measurements: orthorhombic (
Pnma) → orthorhombic (
Cmcm) → tetragonal (
I4/mcm) → cubic (
) at temperatures equal to 995 K and 1105 K, as well as 1140 K, respectively [
92].
For our hybrid-DFT computations, we adopted the classical cubic unit cells of SrZrO
3, PbZrO
3, PbTiO
3, BaTiO
3 and SrTiO
3 perovskites. The cubic unit cells of all five of our ab initio computed ABO
3 perovskites contain five atoms [
93,
94,
95,
96]. They all have exactly the same space group
, as well as the same space group number, 221. In the cubic ABO
3 perovskite matrix, the A-type atom (Sr, Pb or Ba) was located at the cube corner position. Thereby, the ABO
3 perovskite A atom, in our computations, is located at the coordinate system origin and has the following coordinates (0, 0, 0). The B-type ABO
3 perovskite atom (Ti or Zr) is located at the body center location of the cube. The coordinates of the B atom are equal to (½, ½, ½). Finally, the ABO
3 perovskite unit cell’s three oxygen atoms are located at the cube face center positions with the following coordinates (½, ½, 0), (½, 0, ½), (0, ½, ½).
The aim of our research reported herein was to perform necessary additional hybrid-DFT computations for SrZrO3, PbZrO3, PbTiO3, BaTiO3 and SrTiO3 perovskite (001) surfaces, as well as their (001) heterostructures, in order to complete our many years of lasting work in this direction, and to create a theory that explains systematic trends in ABO3 perovskite (001) surface and interface computations. In the first part of our contribution, we present comparative hybrid-DFT computation results dealing with the pristine SrZrO3, PbZrO3, PbTiO3, BaTiO3 and SrTiO3 (001) surfaces. We computed the SrZrO3, PbZrO3, PbTiO3, BaTiO3 and SrTiO3 (001) surface atom relaxations for both possible BO2 and AO (001) surface terminations and compared the calculated surface rumplings s and interlayer distances Δdij with the available experimental data. As a next step, we computed the SrZrO3, PbZrO3, PbTiO3, BaTiO3 and SrTiO3 (001) surface energies, as well as B-O chemical band populations near the BO2-terminated (001) surfaces. Finally, we computed the band gaps for bulk PbTiO3, BaTiO3 and SrTiO3 and their (001) surfaces by means of different exchange-correlation functionals. As a logical next step, we extended our (001) surface calculations to hybrid-DFT computations of considerably less-studied SrTiO3/BaTiO3, SrZrO3/PbZrO3 and PbTiO3/SrTiO3 (001) heterostructures.
The present paper is structured in the following way. Our hybrid-DFT calculations details, dealing with ABO
3 perovskite (001) surfaces and heterostructures, are described in
Section 2.
Section 3.1 presents our computation results dealing with SrZrO
3, PbZrO
3, PbTiO
3, BaTiO
3 and SrTiO
3 (001) surfaces, whereas
Section 3.2 addresses their SrTiO
3/BaTiO
3, SrZrO
3/PbZrO
3 and PbTiO
3/SrTiO
3 (001) heterostructures. In particular, our novel hybrid-DFT computations are devoted to charge density redistribution and changes in the band structure in stoichiometric, as well as non-stoichiometric (001), heterostructures, consisting of BTO, PTO and PZO (001) thin films, with different thicknesses deposited on the TiO
2-, TiO
2- and ZrO
2-terminated STO and SZO (001) substrate. We analyzed our hybrid-DFT calculation results and detected systematic trends common for all STO/BTO and STO/PTO, as well as SZO/PZO (001), heterostructures.
2. Hybrid-DFT Calculation Details
We carried out our hybrid-DFT computations for the PbTiO
3 (PTO), BaTiO
3 (BTO), SrTiO
3 (STO), PbZrO
3 (PZO) and SrZrO
3 (001) bulk, as well as their BO
2- and AO-terminated (001) surfaces, and their different (001) heterostructures, employing the computer code CRYSTAL [
97]. For our hybrid-DFT computations, we employed the nowadays very popular hybrid exchange-correlation functionals B3LYP [
98] or B3PW [
99,
100]. It is worthwhile to note that the hybrid exchange-correlation functionals, for example, B3LYP or B3PW allows achieving an outstanding agreement with the experiment for the band gaps of ABO
3 perovskites as well as related materials, like CaF
2 [
101] and MgF
2 [
102]. In contrast, density functional theory (DFT) strongly underestimates, while the Hartree–Fock (HF) method, as a rule, very strongly overestimates the ab initio calculated band gaps of solids. For example, the experimental CaF
2 direct band gap value at the Γ-point is equal to 12.1 eV [
103]. Our, by means of Perdew–Burke–Ernzerhof (PBE) exchange-correlation functional [
104,
105], extensively used in DFT, ab initio computed CaF
2 Γ-Γ band gap is very strongly underestimated with respect to the experimental value (12.1 eV) and is equal to 8.45 eV. From another side, our ab initio HF [
106] computed CaF
2 Γ-Γ band gap (20.77 eV) is 1.72 times overestimated compared to the experimental CaF
2 band gap value (12.1 eV [
105]). According to our hybrid-DFT computations, the hybrid exchange correlation functionals, like B3LYP (10.85 eV) and B3PW (10.96 eV) allows the achievement of the best possible agreement with the experimental CaF
2 Γ-Γ band gap equal to 12.1 eV. The experimentally measured MgF
2 Γ-Γ band gap is equal to 13.0 eV [
107]. Again, our PBE exchange-correlation functional computed MgF
2 Γ-Γ band gap is considerably underestimated regarding the experimental value of (13.0 eV) and is equal to 6.9 eV. In contrast, our ab initio HF computed MgF
2 Γ-Γ band gap (19.65 eV) very strongly, namely 1.65 times, overestimates the experimentally measured MgF
2 band gap at Γ-point (13.0 eV). The best possible results, according to our hybrid-DFT computations, are possible to achieve by means of the hybrid exchange-correlation functionals B3LYP (9.42 eV) and B3PW (9.48 eV). Since, as it is possible to see from our computation results for CaF
2 and MgF
2 solids, the hybrid exchange-correlation functionals B3LYP and B3PW allows achieving the best possible results for CaF
2 and MgF
2 Γ-Γ band gaps, we performed all of our coming ABO
3 perovskite (001) surface and interface computations using the B3LYP or B3PW functionals. The B3LYP and B3PW hybrid exchange-correlation functionals make use of 20% of the Hartree–Fock method as well as 80% of the density functional Hamiltonian, when incorporated in the CRYSTAL computer code [
97].
The major advantage of the CRYSTAL computer software package, which is of key importance for our hybrid-DFT computations of neutral SrZrO
3, PbZrO
3, PbTiO
3, BaTiO
3 and SrTiO
3 (001) surfaces and interfaces, is the implementation of the isolated 2-dimensional (2D) slab model. In our hybrid-DFT computations, the reciprocal space integration was carried out by examining the Brillouin zone with the 8 × 8 × 8 times expanded Pack–Monkhorst net for the bulk of SrZrO
3, PbZrO
3, PbTiO
3, BaTiO
3 and SrTiO
3 solids, as well as by 8 × 8 × 1 net for their (001) surfaces and interfaces. With a goal of reaching a high accuracy in our ab initio computations, we employed sufficiently large tolerances equal to 7, 8, 7, 7, 14 for the Coulomb overlap, Coulomb penetration and exchange overlap, as well as first exchange pseudo-overlap and the second exchange pseudo-overlap [
97].
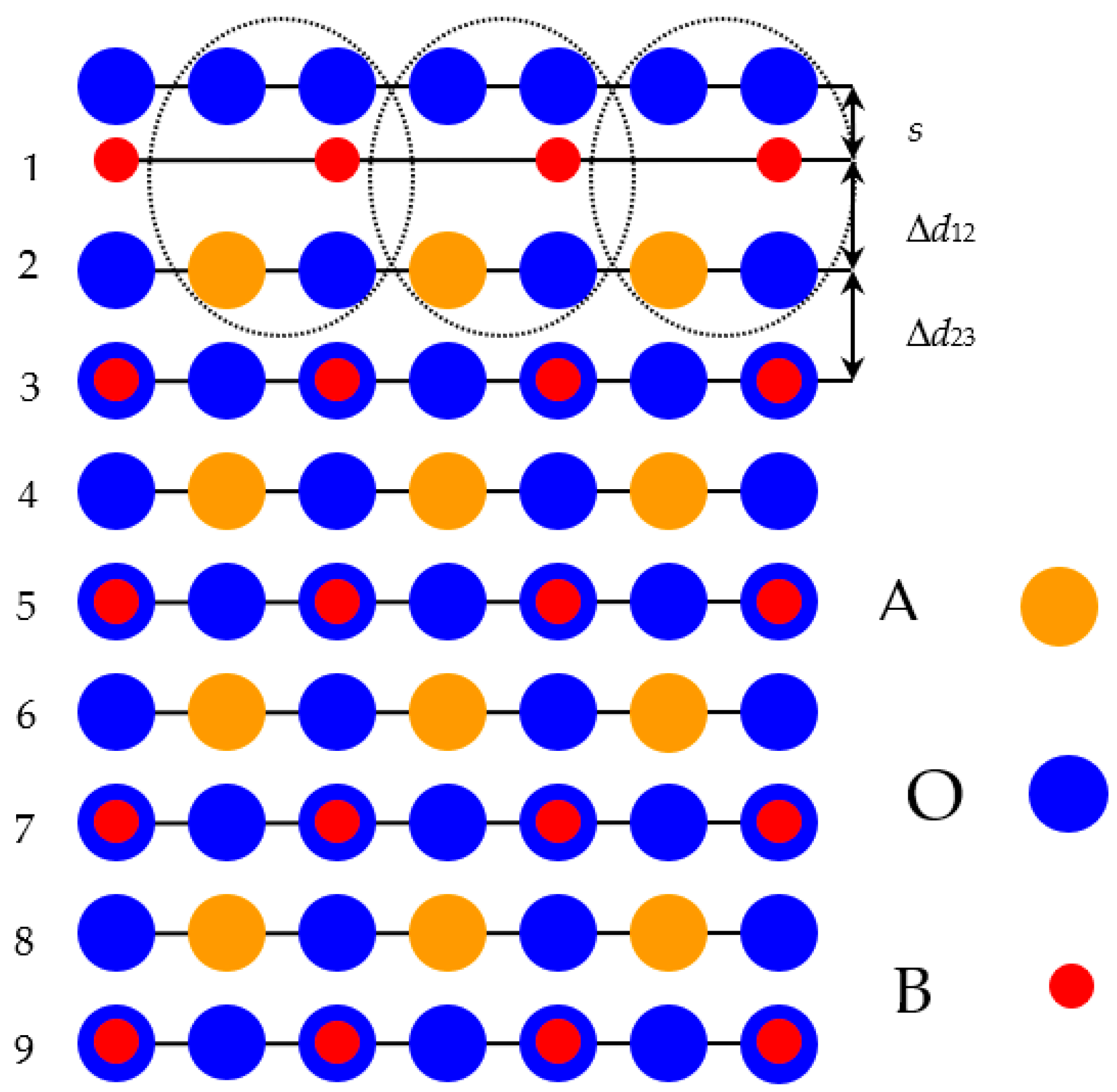
With the aim of computing the neutral BO
2-terminated (001) surfaces of ABO
3 solids, we customized symmetrical slabs. They involved nine neutral as well as alternating BO
2 and AO layers (
Figure 1). All of these nine layers were perpendicular to the [001] crystal direction [
108,
109,
110,
111]. Taking into consideration the standard ionic charges equal to (+2
e) for the A atom and (+4
e) for the B atom, as well as (−2
e) for the O atom, both alternating BO
2 and AO layers are neutral, since they have a summary slab charge equal to zero. The slab, containing nine layers, used by us for the BO
2-terminated ABO
3 perovskite (001) surface computations (
Figure 1) consisted of a supercell containing 23 atoms. Our hybrid-DFT computed BO
2-terminated (001) slabs (
Figure 1) of ABO
3 perovskite were non-stoichiometric. They have a unit cell, used in our hybrid-DFT computations, described by the following chemical formula—A
4B
5O
14 [
112,
113,
114].
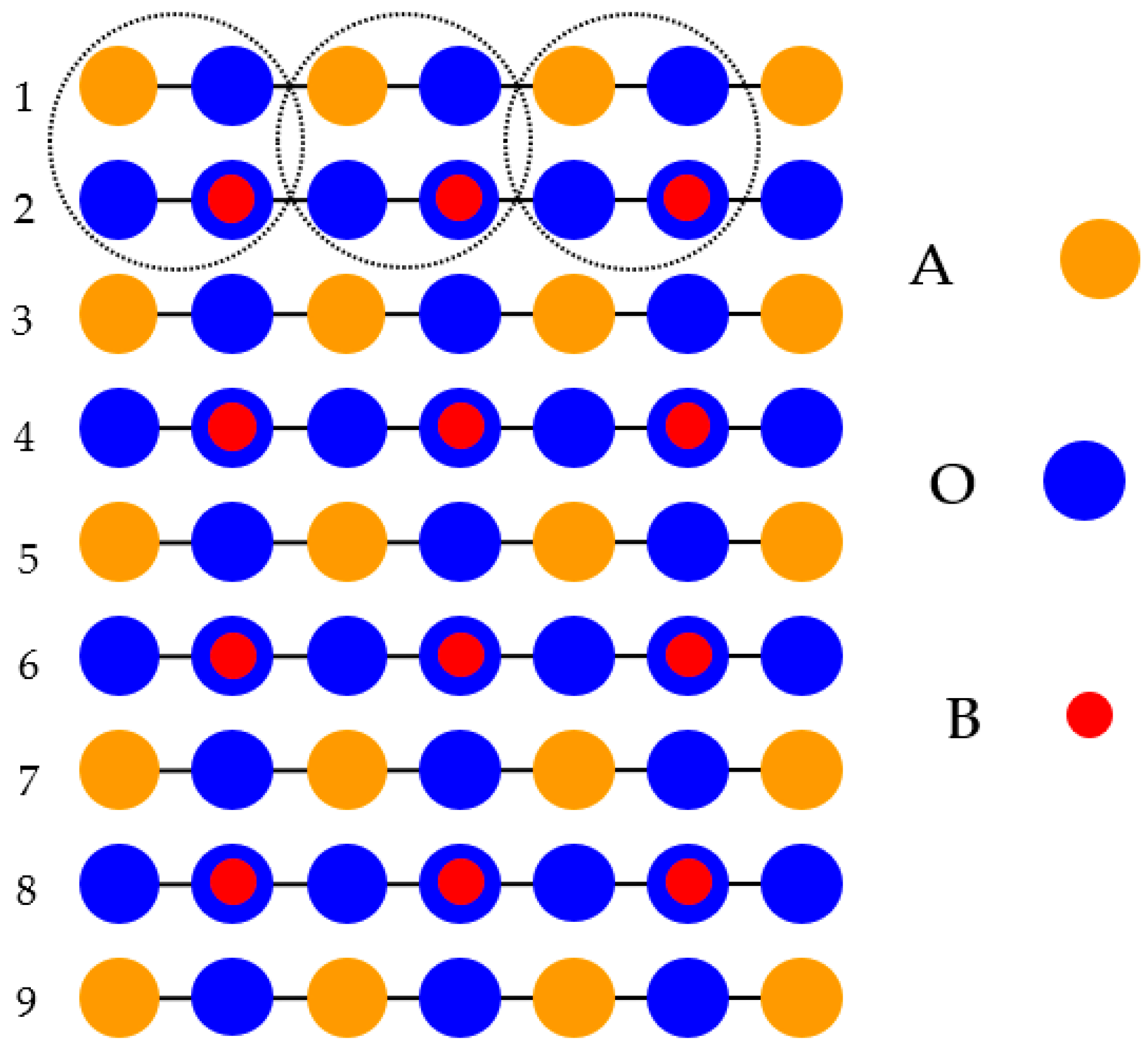
The second slab in our hybdid-DFT computations was terminated from both sides by the AO planes (
Figure 2). It also contained nine alternating layers (
Figure 2) but consisted of a supercell that incorporated only 22 atoms. Furthermore, the AO-terminated nine-layer slab, used in our hybrid-DFT computations, was non-stoichiometric and has the following chemical formula—A
5B
4O
13. For Sr, Ba, Pb, Ti and O atoms [
115], we used the basis sets developed in [
117]. The inner-core electrons for Sr, Ba, Pb, Ti and Zr atoms were described by a small core Hay–Wadt effective pseudopotentials [
97,
116]. We computed the number of light oxygen atoms using the all-electron basis sets [
97,
115]. In order to correctly define the chemical bonding in ABO
3 perovskites, as well as covalency effects, we employed a widely accepted Mulliken population [
117,
118,
119] analysis. The Mulliken population analysis is incorporated in the CRYSTAL computer code [
97], used in all of our hybrid-DFT computations for the ABO
3 solid (001) surfaces and their respective heterostructures (
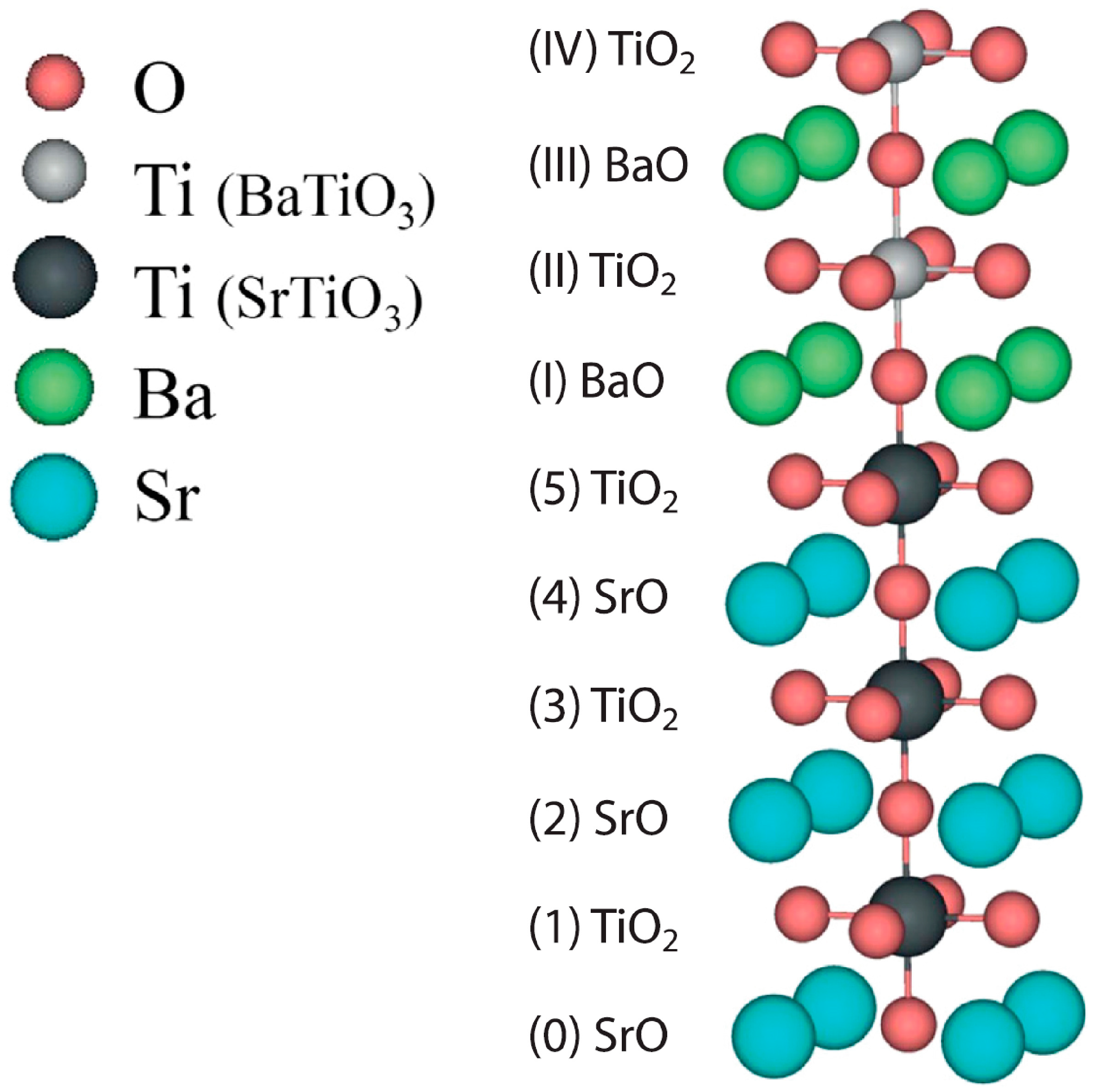
Figure 3).
With ambition to compute the ABO
3 solid (001) surface energies, we opened our hybid-DFT computations with cleavage energies for unrelaxed AO- as well as BO
2-terminated (001) surfaces. In our hybrid-DFT computations, the two nine-layer AO- as well as BO
2-terminated slabs contained 22 and 23 atoms of each of them. These two AO- and BO
2-terminated slabs represented together nine ABO
3 perovskite bulk unit cells. Each ABO
3 perovskite bulk unit cell contained five atoms. AO as well as BO
2-terminated ABO
3 perovskite (001) surfaces arose at the same time moment under crystal cleavage. Therefore, the relevant cleavage energy is shared uniformly among created surfaces. Thereby, our ab initio computed ABO
3 perovskite (001) surface cleavage energy is equal for both AO- as well as BO
2-terminations:
where
Eslabunrel (AO) and
Eslabunrel (BO
2) are our hybrid-DFT computed total energies for unrelaxed AO- and BO
2-terminated ABO
3 perovskite nine-layer slabs.
Ebulk is our hybrid-DFT computed total bulk unit cell energy containing five atoms. Factor 4 in Equation (1) arises from the fact that we created four surfaces during the ABO
3 perovskite cleavage event. As a next step, we computed the relaxation energies for AO- and BO
2-terminated ABO
3 solid (001) surfaces. We relaxed both sides of our nine-layer AO- and BO
2-terminated slabs.
Eslab(Ψ) is our hybrid-DFT computed slab total energy after the geometry relaxation. In our case the symbol Ψ denotes AO- or BO
2-terminated ABO
3 perovskite (001) surface. After all of our hybrid-DFT computed ABO
3 perovskite AO- or BO
2-terminated (001) surface energy is defined as a sum of the cleavage as well as relaxation energies:
As a next step, we will discuss our calculation details for the ABO
3 perovskite (001) heterostructures, using as an example the BaTiO
3/SrTiO
3 (001) interface. At room temperature, the SrTiO
3 substrate has a high-symmetry cubic structure. In our hybrid-DFT computations, we computed both SrTiO
3 and BaTiO
3 perovskites at their cubic, high-symmetry phase with the space group number
. In our hybrid-DFT computations, we modelled the BaTiO
3/SrTiO
3 (001) interface employing the single-slab model. In order to maximally apply the advantages of symmetry, our slabs were symmetrically terminated. In our hybrid-DFT computations, the SrTiO
3 (001) substrate always contained 11 alternating SrO as well as TiO
2 layers. From 1 to 10 BaO and TiO
2 alternating layers were augmented on both sides of the 11-layer TiO
2-terminated SrTiO
3 (001) substrate (
Figure 3).
We allowed all coordinated of atoms to relax in our hybrid-DFT computed BaTiO
3/SrTiO
3 (001) heterostructure. In our hybrid-DFT computations, due to restrictions imposed by the cubic symmetry of the system, atomic displacements are possible only along axis
z. It is worthwhile to note that the mismatch equal to approximately 2.5 percent among BaTiO
3 and SrTiO
3 bulk lattice constants happens at the time of BaTiO
3 epitaxial growth. The joint equilibrium average lattice constant employed in all of our future hybrid-DFT computations for BaTiO
3/SrTiO
3 (001) interfaces is equal to 3.958 Å. The joint lattice constant for the BaTiO
3/SrTiO
3 (001) interface was hybrid-DFT computed by us for the thickest (001) interface, which consisted of the 11-layer SrTiO
3 substrate as well as, from both sides of this substrate, augmented 10 BaTiO
3 layers. In our hybrid-DFT calculations of the shift (Δ
z) for each layer of the BaTiO
3/SrTiO
3 (001) heterostructure, we take into consideration the shift of the previous atomic layer. Therefore, the reference z coordinate for each mono-layer
N is described by the following equation:
where, in Equation (4),
zN-1Me as well as
zN-1O defines the
z coordinates for the cation and anion from the previous atomic monolayer.
4. Conclusions
According to our hybrid-DFT computations for BO
2- and AO-terminated PbTiO
3, BaTiO
3, SrTiO
3, PbZrO
3 and SrZrO
3 (001) surfaces, in most cases, the upper-layer ions relax inwards, towards the bulk, whereas the second-layer ions shift upwards. It is worthwhile to also note that independent calculation results of other authors for ABO
3 perovskite material’s neutral (001) surfaces confirm this systematic trend, that the upper layer ions, in most cases, relax inwards, whereas the second layer ions, again, in most cases, relax upwards [
4,
9,
20,
49,
50,
54,
66,
67,
135,
136,
137,
138].
Our hybrid-DFT computed surface rumpling
s for the BO
2-terminated ABO
3 perovskite (001) surfaces (
Table 5) is positive for most computed ABO
3 perovskites, such as PbZrO
3, PbTiO
3, BaTiO
3 and SrTiO
3 (+0.38, +3.12, +2.73 and +2.12, respectively). Furthermore, the experimentally detected surface rumpling
s for the TiO
2-terminated SrTiO
3 (001) surface matrix by two different experimental methods (LEED [
132] and RHEED [
133]) is positive (
Table 5). Moreover, the agreement between our B3PW computed surface rumpling
s for the TiO
2-terminated SrTiO
3 (001) surface (+2.12) and LEED [
131] experimentally detected respective SrTiO
3 (001) surface rumpling
s (+2.1 ± 2) is almost perfect (
Table 5). Nevertheless, there is a single exception to this systematic trend. Namely, our hybrid-DFT computed surface rumpling
s for the ZrO
2-terminated SrZrO
3 (001) surface is negative (−0.72) and is in line with another ab initio calculation result performed by Wang et al. [
131] (−0.70).
Our hybrid-DFT computed B-O atom chemical bond population values in the bulk of PbTiO
3 (+0.098
e), BaTiO
3 (+0.098
e), SrTiO
3 (+0.088
e), PbZrO
3 (+0.106
e) and SrZrO
3 (+0.092
e) perovskites are considerably enhanced on their BO
2-terminated PbTiO
3 (+0.114
e), BaTiO
3 (+0.126
e), SrTiO
3 (+0.118
e), PbZrO
3 (+0.116
e) and SrZrO
3 (+0.114
e) (001) surfaces (
Table 6). Our hybrid-DFT computed BO
2- and AO-terminated (001) surface energies for PbTiO
3 (0.74 and 0.83 eV), BaTiO
3 (1.07 and 1.19 eV), SrTiO
3 (1.23 and 1.15 eV), PbZrO
3 (0.93 and 1.00 eV) and SrZrO
3 (1.24 and 1.13 eV) are comparable for both (001) surface terminations (
Table 7). Thereby, both BO
2- and AO-terminated (001) surfaces may co-exist in ABO
3 perovskite matrices. It is worthwhile to note that our hybrid-DFT computed ABO
3 perovskite (001) surface energies are always smaller than the (011) [
1,
2] and especially the (111) [
128] surface energies. This means, that, according to our hybrid-DFT computations, the ABO
3 perovskite (001) surfaces are always the most stable, whereas the (011) and especially the (111) ABO
3 perovskite surfaces are considerably less stable. Finally, our hybrid-DFT computed bulk Γ-Γ band gaps for PbTiO
3, BaTiO
3 and SrTiO
3 perovskites are in fair agreement with the experimentally available data. For BO2- and AO-terminated PbTiO
3, BaTiO
3 and SrTiO
3 (001) surfaces (
Table 8), the band gaps are always reduced with regard to the respective bulk Γ-Γ band gaps.
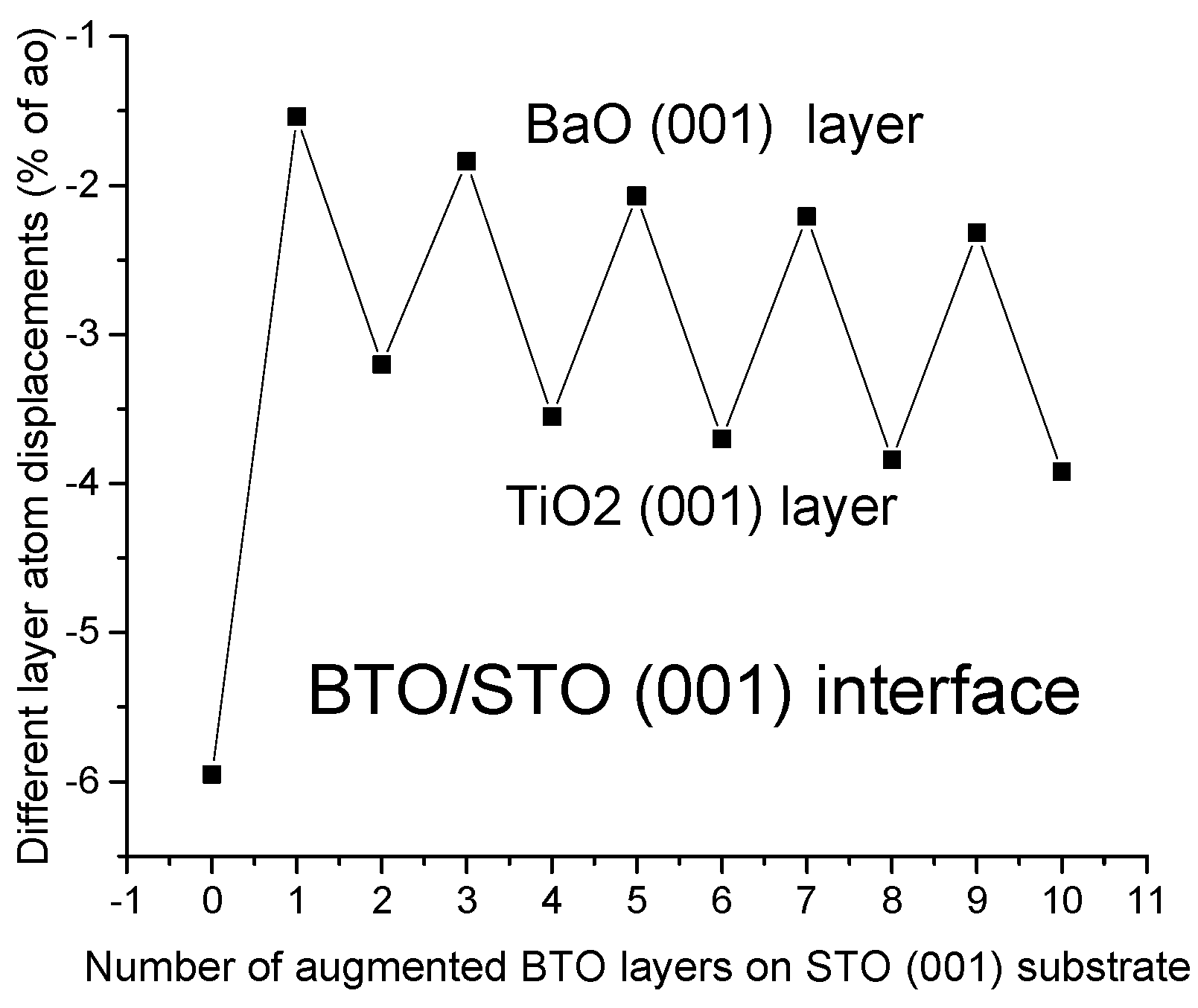
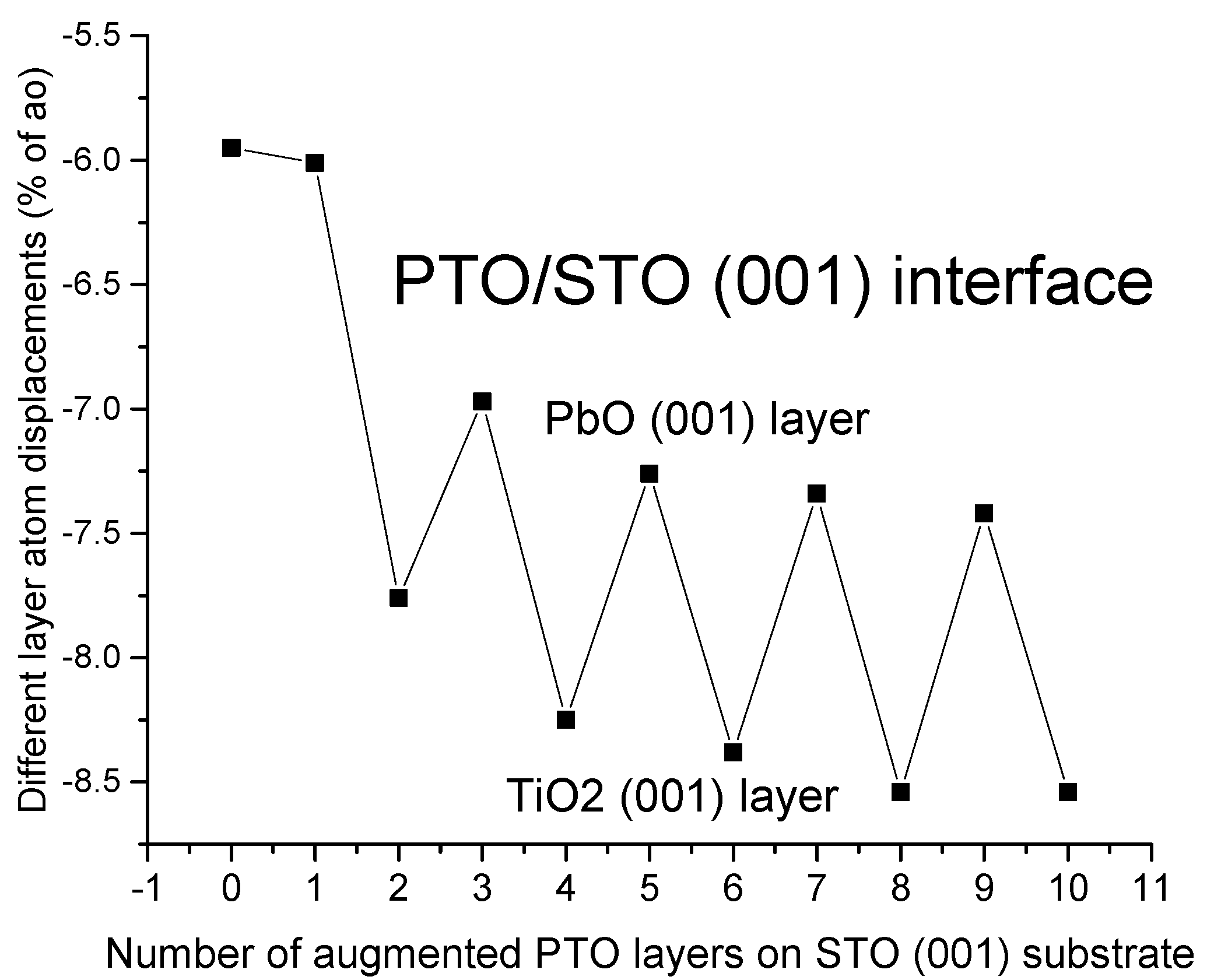
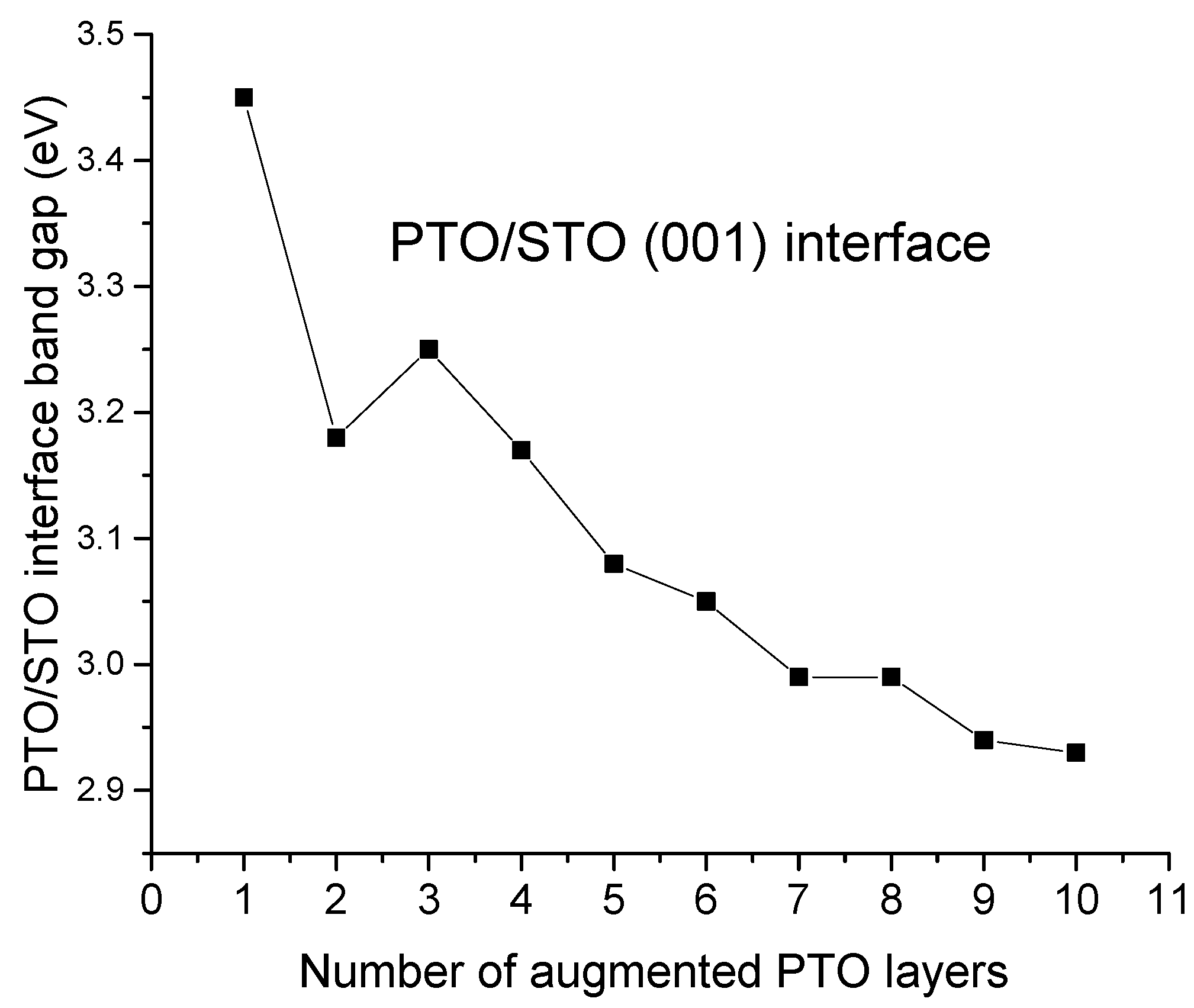
For our B3PW computed BTO/STO as well as PTO/STO (001) interfaces, the average augmented upper-layer atom relaxation magnitudes increased by the number of augmented BTO or PTO (001) layers but always independently from the number of augmented layers, which were stronger for TiO
2-terminated than BaO- or PbO-terminated upper augmented layers. All of our B3PW computed PTO/STO (001) interface upper augmented layer average atom displacement magnitudes Δ
z are between (−6.01% of
a0) for the first augmented layer and (−8.54% of
a0) for 10 augmented layers. In contrast to the PTO/STO (001) interfaces, for our hybrid-DFT computed BTO/STO (001) interfaces, the upper augmented layer average atom displacement magnitudes Δ
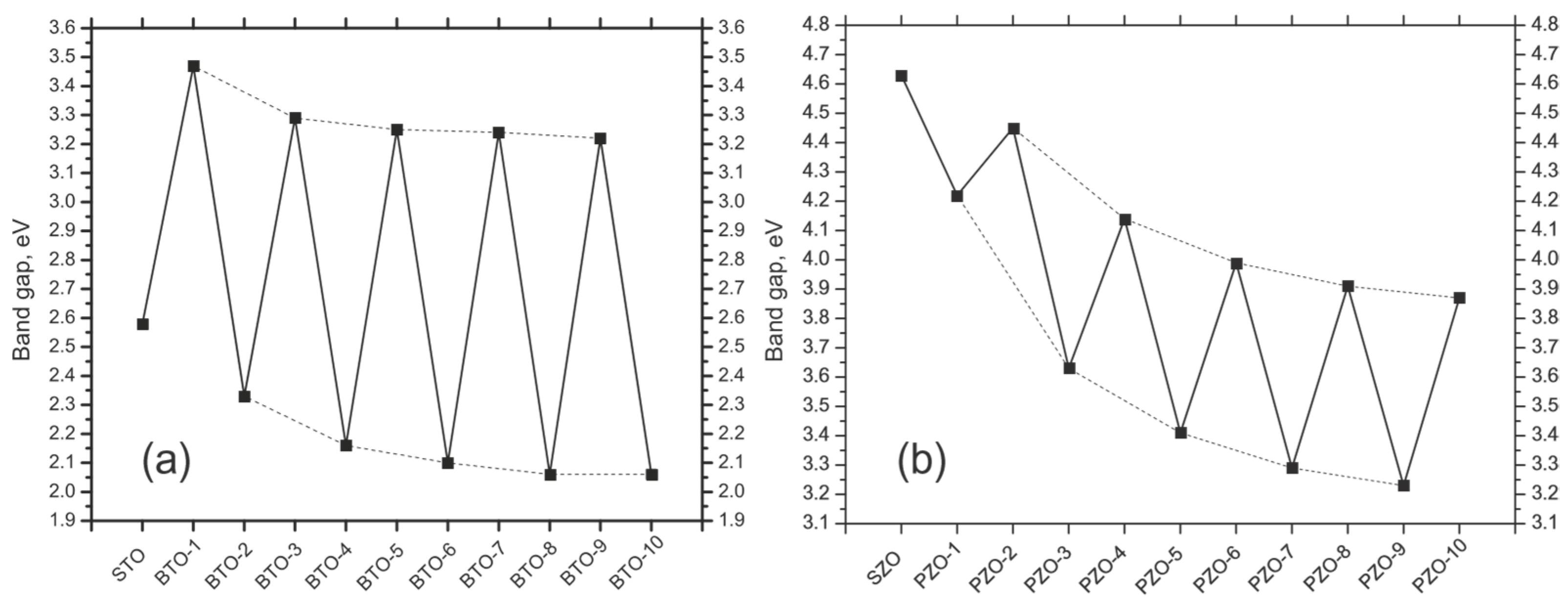
z are considerably smaller, and they are in the range between the (−1.54% of
a0 (1 layer) to −3.92% of
a0 (10 layers), respectively). Our B3PW computed BTO/STO, as well as SZO/PZO (001) interface Γ-Γ band gaps, very strongly depends on the upper augmented layer BO
2- or AO-termination but considerably less so on the number of augmented (001) layers [
135,
139].
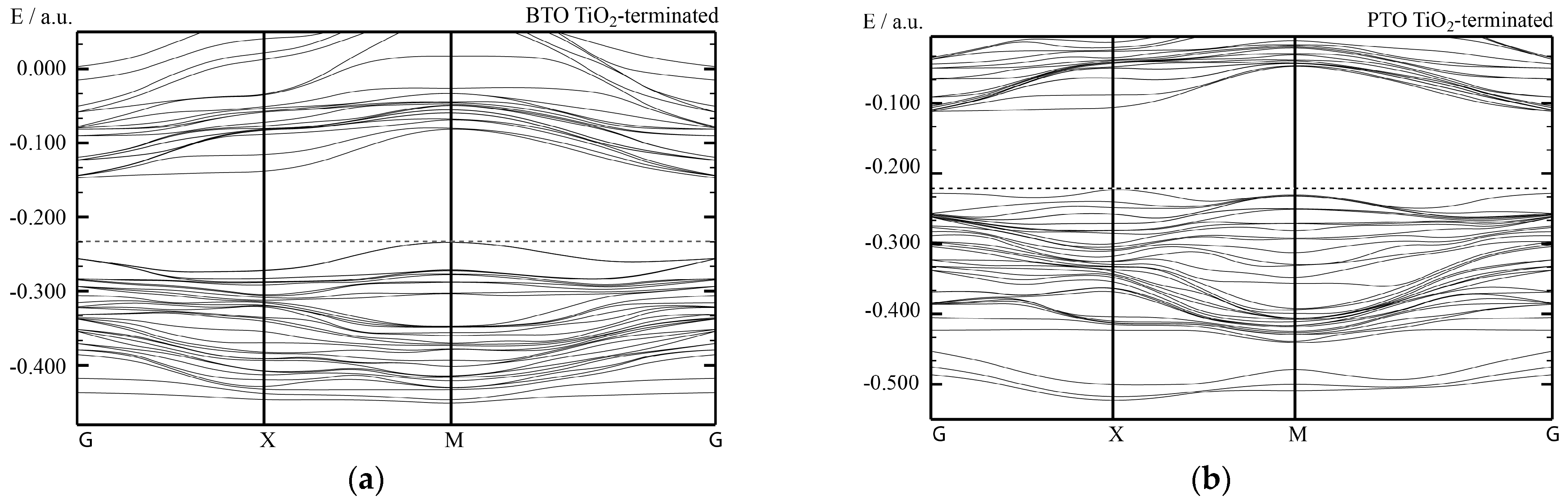
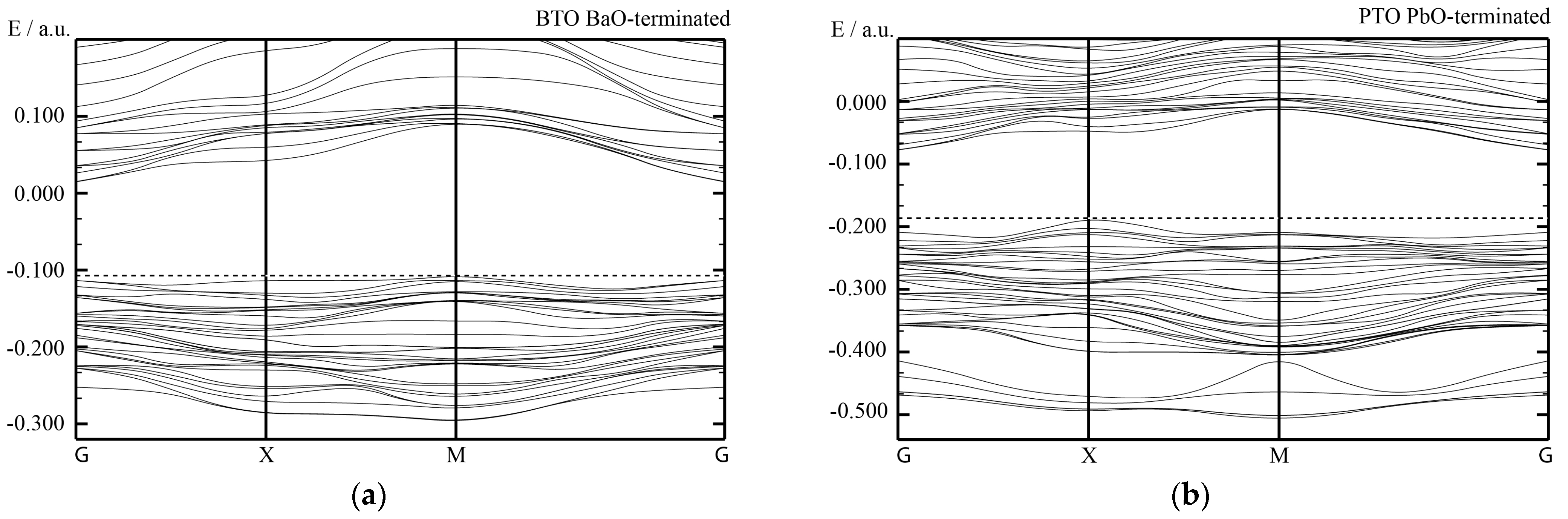
Summing up, all of our hybrid-DFT computed BTO/STO and PTO/STO, as well as PZO/SZO (001) heterostructures, are semiconducting. In general agreement with available experimental data [
84], according to our hybrid-DFT computations, the (001) interface layer does not considerably influence the electronic structure of our studied heterostructures. At the same time, the termination of the deposited BTO and PTO, as well as PZO (001), thin films atop STO or SZO (001) substrates, respectively, may shift the band edges regarding the vacuum level and thereby reduce the (001) heterostructure band gap [
84].


 ,
, 












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}