
BiGAMi: Bi-Objective Genetic Algorithm Fitness Function for Feature Selection on Microbiome Datasets




Abstract
:1. Introduction
2. Materials and Methods
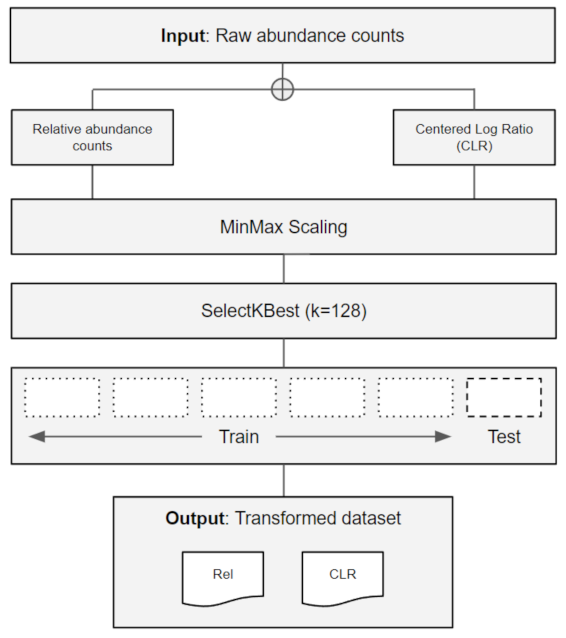
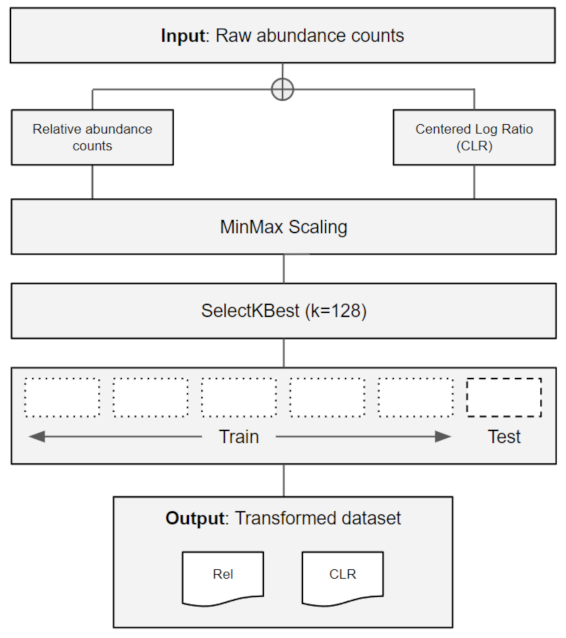
2.1. Data Retrieval and Pre-Processing
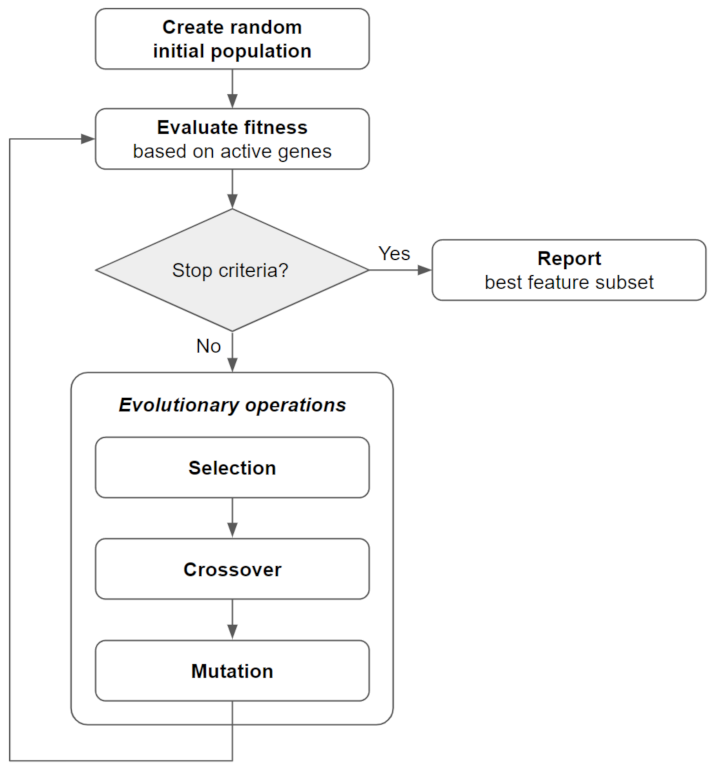
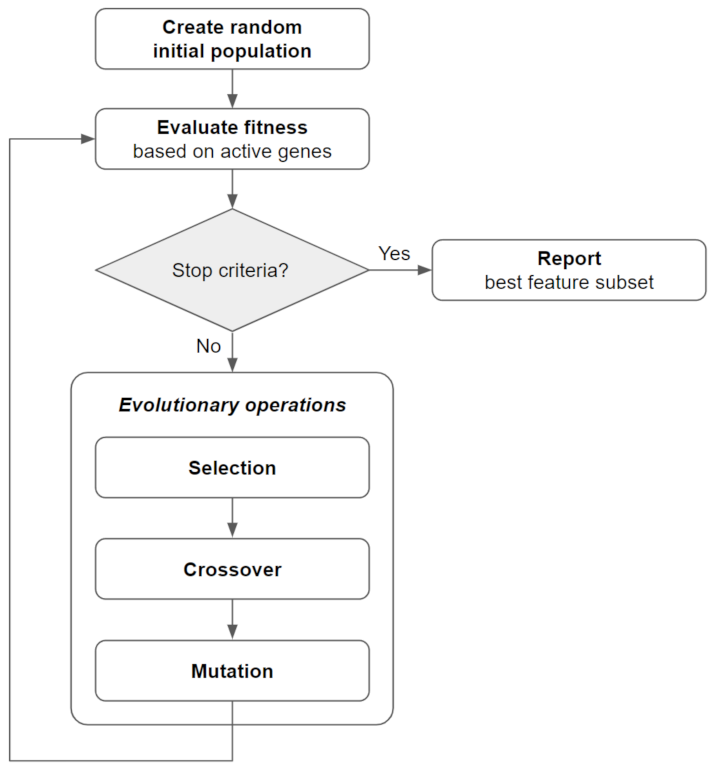
2.2. Bi-Objective Genetic Algorithm Fitness Function and Implementation (BiGAMi)
2.3. Other Feature Selection Implementations
3. Results
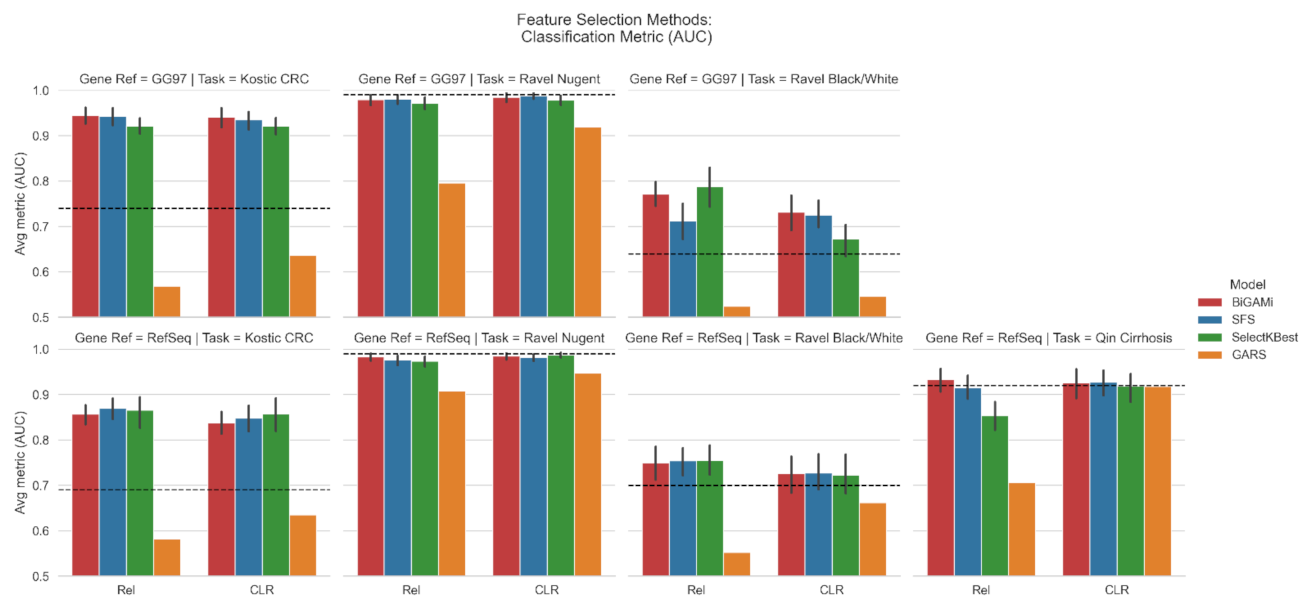
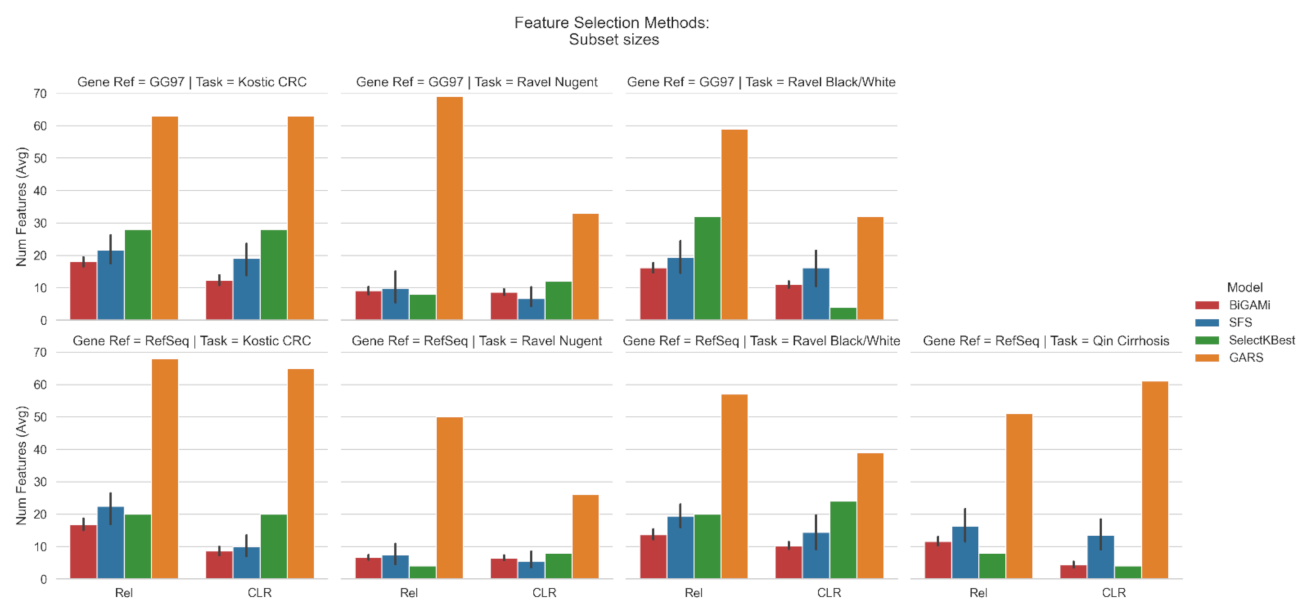
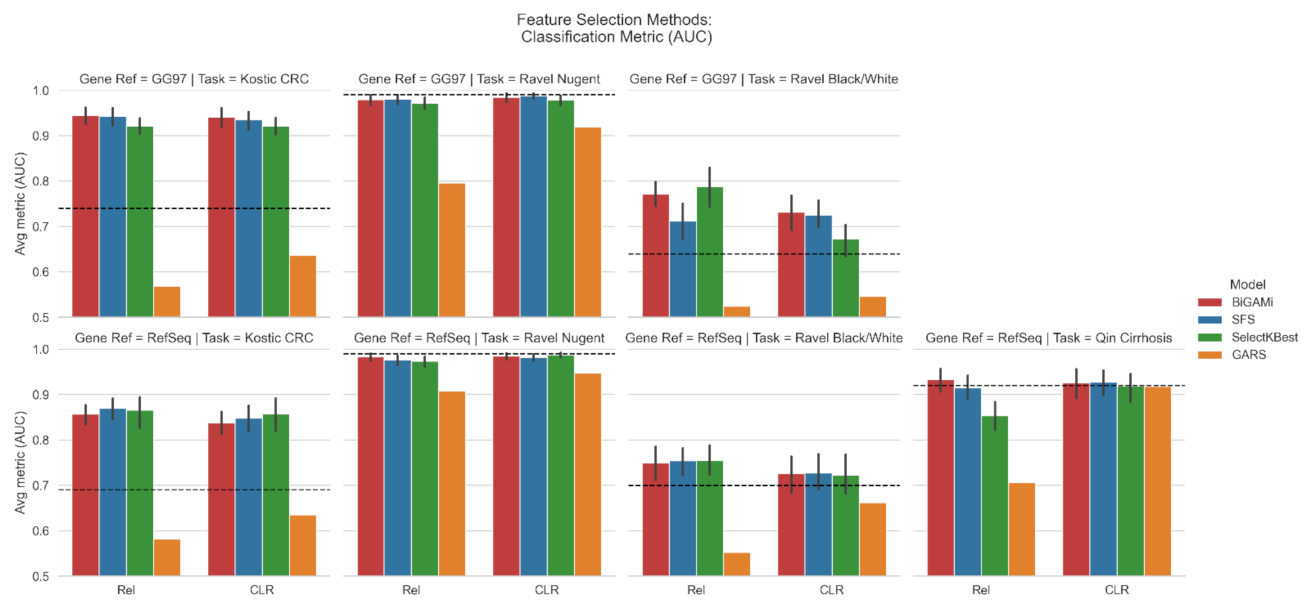
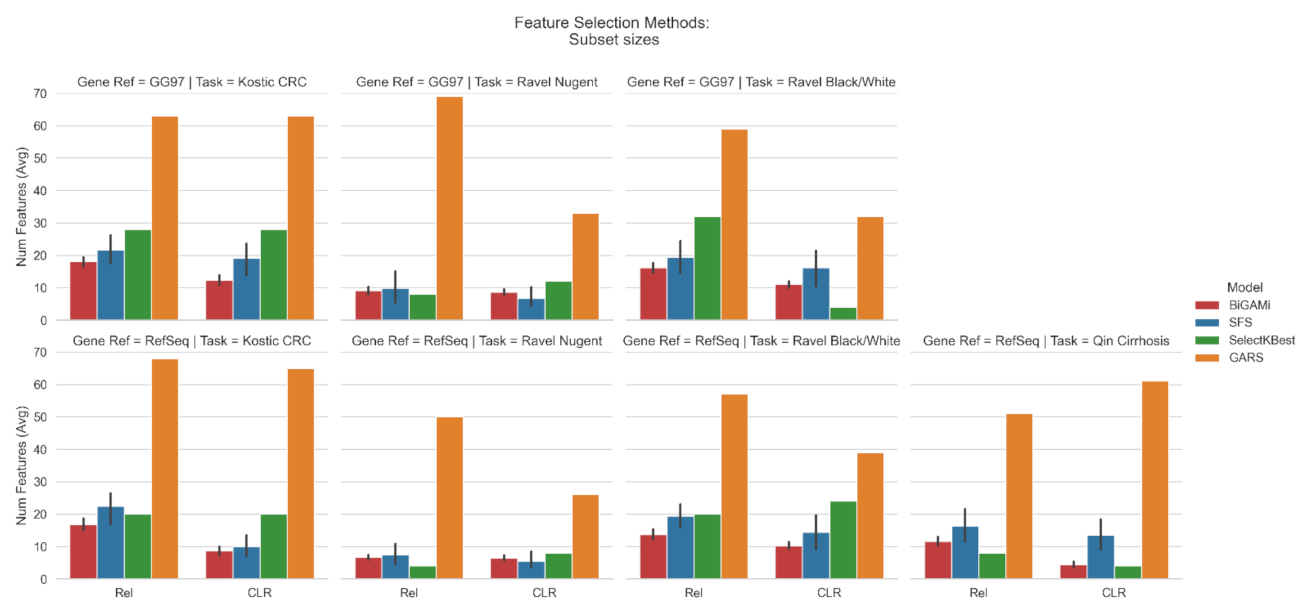
3.1. Feature Selection Using BiGAMi
3.2. Taxonomy Annotation of Feature Subsets
4. Discussion
4.1. BiGAMi Drastically Reduced Microbiome Features for Classification Tasks
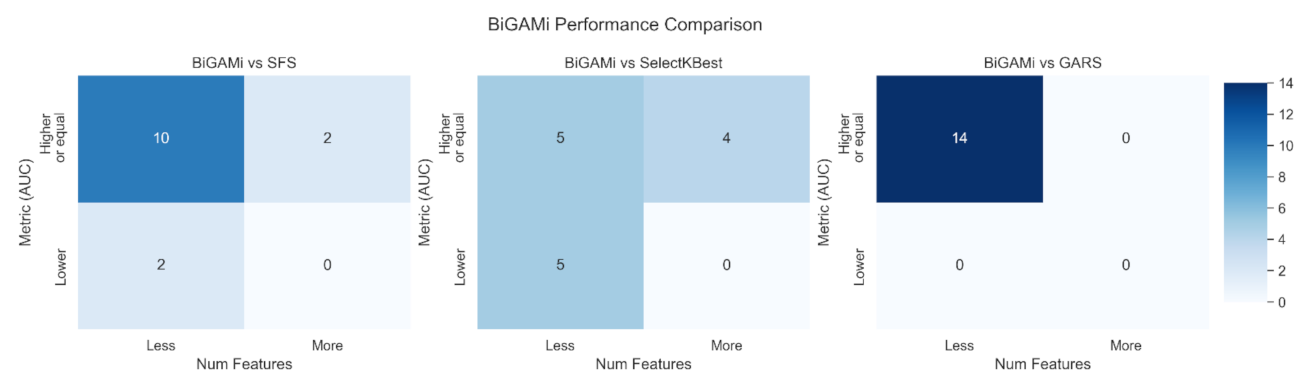
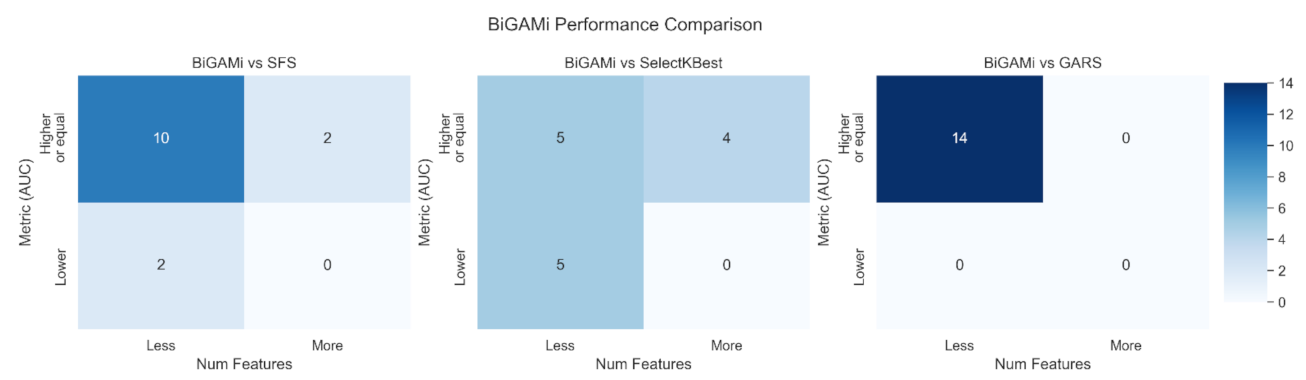
4.2. BiGAMi Outperforms Other Feature Selection Methods
4.3. BiGAMi Selects Features with Relevant Microbiological Role
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Statnikov, A.; Henaff, M.; Narendra, V.; Konganti, K.; Li, Z.; Yang, L.; Pei, Z.; Blaser, M.J.; Aliferis, C.F.; Alekseyenko, A.V. A Comprehensive Evaluation of Multicategory Classification Methods for Microbiomic Data. Microbiome 2013, 1, 11. [Google Scholar] [CrossRef] [PubMed]
- Moitinho-Silva, L.; Steinert, G.; Nielsen, S.; Hardoim, C.C.P.; Wu, Y.-C.; McCormack, G.P.; López-Legentil, S.; Marchant, R.; Webster, N.; Thomas, T.; et al. Predicting the HMA-LMA Status in Marine Sponges by Machine Learning. Front. Microbiol. 2017, 8, 752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cuadrat, R.R.C.; Sorokina, M.; Andrade, B.G.; Goris, T.; Dávila, A.M.R. Global Ocean Resistome Revealed: Exploring Antibiotic Resistance Gene Abundance and Distribution in TARA Oceans Samples. GigaScience 2020, 9, giaa046. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Fang, X.; Zhou, Y.; Dou, L.; Dou, T. Machine Learning-Based Investigation of the Relationship between Gut Microbiome and Obesity Status. Microbes Infect. 2022, 24, 104892. [Google Scholar] [CrossRef] [PubMed]
- Wirbel, J.; Zych, K.; Essex, M.; Karcher, N.; Kartal, E.; Salazar, G.; Bork, P.; Sunagawa, S.; Zeller, G. Microbiome Meta-Analysis and Cross-Disease Comparison Enabled by the SIAMCAT Machine-Learning Toolbox. Genome Biol. 2020, 22, 93. [Google Scholar] [CrossRef]
- Qin, N.; Yang, F.; Li, A.; Prifti, E.; Chen, Y.; Shao, L.; Guo, J.; Le Chatelier, E.; Yao, J.; Wu, L.; et al. Alterations of the Human Gut Microbiome in Liver Cirrhosis. Nature 2014, 513, 59–64. [Google Scholar] [CrossRef]
- Wu, H.; Cai, L.; Li, D.; Wang, X.; Zhao, S.; Zou, F.; Zhou, K. Metagenomics Biomarkers Selected for Prediction of Three Different Diseases in Chinese Population. BioMed Res. Int. 2018, 2018, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Beck, D.; Foster, J.A. Machine Learning Techniques Accurately Classify Microbial Communities by Bacterial Vaginosis Characteristics. PLoS ONE 2014, 9, e87830. [Google Scholar] [CrossRef] [Green Version]
- Tap, J.; Derrien, M.; Törnblom, H.; Brazeilles, R.; Cools-Portier, S.; Doré, J.; Störsrud, S.; Le Nevé, B.; Öhman, L.; Simrén, M. Identification of an Intestinal Microbiota Signature Associated with Severity of Irritable Bowel Syndrome. Gastroenterology 2017, 152, 111–123.e8. [Google Scholar] [CrossRef] [Green Version]
- Marcos-Zambrano, L.J.; Karaduzovic-Hadziabdic, K.; Loncar Turukalo, T.; Przymus, P.; Trajkovik, V.; Aasmets, O.; Berland, M.; Gruca, A.; Hasic, J.; Hron, K.; et al. Applications of Machine Learning in Human Microbiome Studies: A Review on Feature Selection, Biomarker Identification, Disease Prediction and Treatment. Front. Microbiol. 2021, 12, 313. [Google Scholar] [CrossRef]
- Shankar, J.; Szpakowski, S.; Solis, N.V.; Mounaud, S.; Liu, H.; Losada, L.; Nierman, W.C.; Filler, S.G. A Systematic Evaluation of High-Dimensional, Ensemble-Based Regression for Exploring Large Model Spaces in Microbiome Analyses. BMC Bioinform. 2015, 16, 31. [Google Scholar] [CrossRef] [PubMed]
- Bajaj, J.S.; Acharya, C.; Sikaroodi, M.; Gillevet, P.M.; Thacker, L.R. Cost-effectiveness of Integrating Gut Microbiota Analysis into Hospitalisation Prediction in Cirrhosis. GastroHep 2020, 2, 79–86. [Google Scholar] [CrossRef] [PubMed]
- Lopes, D.R.G.; de Souza Duarte, M.; La Reau, A.J.; Chaves, I.Z.; de Oliveira Mendes, T.A.; Detmann, E.; Bento, C.B.P.; Mercadante, M.E.Z.; Bonilha, S.F.M.; Suen, G.; et al. Assessing the Relationship between the Rumen Microbiota and Feed Efficiency in Nellore Steers. J. Anim. Sci. Biotechnol. 2021, 12, 79. [Google Scholar] [CrossRef] [PubMed]
- Andrade, B.G.N.; Bressani, F.A.; Cuadrat, R.R.C.; Tizioto, P.C.; Oliveira, P.S.N.D.; Mourão, G.B.; Coutinho, L.L.; Reecy, J.M.; Koltes, J.E.; Walsh, P.; et al. The Structure of Microbial Populations in Nelore GIT Reveals Inter-Dependency of Methanogens in Feces and Rumen. J. Anim. Sci. Biotechnol. 2020, 11, 1–10. [Google Scholar] [CrossRef]
- Bashiardes, S.; Zilberman-Schapira, G.; Elinav, E. Use of Metatranscriptomics in Microbiome Research. Bioinform. Biol. Insights 2016, 10, 19–25. [Google Scholar] [CrossRef] [Green Version]
- Long, S.; Yang, Y.; Shen, C.; Wang, Y.; Deng, A.; Qin, Q.; Qiao, L. Metaproteomics Characterizes Human Gut Microbiome Function in Colorectal Cancer. NPJ Biofilms Microbiomes 2020, 6, 14. [Google Scholar] [CrossRef]
- Bellman, R.E. Adaptive Control Processes: A Guided Tour; Princeton University Press: Princeton, NJ, USA, 1961; ISBN 978-1-4008-7466-8. [Google Scholar]
- Oh, M.; Zhang, L. DeepMicro: Deep Representation Learning for Disease Prediction Based on Microbiome Data. Sci. Rep. 2020, 10, 6026. [Google Scholar] [CrossRef] [Green Version]
- Bang, S.; Yoo, D.; Kim, S.-J.; Jhang, S.; Cho, S.; Kim, H. Establishment and Evaluation of Prediction Model for Multiple Disease Classification Based on Gut Microbial Data. Sci. Rep. 2019, 9, 10189. [Google Scholar] [CrossRef] [Green Version]
- Dorado-Morales, P.; Vilanova, C.; Garay, C.P.; Martí, J.M.; Porcar, M. Unveiling Bacterial Interactions through Multidimensional Scaling and Dynamics Modeling. Sci. Rep. 2015, 5, 18396. [Google Scholar] [CrossRef] [Green Version]
- Leong, C.; Haszard, J.J.; Heath, A.-L.M.; Tannock, G.W.; Lawley, B.; Cameron, S.L.; Szymlek-Gay, E.A.; Gray, A.R.; Taylor, B.J.; Galland, B.C.; et al. Using Compositional Principal Component Analysis to Describe Children’s Gut Microbiota in Relation to Diet and Body Composition. Am. J. Clin. Nutr. 2019, 111, nqz270. [Google Scholar] [CrossRef]
- Segata, N.; Izard, J.; Waldron, L.; Gevers, D.; Miropolsky, L.; Garrett, W.S.; Huttenhower, C. Metagenomic Biomarker Discovery and Explanation. Genome Biol. 2011, 12, R60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Albadr, M.A.; Tiun, S.; Ayob, M.; AL-Dhief, F. Genetic Algorithm Based on Natural Selection Theory for Optimization Problems. Symmetry 2020, 12, 1758. [Google Scholar] [CrossRef]
- Holland, J.H. Adaptation in Natural and Artificial Systems: An Introductory Analysis with Applications to Biology, Control, and Artificial Intelligence; Complex Adaptive Systems, 1st ed.; MIT Press: Cambridge, MA, USA, 1992; ISBN 978-0-262-08213-6. [Google Scholar]
- Talbi, E.-G. Metaheuristics: From Design to Implementation; Wiley: Hoboken, NJ, USA, 2009; ISBN 978-0-470-27858-1. [Google Scholar]
- Carter, J.; Beck, D.; Williams, H.; Dozier, G.; Foster, J.A. GA-Based Selection of Vaginal Microbiome Features Associated with Bacterial Vaginosis. In Proceedings of the 2014 Annual Conference on Genetic and Evolutionary Computation, Vancouver, BC, Canada, 12–16 July 2014; ACM: Vancouver, BC, Canada, 2014; pp. 265–268. [Google Scholar]
- Chiesa, M.; Maioli, G.; Colombo, G.I.; Piacentini, L. GARS: Genetic Algorithm for the Identification of a Robust Subset of Features in High-Dimensional Datasets. BMC Bioinform. 2020, 21, 54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, P.; West, N.P.; Chen, P.-Y.; Thang, M.W.C.; Price, G.; Cripps, A.W.; Cox, A.J. Selection of Microbial Biomarkers with Genetic Algorithm and Principal Component Analysis. BMC Bioinform. 2019, 20, 413. [Google Scholar] [CrossRef] [PubMed]
- Vangay, P.; Hillmann, B.M.; Knights, D. Microbiome Learning Repo (ML Repo): A Public Repository of Microbiome Regression and Classification Tasks. GigaScience 2019, 8, giz042. [Google Scholar] [CrossRef]
- Kostic, A.D.; Gevers, D.; Pedamallu, C.S.; Michaud, M.; Duke, F.; Earl, A.M.; Ojesina, A.I.; Jung, J.; Bass, A.J.; Tabernero, J.; et al. Genomic Analysis Identifies Association of Fusobacterium with Colorectal Carcinoma. Genome Res. 2012, 22, 292–298. [Google Scholar] [CrossRef] [Green Version]
- Ravel, J.; Gajer, P.; Abdo, Z.; Schneider, G.M.; Koenig, S.S.K.; McCulle, S.L.; Karlebach, S.; Gorle, R.; Russell, J.; Tacket, C.O.; et al. Vaginal Microbiome of Reproductive-Age Women. Proc. Natl. Acad. Sci. USA 2011, 108, 4680–4687. [Google Scholar] [CrossRef] [Green Version]
- McDonald, D.; Price, M.N.; Goodrich, J.; Nawrocki, E.P.; DeSantis, T.Z.; Probst, A.; Andersen, G.L.; Knight, R.; Hugenholtz, P. An Improved Greengenes Taxonomy with Explicit Ranks for Ecological and Evolutionary Analyses of Bacteria and Archaea. ISME J. 2012, 6, 610–618. [Google Scholar] [CrossRef]
- O’Leary, N.A.; Wright, M.W.; Brister, J.R.; Ciufo, S.; Haddad, D.; McVeigh, R.; Rajput, B.; Robbertse, B.; Smith-White, B.; Ako-Adjei, D.; et al. Reference Sequence (RefSeq) Database at NCBI: Current Status, Taxonomic Expansion, and Functional Annotation. Nucleic Acids Res. 2016, 44, D733–D745. [Google Scholar] [CrossRef] [Green Version]
- Pedregosa, F.; Varoquaux, G.; Gramfort, A.; Michel, V.; Thirion, B.; Grisel, O.; Blondel, M.; Prettenhofer, P.; Weiss, R.; Dubourg, V.; et al. Scikit-Learn: Machine Learning in Python. J. Mach. Learn. Res. 2011, 12, 2825–2830. [Google Scholar]
- De Rainville, F.-M.; Fortin, F.-A.; Gardner, M.-A.; Parizeau, M.; Gagné, C. DEAP: A Python Framework for Evolutionary Algorithms. In Proceedings of the Fourteenth International Conference on Genetic and Evolutionary Computation Conference Companion—GECCO Companion ’12, Philadelphia, PA, USA, 7–11 July 2012; ACM Press: Philadelphia, PA, USA, 2012; p. 85. [Google Scholar]
- Ferri, F.J.; Pudil, P.; Hatef, M.; Kittler, J. Comparative Study of Techniques for Large-Scale Feature Selection. Mach. Intell. Pattern Recognit. 1994, 16, 403–413. [Google Scholar]
- Gloor, G.B.; Macklaim, J.M.; Pawlowsky-Glahn, V.; Egozcue, J.J. Microbiome Datasets Are Compositional: And This Is Not Optional. Front. Microbiol. 2017, 8, 2224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Praus, P. Robust Multivariate Analysis of Compositional Data of Treated Wastewaters. Environ. Earth Sci. 2019, 78, 248. [Google Scholar] [CrossRef]
- Van den Boogaart, K.G.; Tolosana-Delgado, R. Analyzing Compositional Data with R. In Analyzing Compositional Data with R; Springer: Berlin/Heidelberg, Germany, 2013; pp. 95–175. [Google Scholar]
- Mallick, H.; Rahnavard, A.; McIver, L.J.; Ma, S.; Zhang, Y.; Nguyen, L.H.; Tickle, T.L.; Weingart, G.; Ren, B.; Schwager, E.H.; et al. Multivariable Association Discovery in Population-Scale Meta-Omics Studies. PLoS Comput. Biol. 2021, 17, e1009442. [Google Scholar] [CrossRef]
- Mandal, S.; Van Treuren, W.; White, R.A.; Eggesbø, M.; Knight, R.; Peddada, S.D. Analysis of Composition of Microbiomes (ANCOM): A Novel Method for Studying Microbial Composition. Microb. Ecol. Health Dis. 2015, 26, 27663. [Google Scholar]
- Delgado, R.T.; Talebi, H.; Khodadadzadeh, M.; Boogaart, K.G. van den On Machine Learning Algorithms and Compositional Data. In Proceedings of the 8th International Workshop on Compositional Data Analysis (CoDaWork2019), Terrassa, Spain, 3–8 June 2019; Universitat Politècnica de Catalunya: Barcelona, Spain, 2019; pp. 172–175, ISBN 978-84-947240-2-2. [Google Scholar]
- Michel-Mata, S.; Wang, X.-W.; Liu, Y.-Y.; Angulo, M.T. Predicting Microbiome Compositions from Species Assemblages through Deep Learning. iMeta 2022, 1, e3. [Google Scholar] [CrossRef]
- Tepanosyan, G.; Sahakyan, L.; Maghakyan, N.; Saghatelyan, A. Combination of Compositional Data Analysis and Machine Learning Approaches to Identify Sources and Geochemical Associations of Potentially Toxic Elements in Soil and Assess the Associated Human Health Risk in a Mining City. Environ. Pollut. 2020, 261, 114210. [Google Scholar] [CrossRef]
- Zhong, M.; Xiong, Y.; Ye, Z.; Zhao, J.; Zhong, L.; Liu, Y.; Zhu, Y.; Tian, L.; Qiu, X.; Hong, X. Microbial Community Profiling Distinguishes Left-Sided and Right-Sided Colon Cancer. Front. Cell. Infect. Microbiol. 2020, 10, 498502. [Google Scholar] [CrossRef]
- Gao, R.; Gao, Z.; Huang, L.; Qin, H. Gut Microbiota and Colorectal Cancer. Eur. J. Clin. Microbiol. Infect. Dis. 2017, 36, 757–769. [Google Scholar] [CrossRef]
- Yang, J.; McDowell, A.; Kim, E.K.; Seo, H.; Lee, W.H.; Moon, C.-M.; Kym, S.-M.; Lee, D.H.; Park, Y.S.; Jee, Y.-K.; et al. Development of a Colorectal Cancer Diagnostic Model and Dietary Risk Assessment through Gut Microbiome Analysis. Exp. Mol. Med. 2019, 51, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Flemer, B.; Lynch, D.B.; Brown, J.M.R.; Jeffery, I.B.; Ryan, F.J.; Claesson, M.J.; O’Riordain, M.; Shanahan, F.; O’Toole, P.W. Tumour-Associated and Non-Tumour-Associated Microbiota in Colorectal Cancer. Gut 2017, 66, 633–643. [Google Scholar] [CrossRef] [PubMed]
- Xu, K.; Jiang, B. Analysis of Mucosa-Associated Microbiota in Colorectal Cancer. Med. Sci. Monit. 2017, 23, 4422–4430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chee, W.J.Y.; Chew, S.Y.; Than, L.T.L. Vaginal Microbiota and the Potential of Lactobacillus Derivatives in Maintaining Vaginal Health. Microb. Cell Fact. 2020, 19, 203. [Google Scholar] [CrossRef] [PubMed]
- Morrill, S.; Gilbert, N.M.; Lewis, A.L. Gardnerella Vaginalis as a Cause of Bacterial Vaginosis: Appraisal of the Evidence from In Vivo Models. Front. Cell. Infect. Microbiol. 2020, 10, 168. [Google Scholar] [CrossRef]
- Diop, K.; Dufour, J.-C.; Levasseur, A.; Fenollar, F. Exhaustive Repertoire of Human Vaginal Microbiota. Hum. Microbiome J. 2019, 11, 100051. [Google Scholar] [CrossRef]
- Fettweis, J.M.; Brooks, J.P.; Serrano, M.G.; Sheth, N.U.; Girerd, P.H.; Edwards, D.J.; Strauss, J.F.; the Vaginal Microbiome Consortium; Jefferson, K.K.; Buck, G.A. Differences in Vaginal Microbiome in African American Women versus Women of European Ancestry. Microbiology 2014, 160, 2272–2282. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Ji, F.; Guo, J.; Shi, D.; Fang, D.; Li, L. Dysbiosis of Small Intestinal Microbiota in Liver Cirrhosis and Its Association with Etiology. Sci. Rep. 2016, 6, 34055. [Google Scholar] [CrossRef]
- Yang, L.; Bian, X.; Wu, W.; Lv, L.; Li, Y.; Ye, J.; Jiang, X.; Wang, Q.; Shi, D.; Fang, D.; et al. Protective Effect of Lactobacillus Salivarius Li01 on Thioacetamide-induced Acute Liver Injury and Hyperammonaemia. Microb. Biotechnol. 2020, 13, 1860–1876. [Google Scholar] [CrossRef]
- Jensen, A.; Grønkjær, L.L.; Holmstrup, P.; Vilstrup, H.; Kilian, M. Unique Subgingival Microbiota Associated with Periodontitis in Cirrhosis Patients. Sci. Rep. 2018, 8, 10718. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Task I | Task II | Task III | Task IV | |
|---|---|---|---|---|
| Dataset | Kostic [30] | Ravel [31] | Ravel [31] | Qin [6] |
| Year | 2012 | 2011 | 2011 | 2014 |
| Description | Healthy vs. Tumor Colon Biopsy Tissues | Low vs. High Vaginal Nugent Score | Black vs. White phenotype classification | Cirrhosis vs. healthy |
| Topic area | Colorectal Cancer | Vaginal | Vaginal | Cirrhosis |
| Classification targets | Healthy, Tumor | Low, High | Black, White | Cirrhosis, Healthy |
| Number of samples | 190 | 342 | 200 | 130 |
| Number of subjects | 95 | 342 | 200 | 130 |
| Number of OTUs GG97 | 3228 | 1093 | 1093 | n/a |
| Number of OTUs RefSeq | 908 | 586 | 586 | 8483 |
| Parameter | Description | Value |
|---|---|---|
| n_searches | Number of individual GA runs | 25 |
| pop_size | GA population size | 250 |
| max_iter | Number of GA iterations/generations | 10 |
| bestN | Elitism concept | 1 |
| crossover | Crossover strategy | 1p (One-point) |
| CXPB | Crossover probability | 0.8 |
| MUPB | Mutation probability | 0.8 |
| init | GA individual initialization strategy | zero |
| init_ind_length | Average number of enabled GA individual chromosomes | 10 |
| select | GA crossover selection strategy | Tournament (size = 3) |
| mutate | GA mutation operation | mutFlipOne (custom) |
| Task | Database | Total OTUs | Baseline AUC | Input Data | SGD AUC | SFS AUC/OTUs | SelectKBest AUC/OTUs | GARS AUC/OTUs | BiGAMi AUC/OTUs (Ours) |
|---|---|---|---|---|---|---|---|---|---|
| (I) | GG97 | 3228 | 0.74 | Rel | 0.85 | 0.94/21.6 | 0.92/28 | 0.57/63 | 0.95/18.1 |
| CLR | 0.9 | 0.94/19.0 | 0.92/28 | 0.64/63 | 0.94/12.3 | ||||
| RefSeq | 908 | 0.69 | Rel | 0.8 | 0.87/22.4 | 0.87/20 | 0.58/68 | 0.86/16.8 | |
| CLR | 0.85 | 0.85/10.0 | 0.86/20 | 0.64/65 | 0.84/8.2 | ||||
| (II) | GG97 | 1093 | 0.99 | Rel | 0.96 | 0.98/9.8 | 0.97/8 | 0.80/69 | 0.98/9.2 |
| CLR | 0.97 | 0.99/6.6 | 0.98/12 | 0.92/33 | 0.99/8.6 | ||||
| RefSeq | 586 | 0.99 | Rel | 0.96 | 0.98/7.4 | 0.97/4 | 0.91/50 | 0.98/6.6 | |
| CLR | 0.96 | 0.98/5.5 | 0.99/8 | 0.95/26 | 0.98/6.5 | ||||
| (III) | GG97 | 1093 | 0.64 | Rel | 0.65 | 0.71/19.4 | 0.79/32 | 0.52/59 | 0.77/16.2 |
| CLR | 0.63 | 0.73/16.2 | 0.67/4 | 0.55/32 | 0.73/11.1 | ||||
| RefSeq | 586 | 0.7 | Rel | 0.6 | 0.75/19.4 | 0.76/20 | 0.55/57 | 0.75/13.6 | |
| CLR | 0.61 | 0.73/14.4 | 0.72/24 | 0.62/39 | 0.73/10.2 | ||||
| (IV) | RefSeq | 8483 | 0.92 | Rel | 0.82 | 0.92/16.4 | 0.85/8 | 0.71/51 | 0.93/11.6 |
| CLR | 0.83 | 0.93/13.5 | 0.92/4 | 0.92/61 | 0.93/4.3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Leske, M.; Bottacini, F.; Afli, H.; Andrade, B.G.N. BiGAMi: Bi-Objective Genetic Algorithm Fitness Function for Feature Selection on Microbiome Datasets. Methods Protoc. 2022, 5, 42. https://doi.org/10.3390/mps5030042
Leske M, Bottacini F, Afli H, Andrade BGN. BiGAMi: Bi-Objective Genetic Algorithm Fitness Function for Feature Selection on Microbiome Datasets. Methods and Protocols. 2022; 5(3):42. https://doi.org/10.3390/mps5030042
Chicago/Turabian StyleLeske, Mike, Francesca Bottacini, Haithem Afli, and Bruno G. N. Andrade. 2022. "BiGAMi: Bi-Objective Genetic Algorithm Fitness Function for Feature Selection on Microbiome Datasets" Methods and Protocols 5, no. 3: 42. https://doi.org/10.3390/mps5030042
APA StyleLeske, M., Bottacini, F., Afli, H., & Andrade, B. G. N. (2022). BiGAMi: Bi-Objective Genetic Algorithm Fitness Function for Feature Selection on Microbiome Datasets. Methods and Protocols, 5(3), 42. https://doi.org/10.3390/mps5030042