1. Introduction
Alpha-hemoglobin-stabilizing protein (AHSP) is a molecular chaperone protein that sequesters free alpha-hemoglobin (
αHb). AHSP was first experimentally measured by the Clinton lab in 2001 [
1] followed by the seminal report elucidating its functional role in 2002 by the Weiss lab [
2]. During the formation of the hemoglobin tetramer, or hemoglobinization, two units of both
αHb and beta-hemoglobin (βHb) must coordinate bond with a ferrous iron (Fe) prosthetic group in order to create a functional hemoglobin protein. Unlike βHb, free
αHb can neither bind to a heme nor self-associate, but instead aggregates onto the cell membrane.
αHb precipitation results in apoptosis of immature erythroid cells and reduced lifespan of erythrocytes [
3,
4].
Due to the high tissue specificity of hemoglobin (Hb), the body’s primary means of transporting and utilizing oxygen, AHSP is predominantly expressed in hematopoietic tissues and erythroid progenitors [
3]. Much of AHSP’s function was studied in hereditary anemia; it has not been considered in other anemia such as anemia of inflammation/critical illness, which is refractory to erythropoietin administration [
5,
6]. Our lab has found implications of AHSP in burn patients during late stage erythropoiesis (manuscript under review) while investigating peripheral blood mononuclear cells (PBMC) derived erythroblasts in ex vivo cultures [
7]. The pattern of AHSP protein expression during erythropoiesis is not fully understood, but gene expression during erythroid maturation is reported by Dos Santos at al. [
8]. Thus far, little is known regarding AHSP as it is still a relatively new protein in the field of erythropoiesis. In this methodological study we have portrayed a stage specific expression pattern of AHSP in erythroblast chronology. Results underscore AHSP gene expression, protein expression and localization over its developmental trajectory in ex vivo culturing from human bone marrow cells and human PBMCs. For validation, we have also compared our ex vivo observations with in vivo early and late stage erythroblasts obtained from murine bone marrow.
3. Discussion
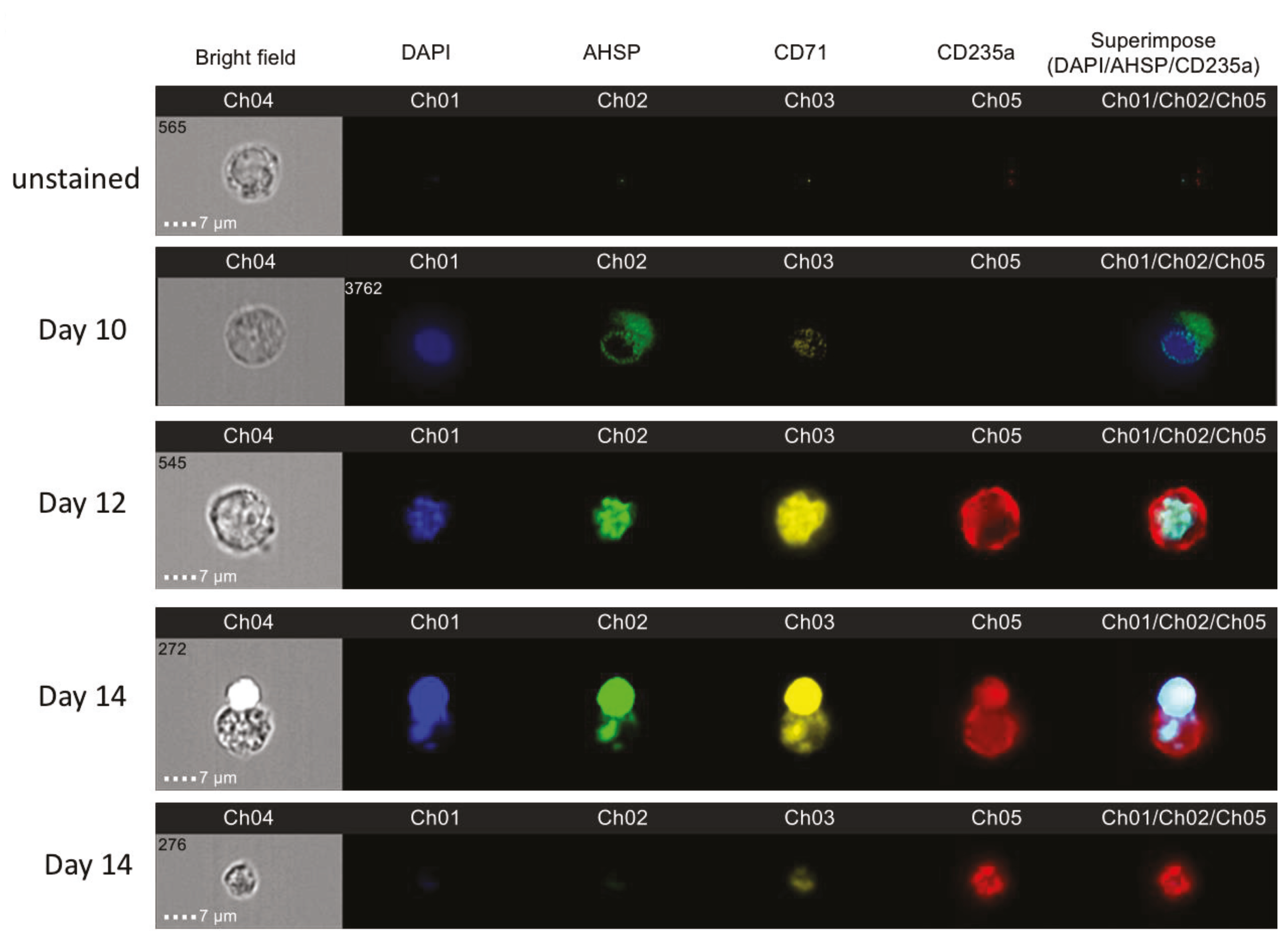
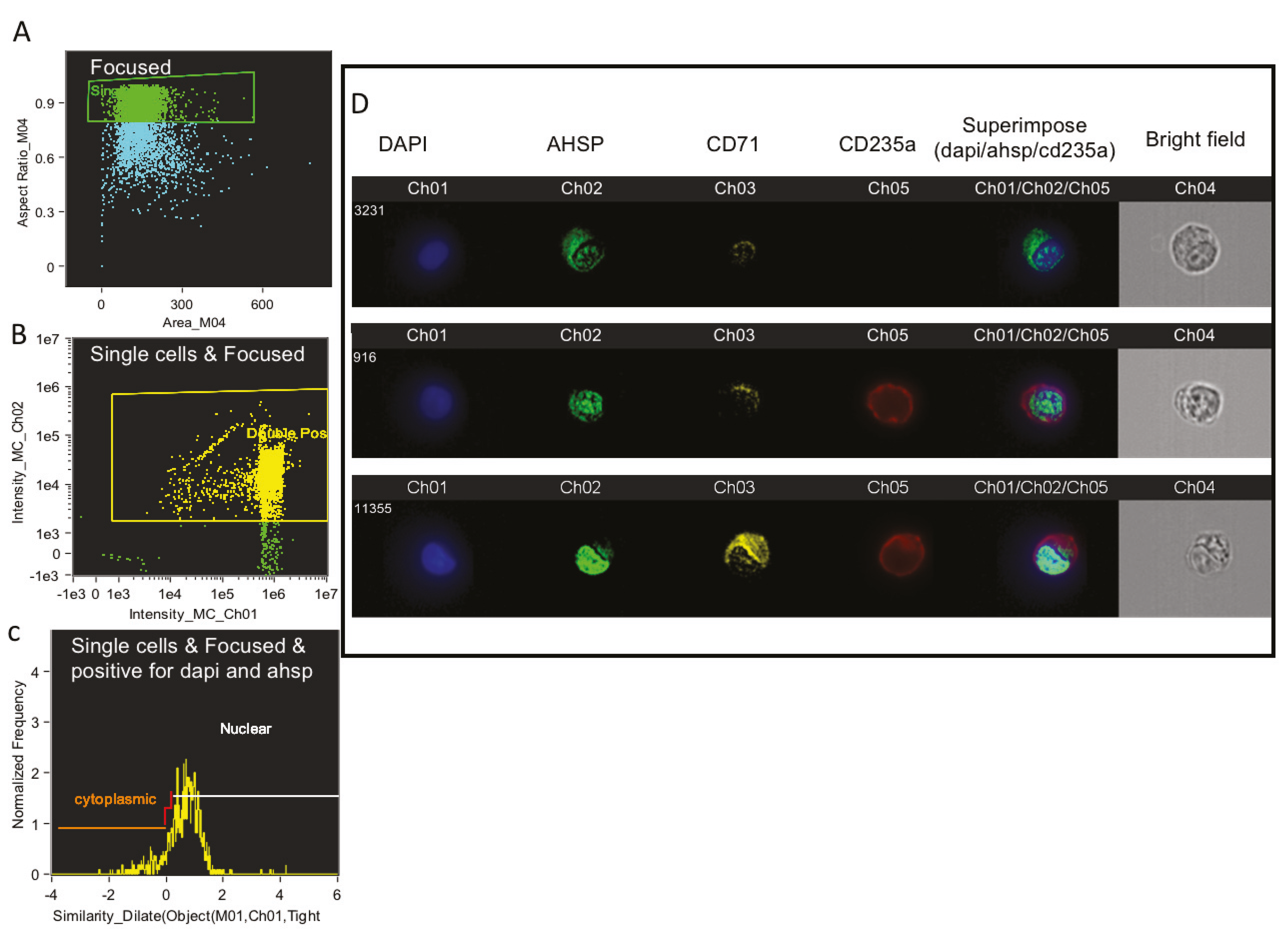
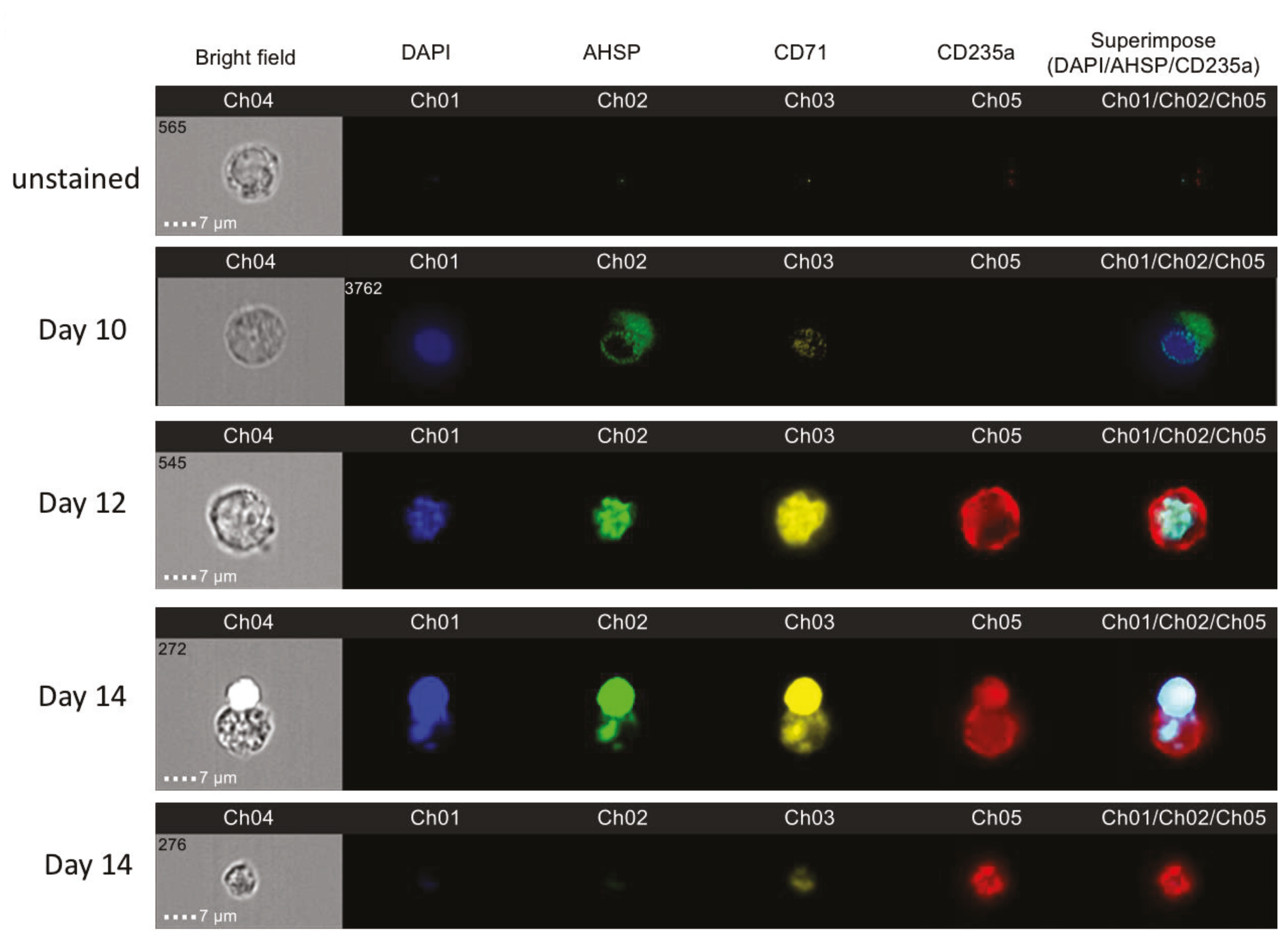
We identified AHSP protein expression throughout erythropoiesis initially present in the cytoplasm and then culminating in the nucleus. With the use of imaging flow cytometry, we were able to provide evidence for the visual localization of AHSP throughout ex vivo red blood cell development. Remarkably, during erythroblast development using biphasic cell culturing, AHSP did not remain in its original localization, the cytoplasm. Instead, it began to translocate from the cytoplasm to the nucleus (on day 12) and eventually all of the AHSP was colocalized to the nucleus (on day 14). To our knowledge this is the first publication depicting AHSP protein expression from two different sources namely naïve bone marrow-derived and PBMC-derived erythroblasts. Moreover, we concurrently validated these ex vivo contrived observations in in vivo murine total bone marrow cells and confirmed that among nucleated erythroblasts, late erythroblasts expressed higher protein and gene expression for AHSP compared to early erythroblasts.
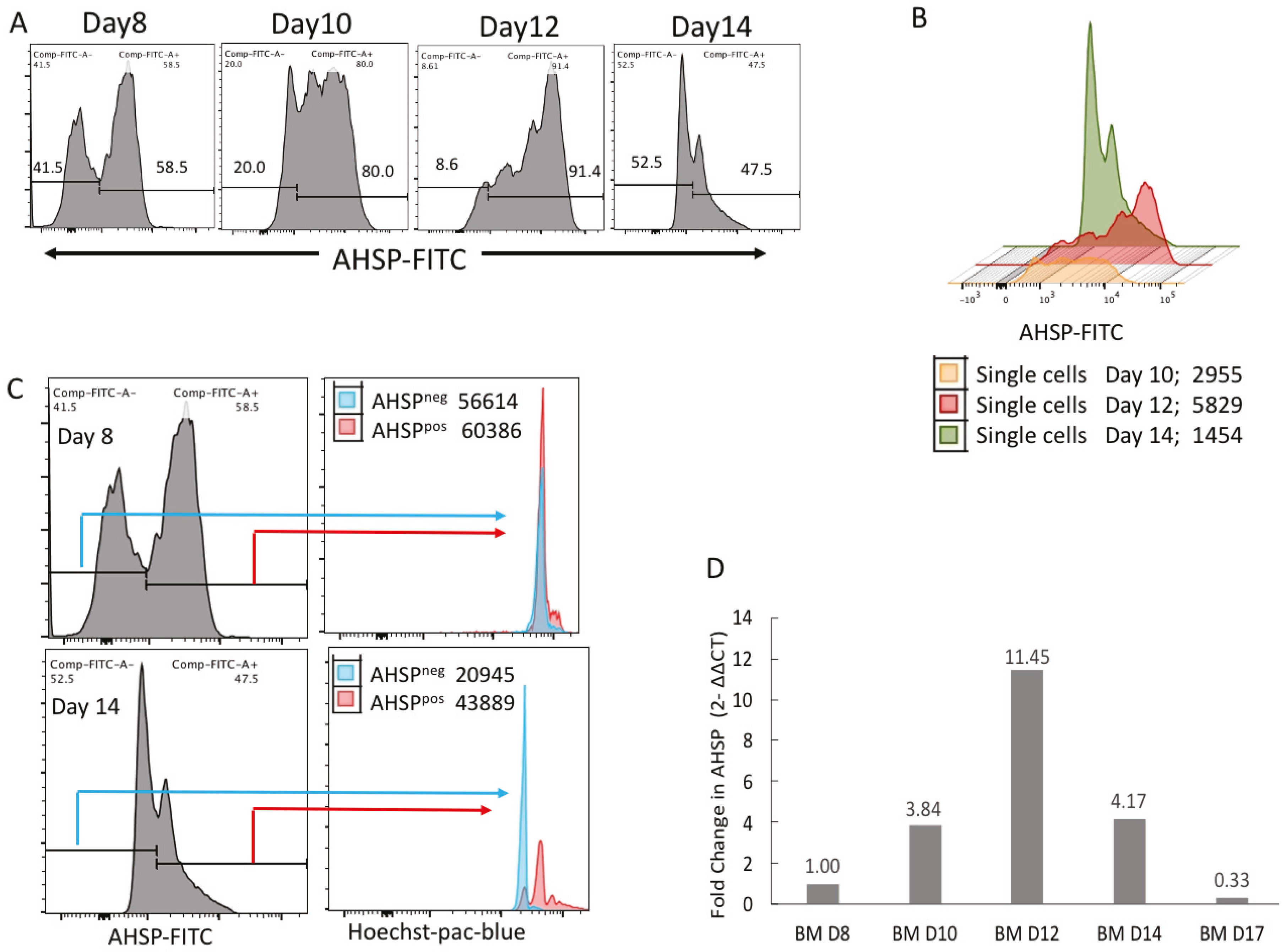
Examination of erythroblasts from bi phasic culture day 8 through culture day 14 illustrated in this manuscript was preceded by a preliminary analysis of HuBM obtained from four adult donor samples. Flow cytometry results depicting the percentage of AHSP
+ cells (mean ± SEM) from culture day 9 and day 11 are shown in bar graphs in the
Supplemental Figure S1. Based on this preliminary reproducible observation, we performed multiple techniques with samples from multiple culture days using the same starting donor HuBM for data representation defined in this manuscript.
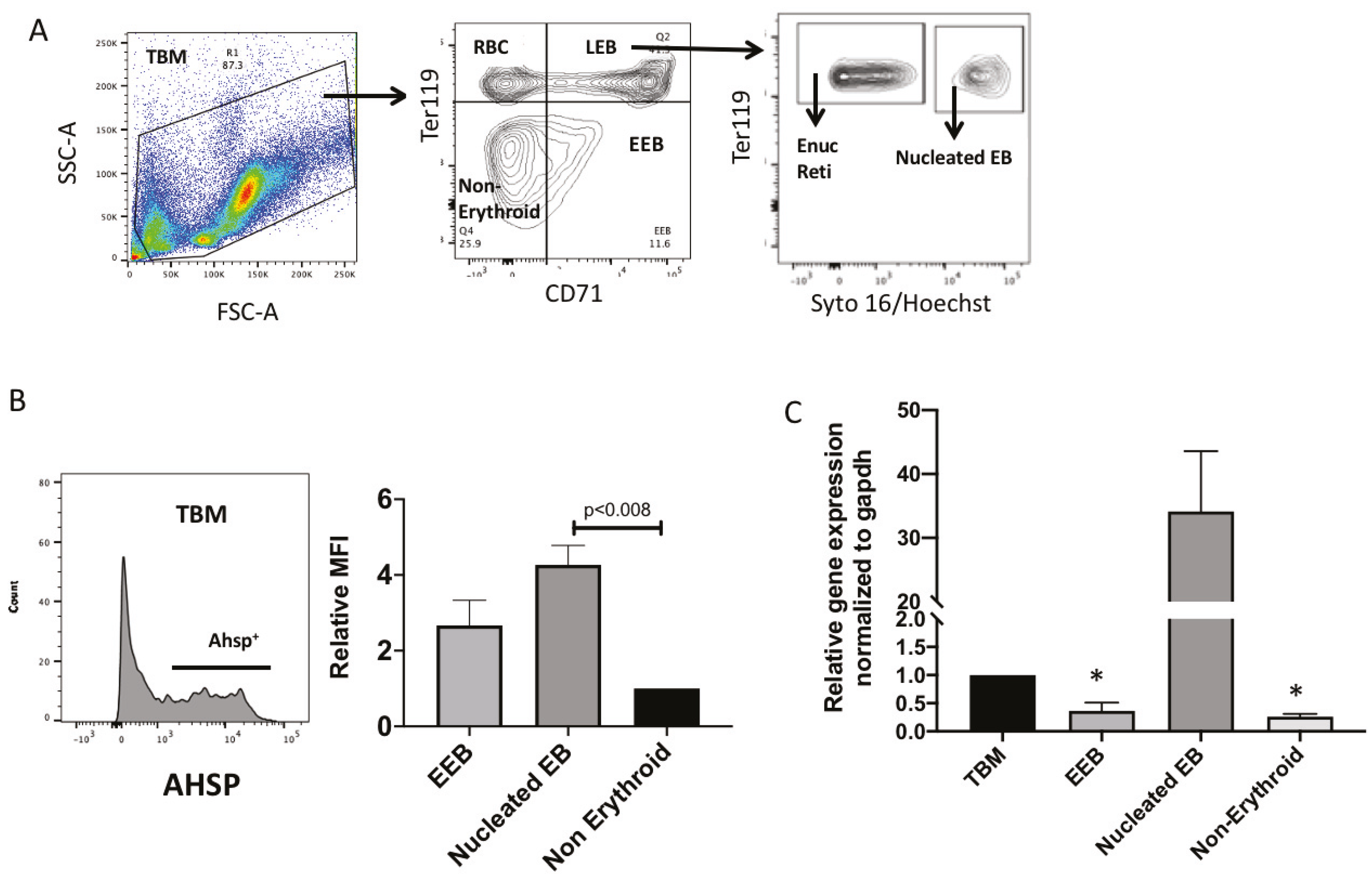
Furthermore, surface antigens transferrin 1 (CD71) and glycophorin (CD235a) can be used to define the several erythroblast subtypes namely, CfuE, ProE, IntE (BasoE), PolyE, OrthoE and reticulocytes from day 9 and day 11 HuBM derived cultures [
7,
11] with slight modifications as in
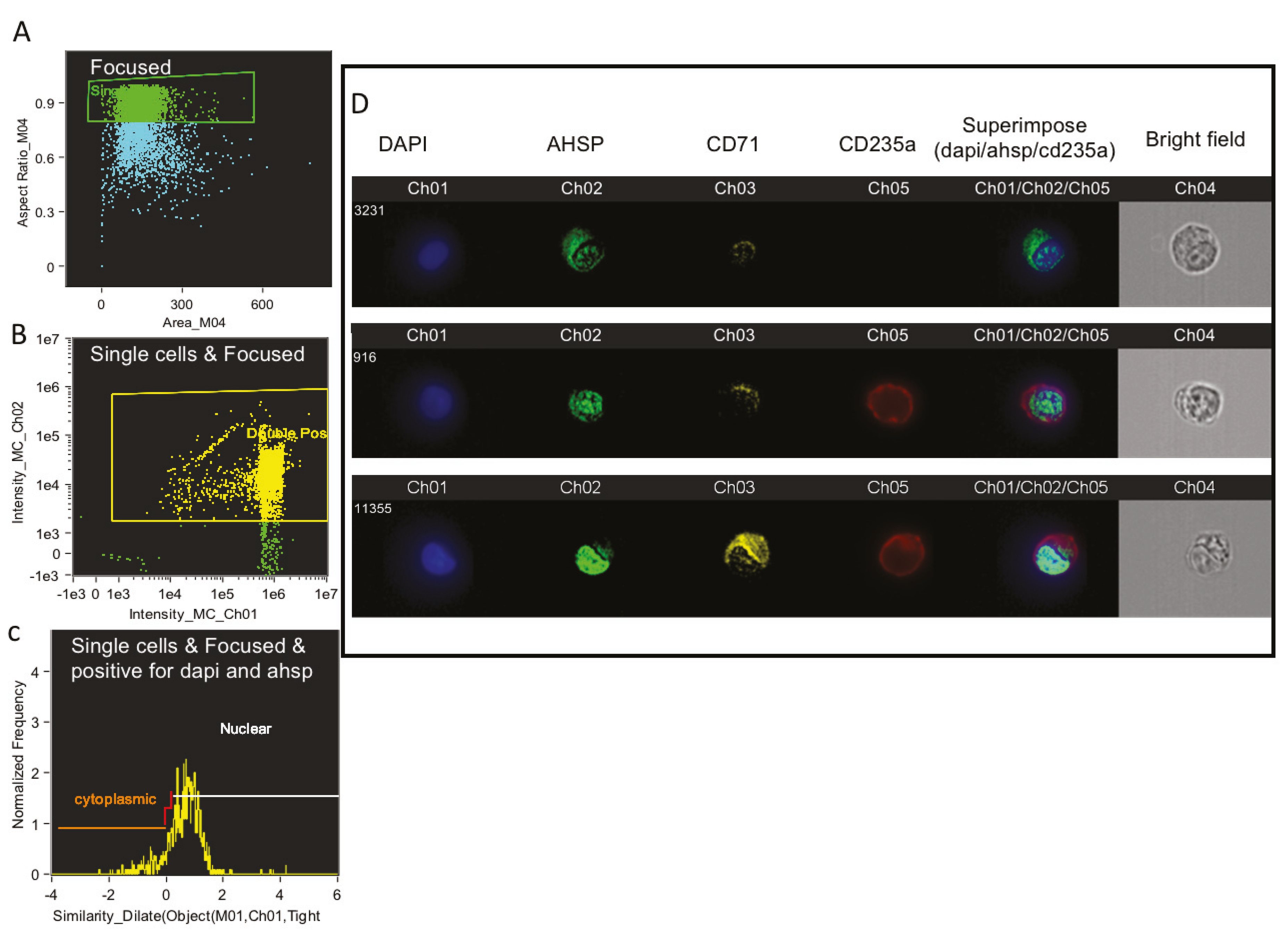
Supplemental Figure S2A,B. Based on the CD71/CD235a gating one can report the MFI of AHSP to inform subset based expression of the chaperone protein in the erythroblasts. Rather, we sought to image stream technology since the primary focus of this manuscript is to highlight the sub cellular localization of AHSP. Thus, we believe, just providing the MFI of AHSP from CD71/CD235a gating will not be sufficient. Additionally, we utilized multispectral imaging flow cytometry (MIFC) to further confirm the cytoplasmic and nuclear expression and localization of AHSP.
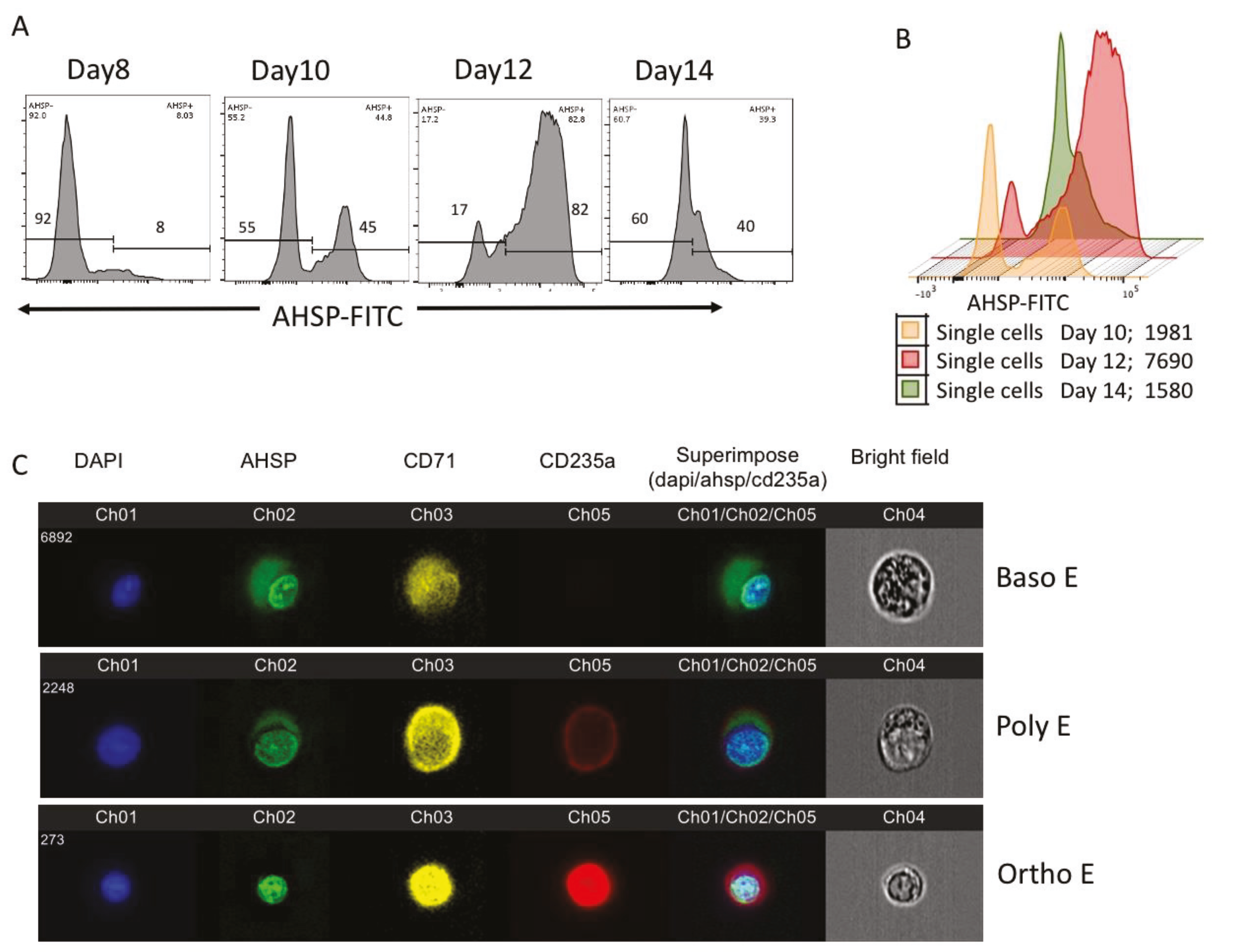
We identified that AHSP gene transcripts began to be expressed at low levels and then peaked with late maturation, declining to low levels in mature reticulocytes. Similarly, the translated protein progressively increased from early to late erythroblasts and peaks in nucleated BasoE, PolyE and OrthoE subsets, significantly decreasing during terminal maturation, tracking the same pattern as gene expression. This confirms the pattern of AHSP gene expression relayed by Dos Santos Oresco et al. 2002, though the specific peak of expression is two days later in our study, specifically day 12 and not day 8–10 range that Dos Santos Oresco reports [
8]. This difference may be due to the later refined methodology of culturing PBMCs with serum free media and only adding cytokines specific to erythroblast differentiation. There was an additional effort to track AHSP gene expression in Varricchio et al. in 2010 but they did not identify a distinct decrease in late maturation stage of erythroblasts [
12]. Indeed, they were unable to identify a statistical difference in the increase in AHSP mRNA perhaps due to, as they remark, the greater variability in their human blood donor samples.
In our current study, we observed erythroblast development periodically every other day from phase 2 of biphasic cultures from either human bone marrow or PBMC derived erythroblasts. The beginning of AHSP protein expression coincides with its mRNA expression on day 8 of culturing when AHSP transcription and protein was significantly detectable. Similarly, AHSP mRNA levels and AHSP protein levels peak at day 12 of ex vivo culturing. AHSP expression is so developmentally synchronized in the erythroid lineage that Yu et al., 2016 have proposed that expression of AHSP be used for the, “detection of erythroid elements within the bone marrow” [
13]. Such a practice may miss early and later maturation stages of erythroid cells.
It is important to note that day 14 was also the beginning of the period of late maturation when the nucleus was ejected from the maturing reticulocyte. We were able to distinctly capture the process of ejection of a nucleus at this time point and can identify that AHSP localizes with the nucleus being ejected. Additionally, when we image mature condensed red blood cells without a nucleus there was also correspondingly an absence of AHSP. It is known that AHSP stabilizes newly formed
αHb effectively, preserving
αHb while preventing aggregation and reactive oxygen species mediated stress [
2,
14] by limiting
αHb reactivity through reversibly binding to the same location as βHb [
15]. Therefore, we could interpret that excess free
αHb is sequestered by AHSP and translocated to the nucleus in maturing erythroblasts when effective hemoglobinization was accomplished.
This notion is further strengthened by probing AHSP negative cells on Day 8 and Day 14 based on nuclear staining by flow cytometry. These results reveal that during early stages of erythroid development, all of the erythroid progenitors were expressing nuclei, but only a few were expressing AHSP at low levels. Progressively by Day 12 about 2-fold more erythroblasts expressed AHSP. This then reversed back to very few cells expressing AHSP by Day 14 and the very same erythroblasts were devoid of nucleus. These results were reconfirmed by AMNIS image stream attesting nuclear localization during late OrthoE stage and eventually ejected with the nucleus in reticulocytes.
A recent applicable study on the quantification of the proteins in the extruded nucleus versus the cytoplasm of reticulocytes unfortunately did not mention AHSP [
16]. Interestingly, hemoglobin subunit alpha, which is the binding partner of AHSP, was detected in both the nucleus and the cytoplasm [
13]. Though at this juncture AHSP would have been outcompeted by hemoglobin beta in binding to hemoglobin alpha [
15] functionally, it can be deduced that mature red blood cells need hemoglobin and not necessarily AHSP. The lack of AHSP in the reticulocytes may reflect its completed role in the developing erythroblast whereas hemoglobin remains essential for the red cell’s function. Autonomously in the arterial endothelium, free alpha hemoglobin is escorted to endothelial nitric oxide synthase (eNOS) by AHSP during nitric oxide degradation [
17].
Therefore, the above study highlights free αHb exclusive chaperone function of AHSP outside of red blood cells besides the crucial role of AHSP in stable hemoglobinization during erythroid maturation.
Nonetheless, in recognizing that AHSP is comparable to the heat shock family of proteins as members of chaperoning erythropoiesis proteins, Bell et al., 2013 identified that most heat shock proteins (hsp) are located in both the extruded nucleus and the mature reticulocyte except for hsp 90-alpha and hsp 4, which were mainly identified in the reticulocyte [
3,
16,
18]. Though AHSP does not follow this pattern of localization, there were various other proteins, which were more localized with the extruded nucleus. The authors noted these proteins were related to the endoplasmic reticulum (ER). Bell et al. state that the majority of ER is extruded along with the nucleus. Previous research from the Weiss lab has identified the association of AHSP with ER and readily binding to the nascent transcribed AHSP to facilitate AHSP’s functional conformation [
15]. Perhaps AHSP remains distinctly associated with the expansive ER thus being removed from the maturing red blood cell jointly with the ER. Future experiments are needed to clarify whether this is the case and may consist of colocalization assays with the erythroid ER.
4. Methods
4.1. Human Bone Marrow Cells
BM aspirate from an unidentified healthy male donor of 24 years age was obtained as a 1 mL excess portion from a clinical use of the main aspirate not related to this study. Samples were stored at −80 °C in 70% freezing media consisting of 90% fetal bovine serum with 10% dimethyl sulfoxide at 4 × 106 cells per 1.5 mL cryotube (Sigma-Aldrich, St. Louis, MO, USA) until day of plating for cell culture.
4.2. Human Peripheral Blood Mononuclear Cells
Blood samples were collected from healthy adults over the age of 18 years enrolled as part of a larger related overarching study and were utilized towards this study. Institutional review board approval was obtained from the Loyola University Health Sciences division for use of human participants. Written informed consent to utilize samples for research purposes was obtained. Blood was drawn by a registered nurse in a purple vacutainer EDTA tube containing Ficoll-Hypaque (Bioscience, Franklin Lakes, NJ, USA). Within 30 min of blood draw, the vacutainer was centrifuged at 2900 rpm for 25 min at 22 °C to isolate PBMCs via gradient-density centrifugation. In 90% fetal bovine serum with 10% dimethyl sulfoxide (Sigma-Aldrich, St. Louis, MO, USA) 4 × 106 PBMC cells were resuspended and stored at 80 °C until the day of culturing. Prior to plating, cells were assessed by Trypan blue exclusion for viability.
4.3. Cell Culture of Human Cells
Cells (bone marrow and PBMCs) were thawed in a 37 °C water bath for 1.5 min and then washed with Iscove’s Modified Dulbecco’s Medium (IMDM). The ACK (ammonium-chloride-potassium) lysing buffer was used to remove red blood cells from unfractionated bone marrow samples. Cells were then washed and placed in an expansion medium containing serum-free expansion medium (SFEM; Stem Cell Technologies, Vancouver, BC, Canada) supplemented with granulocyte macrophage colony-stimulating factor (20 ng/mL; eBioscience, San Diego, CA), stem cell factor (SCF; 45 ng/mL; Stem Cell Technologies) and interleukin-3 (20 ng/mL). In the last step, plasma from a healthy control study donor was added at 5% concentration per well, followed by incubation at 37 °C with 5% CO2.
On day 7, non-adherent cells were washed in IMDM and 0.05 × 106 cells/well of a 12 well plate were seeded in SFEM supplemented with human recombinant Epo (1 U/mL; Stem Cell Technologies), holotransferrin (0.3 mg/mL; R&D Systems, Minneapolis, MN) and stem cell factor (10 ng/mL; Stem Cell Technologies) and 5% plasma from a control subject was added followed by incubation at 37 °C with 5% CO2.
4.4. Fluorescence-Activated Cell Sorting (FACS)
On day 8, 10, 12, 14 and 16 time points, non-adherent cells were collected and washed in IMDM. Cells were stained with cell surface antibodies, anti CD235a cat# 551336 (BDbiosciences, San Jose, CA, USA) and anti CD71 cat# 555537 (BDbiosciences, San Jose, CA, USA) for 30 min at 4 °C. Next cells were fixed and permeabilized with 200 ul of Cytospin/Cytoperm (BD Biosciences San Jose, CA) followed by intracellular staining using anti AHSP cat# sc515436 (Santa Cruz Biotechnology, Dallas, TX, USA) for 2 h at room temperature, and then with Hoechst 33342 nuclear stain cat# H3570 (Thermofisher Invitrogen, Waltham, MA) for 10 min at room temperature. Cells were then analyzed by flow cytometry using BD LSRFortessa and gated with FlowJo software.
4.5. Amnis Single Cell Imaging
After 100,000 cells were analyzed per well using FACS, 10,000 of the remaining stained cells were run in the Amnis ImageStreamX MKII flow cytometer (Amnis Corporation, Seattle, WA, USA). For each individual cell the image of bright field view as well the fluorescence imagery of anti CD235a, anti CD71, anti AHSP and Hoechst nuclear stain were recorded at 60× magnification. Hoechst, FITC, PE and APC were excited with a 100 mW of 488 nm laser and 150 mW of 647 laser. Fluorescence was collected on channel one (430−505), channel two (488−505 nm), channel three (505−560 nm), channel five (560−595 nm) and channel four brightfield respectively. Ideas software (AMNIS Corporation) was utilized to analyze the images of anti CD235a in red, anti CD71 in yellow, anti AHSP in green and Hoechst nuclear stain in blue consistently using the same compensation matrix for each sample. Cells were gated on camera’s focus, followed by gating for the exclusion of debris. Next we gated for erythroblasts using anti CD71 and anti CD235a staining and finally graphed the intensity of fluorescence imagery of anti AHSP. The IDEAS software was used to overlay Hoechst nuclear stain, anti AHSP and anti CD235a.
4.6. Murine Animals for Bone Marrow
B6D2F1 mice weighing 25–30 g (Jackson Laboratories; Barr Harbor, ME, USA) were allowed to acclimatize for seven days following arrival at our Comparative Medicine Facility under a controlled temperature (20−22 °C) and humidity (20−40%) environment with a 12-h light—dark cycle. All experimental protocols were approved by the IACUC (Institutional Animal Care and Use Committee) of Loyola University Chicago Health Sciences Division.
4.7. Murine Bone Marrow Cell Extraction
Healthy naïve mice were sacrificed and on the same day bone marrow cells were harvested from the femurs of each mouse by flushing the marrow via a 25-guage needle through the proximal end of the femur and eluted into McCoy’s medium (Invitrogen, Carlsbad, CA, USA). Total bone marrow cells were then separated into subpopulations followed by gene expression analysis or staining for intracellular and surface erythropoietic cell markers (see details in separate sections below). For both procedures, murine bone marrow cells were stained with the following fluorochrome conjugated monoclonal antibodies specific for mouse: PE-CD71 C2 clone, cat # 553267 (BD Pharmingen, San Diego, CA, USA), PerCP.Cy5.5-Ter119 clone Ter-119; cat # 45-5921-82 (eBioscience, San Diego, CA, USA), Syto16—FITC; cat # S7578 (Fischer Scientific, Waltham, MA, USA) and CD16/CD43 Mouse BD FC Block clone 2.4G2 RUO; cat # 553141 (BD Pharmingen, San Diego, CA, USA), FITC-AHSP G-5 clone; cat # sc-515139-FITC (Santa Cruz Biotechnology, Dallas, TX, USA), Hoechst 33342 cat # H3570 (Thermofisher Invitrogen, Waltham, MA, USA) and OneComp eBeads; cat # 01-1111-41 (Invitrogen/Fischer Scientific, Waltham, MA, USA).
4.8. Murine Bone Marrow Cell Population Sorting
Briefly, 30 × 106 cells total bone marrow (TBM) cells were labeled with the fluorochrome conjugated antibodies for 30 min at 4 °C in the dark. Cells were washed and resuspended in IMDM for sort purification as follows (FACS Aria). TBM cells were gated as non-erythroid (CD71neg Ter119neg), RBCs (CD71neg Ter119pos), early erythroblasts (CD71posTer119neg) and late erythroblasts (CD71pos Ter119pos). Late erythroblasts were then separated into nucleated (Syto16pos) erythroblasts and reticulocytes (Syto16neg). About 50 × 104 sorted cells from each subset were subjected to RNA extraction (See RT-qPCR methods section).
4.9. Murine Bone Marrow AHSP Staining
Briefly, 1 × 106 cells total bone marrow (TBM) cells were labeled with the fluorochrome conjugated antibodies as mentioned in the previous section for 30 min at 4 °C in the dark. Cells were washed, fixed and permeabilized using Cytospin/Cytoperm (BD Biosciences San Jose, CA) following manufacture’s protocol followed by intracellular staining using anti AHSP for 2 h at room temperature. Hoechst nuclear stain was added for the last 10 min, washed and resuspended in PBS, and immediately analyzed by flow cytometry using BD LSR Fortessa and gated with FlowJo software. Positive and negative gates were set with FMO (fluorescent minus one) controls and unstained cells respectively. Flurochrome compensation was performed using OneComp eBeads.
4.10. Real Time Quantitative Polymerase-Chain Reaction (RT-qPCR)
Messenger ribonucleic acid (mRNA) was extracted from original bone marrow cells and PBMCs at various stages of erythropoietic maturation, day 8, day 10, day 12, day 14 and day 17 of cell culture, using the RNeasy kit (Qiagen, Hilden, Germany) as described by the manufacturer. RNA was transcribed into cDNA using the High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Foster City, CA) and the Applied Biosystems Veriti 96-Well Fast Thermocycler 0.1 mL PCR machine. cDNA was appropriately diluted and combined in separate wells with three master mixes consisting of TaqMan Fast Advanced Master Mix (Applied Biosystems) and respective Taqman Gene Expression Assays (20X) consisting of either human AHSP:FAM cat #4448892 (Thermofisher Fischer Scientific, Waltham, MA), GAPDH:FAM cat #4453320 (Thermofisher Fischer Scientific, Waltham, MA, USA) and GATA-1:FAM cat #4453320 (Thermofisher Fischer Scientific, Waltham, MA, USA), or murine AHSP:FAM cat #4331182 (Thermofisher Fischer Scientific, Waltham, MA, USA), GATA1:FAM cat #4331182 (Thermofisher Fischer Scientific, Waltham, MA, USA) and GAPDH:VIC cat #4448489 (Thermofisher Fischer Scientific, Waltham, MA, USA) for respective samples. GAPDH was included as a normalization control for each analysis. The reaction took place in the StepOnePlus Real-Time PCR System (Applied Biosystems). Target gene cycle threshold (Ct) values were normalized to GAPDH Ct values using the ΔΔCt method. Results are expressed as fold change over day 8 BM derived erythroblasts’ gene expression.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}