Extraction of Microbial and Host DNA, RNA, and Proteins from Oak Bark Tissue


Abstract
:1. Introduction
2. Experimental Design
Materials
- DNeasy Plant Mini kit (Qiagen, Venlo, Netherlands)
- Qubit dsDNA HS assay kit (Thermo Fisher, Waltham, MA, US)
- NEBnext microbiome DNA enrichment kit (New England Biolabs, Ipswich, MA, US)
- Genomic DNA Clean and Concentrator kit (Zymo Research, Irvine, CA, US)
- RNA extraction buffer (4 M guanidine thiocyanate, 0.2 M sodium acetate pH 5.0, 25 mM EDTA, 2.5% (w/v) polyvinylpyrrolidone, and 1% (v/v) β-mercaptoethanol)
- 20% sodium lauroyl sarcosinate
- RNeasy plant mini kit (Qiagen, Venlo, Netherlands)
- DNease I (Qiagen)
- RNeasy MinElute Cleanup kit (Qiagen)
- Ribo-Zero ribosomal RNA (rRNA) Removal kits for plant seed/root and for bacteria (Illumina, San Diego, CA, US)
- Qubit RNA HS assay kit (Thermo Fisher)
- Protein solubilization buffer (50 mM Tris-HCl, 25 mM EDTA, 500 mM thiourea, 0.5% DTT)
- Ice cold 20% trichloric acid in acetone with 0.5% DTT
- Ice cold acetone
- 3% SDS solution
3. Procedure
- Use approximately 50 mg of oak tissue homogenized with a mortar and pestle under freezing conditions in liquid nitrogen. Transfer homogenized tissue into DNA extraction kit tube.
- Use the DNeasy Plant Mini kit (Qiagen) according to the manufacturer’s instructions.
- In order to enrich microbiome DNA, deplete the host DNA from the sample using the NEBnext microbiome DNA enrichment kit (New England Biolabs) according to the manufacturer’s instructions.
- Purify the DNA and concentrate using the Genomic DNA Clean and Concentrator kit (Zymo Research) according to the manufacturer’s instructions.
- Store at −20 °C.
- Bark samples collected for transcriptome analysis were immediately frozen in liquid nitrogen for on-site preservation of RNA. For RNA extraction, use approximately 50 mg of oak tissue homogenized with a mortar and pestle under freezing conditions in liquid nitrogen. Briefly, 5 mL of extraction buffer (4 M guanidine thiocyanate, 0.2 M sodium acetate pH 5.0, 25 mM EDTA, 2.5% (w/v) polyvinylpyrrolidone, and 1% (v/v) β-mercaptoethanol) is added to oak tissue kept frozen in a sterilized mortar using liquid nitrogen. The volume of extraction buffer used at this step was increased from the original protocol, as lower volumes of 1−2 mL were not sufficient to produce a significant quantity of RNA.
- The frozen tissue in extraction buffer is further ground until thawed.
- Add an additional 2.5 mL of extraction buffer and 500 μL of 20% sodium lauroyl sarcosinate mixed into the sample. Again, the volume added at this step has been increased from 60 to 500 μL.
- Shake the sample mixture at room temperature for 15 min at 200 rpm on a shaking platform and further process using the RNeasy Plant Mini kit (Qiagen). The incubation time has been increased while the shaking intensity has been decreased in comparison to the original protocol.
- After centrifugation in the QIAShredder column, mix 350 µL of the supernatant with 0.9 volumes of ethanol instead of 0.5 according to the kit protocol, and subsequently follow the manufacturer’s protocol by centrifuging in the RNeasy Mini column.
- The manufacturer’s instructions for the RNeasy Plant Mini kit are followed from this stage onwards.
- Treat the extracted RNA with DNase I (Qiagen) following the RNeasy Plant Mini kit (Qiagen) instructions for DNase treatment.
- The RNA is purified using the RNeasy MinElute Cleanup kit (Qiagen) following the manufacturer’s instructions.
- Deplete rRNA from RNA extracts using a 1:1 combination of the Ribo-Zero rRNA Removal kits for plant seed/root and for bacteria (Illumina) according to the manufacturer’s instructions.
- Purify the rRNA depleted samples again using the RNeasy MinElute Cleanup kit (Qiagen) and store at −80 °C.
- Use approximately 50 mg of oak tissue homogenized with mortar and pestle under freezing conditions in liquid nitrogen. A lower amount of bark tissue is used than in the original protocol (50 mg instead of 300 mg lyophilized powdered bark tissue) due to high levels of proteases and phenolic compounds.
- Add 2 mL solubilization buffer (50 mM Tris-HCl, 25 mM EDTA, 500 mM thiourea, 0.5% DTT) and grind in a mortar and pestle at room temperature.
- Shake the mixture on a platform shaker (150 rpm) for 1 h at ambient temperature.
- Centrifuge samples at 20,000 g for 20 min and extract and store the supernatant at 4 °C. Repeat the procedure using the remaining pellet.
- Extract the supernatant again and pool with the previous supernatant.
- Add ice cold 20% trichloric acid in acetone with 0.5% DTT in a 1:1 ratio to the supernatant pool and precipitate at −20 °C overnight.
- After precipitation, centrifuge the mixture at 20,000− g for 60 min and wash with ice cold acetone (centrifuged at 20,000− g for 30 min).
- Air-dry the pellet, re-suspend in 3% SDS solution and store at −80 °C.
4. Expected Results
Author Contributions
Funding
Conflicts of Interest
References
- Van der Heijden, M.G.A.; Hartmann, M. Networking in the plant microbiome. PLoS Biol. 2016, 14, e1002378. [Google Scholar] [CrossRef] [PubMed]
- Hacquard, S.; Schadt, C.W. Towards a holistic understanding of the beneficial interactions across the Populus microbiome. New Phytol. 2015, 205, 1424–1430. [Google Scholar] [CrossRef] [PubMed]
- Franzosa, E.A.; Hsu, T.; Sirota-Madi, A.; Shafquat, A.; Abu-Ali, G.; Morgan, X.C.; Huttenhower, C. Sequencing and beyond: Integrating molecular “omics” for microbial community profiling. Nat. Rev. Microbiol. 2015, 13, 360–372. [Google Scholar] [CrossRef] [PubMed]
- Rezadoost, M.H.; Kordrostami, M.; Kumleh, H.H. An efficient protocol for isolation of inhibitor-free nucleic acids even from recalcitrant plants. 3 Biotech 2016, 6, 61. [Google Scholar] [CrossRef] [PubMed]
- Kalinowska, E.; Chodorska, M.; Paduch-Cichal, E.; Mroczkowska, K. An improved method for RNA isolation from plants using commercial extraction kits. Acta Biochim. Pol. 2012, 59, 391–393. [Google Scholar] [PubMed]
- Broberg, M.; Doonan, J.; Mundt, F.; Denman, S.; McDonald, J.E. Integrated multi-omic analysis of host-microbiota interactions in acute oak decline. Microbiome 2018, 6. [Google Scholar] [CrossRef] [PubMed]
- Denman, S.; Doonan, J.; Ransom-Jones, E.; Broberg, M.; Plummer, S.; Kirk, S.; Scarlett, K.; Griffiths, A.R.; Kaczmarek, M.; Forster, J.; et al. Microbiome and infectivity studies reveal complex polyspecies tree disease in Acute Oak Decline. ISME J. 2018, 12, 386–399. [Google Scholar] [CrossRef] [PubMed]
- Pagter, M.; Sergeant, K.; Møller, S.M.; Bertram, H.C.; Renaut, J. Changes in the proteome and water state in bark and xylem of Hydrangea paniculata during loss of freezing tolerance. Environ. Exp. Bot. 2014, 106, 99–111. [Google Scholar] [CrossRef]
- Andersen, R.A.; Sowers, J.A. Optimum conditions for bonding of plant phenols to insoluble polyvinylpyrrolidone. Phytochemistry 1968, 7, 293–301. [Google Scholar] [CrossRef]
- Young, C.C.; Burghoff, R.L.; Keim, L.G.; Minak-Bernero, V.; Lute, J.R.; Hinton, S.M. Polyvinylpyrrolidone-Agarose Gel Electrophoresis purification of polymerase chain reaction-amplifiable DNA from soils. Appl. Environ. Microbiol. 1993, 59, 1972–1974. [Google Scholar] [PubMed]
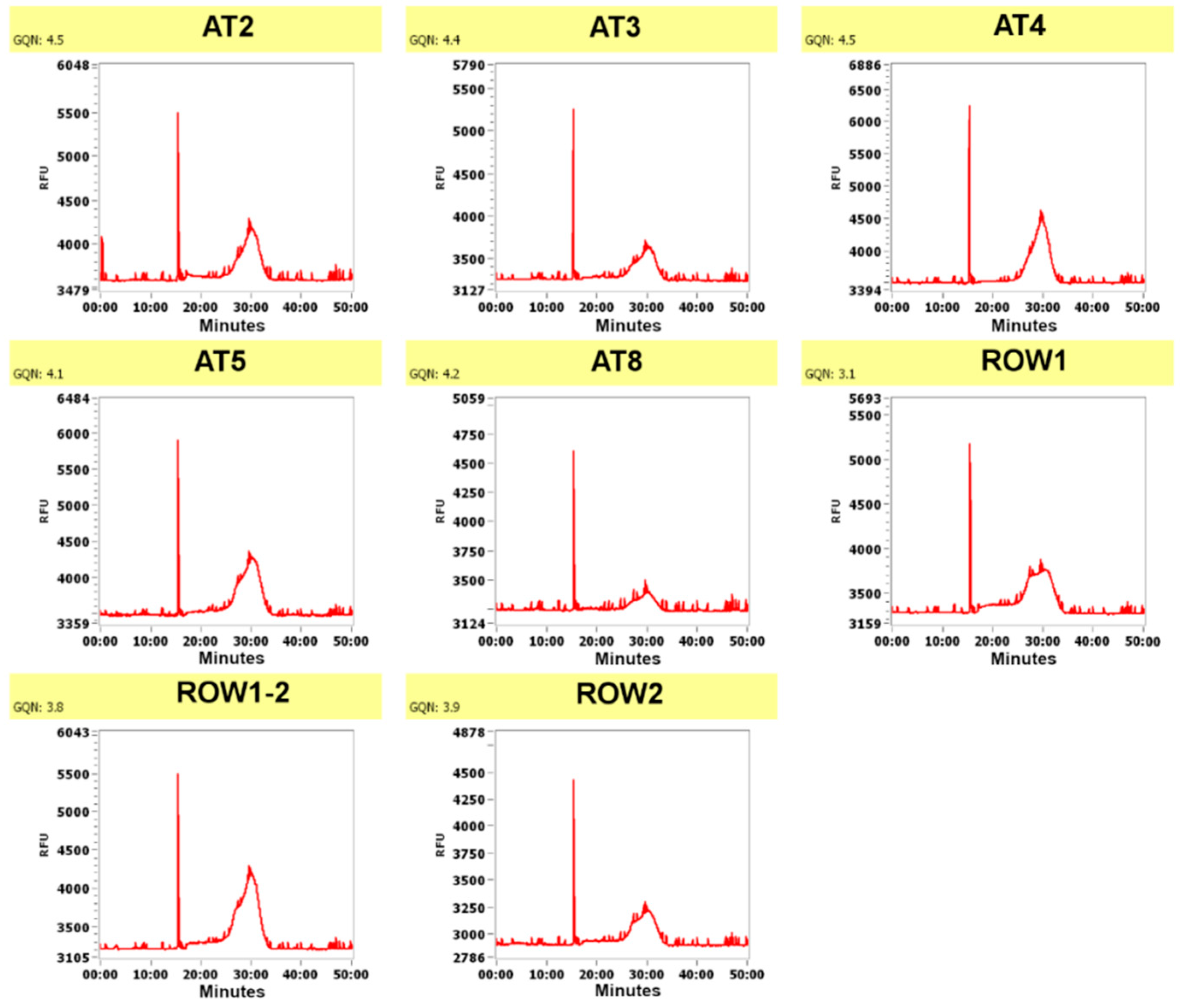
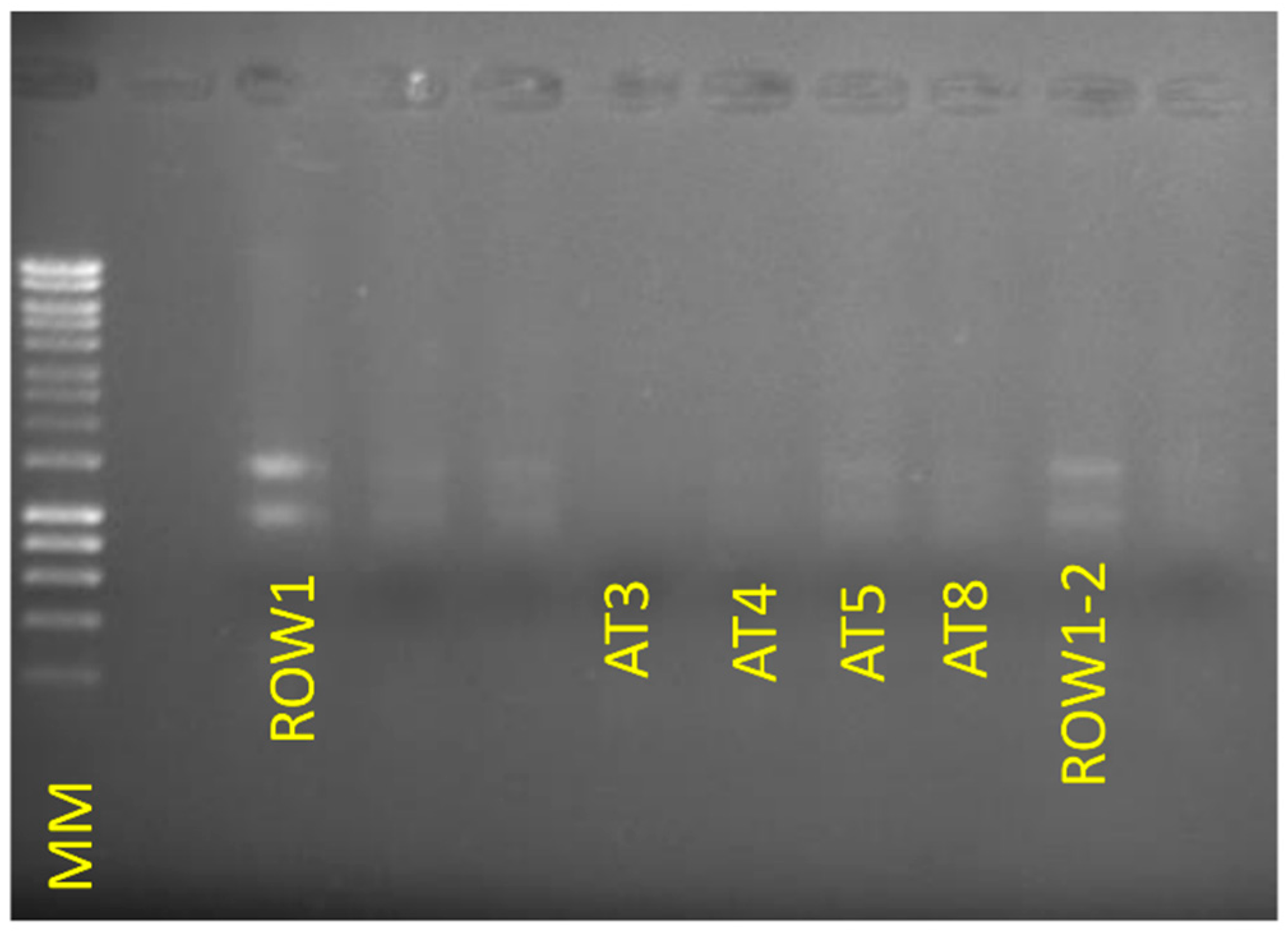
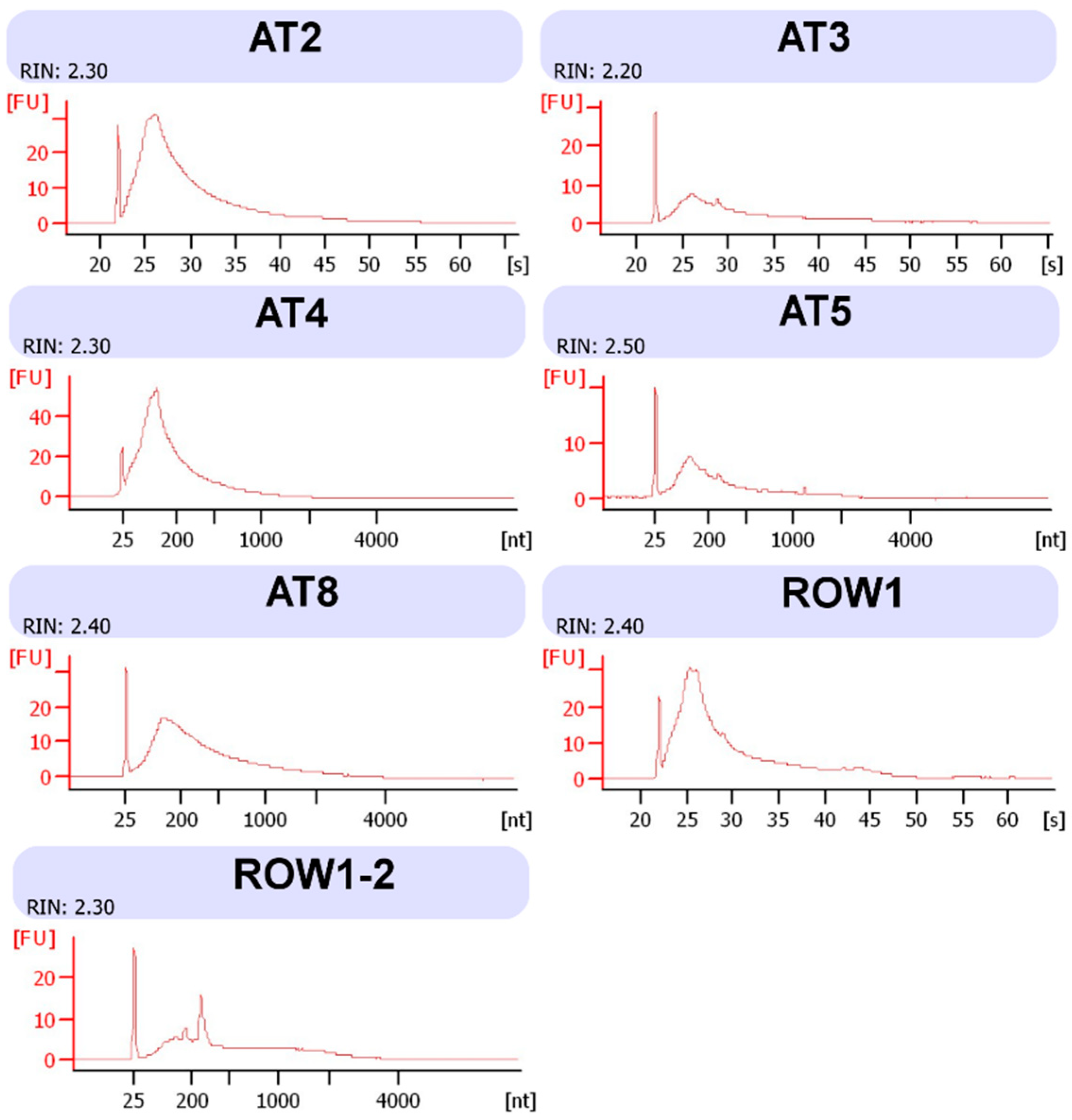

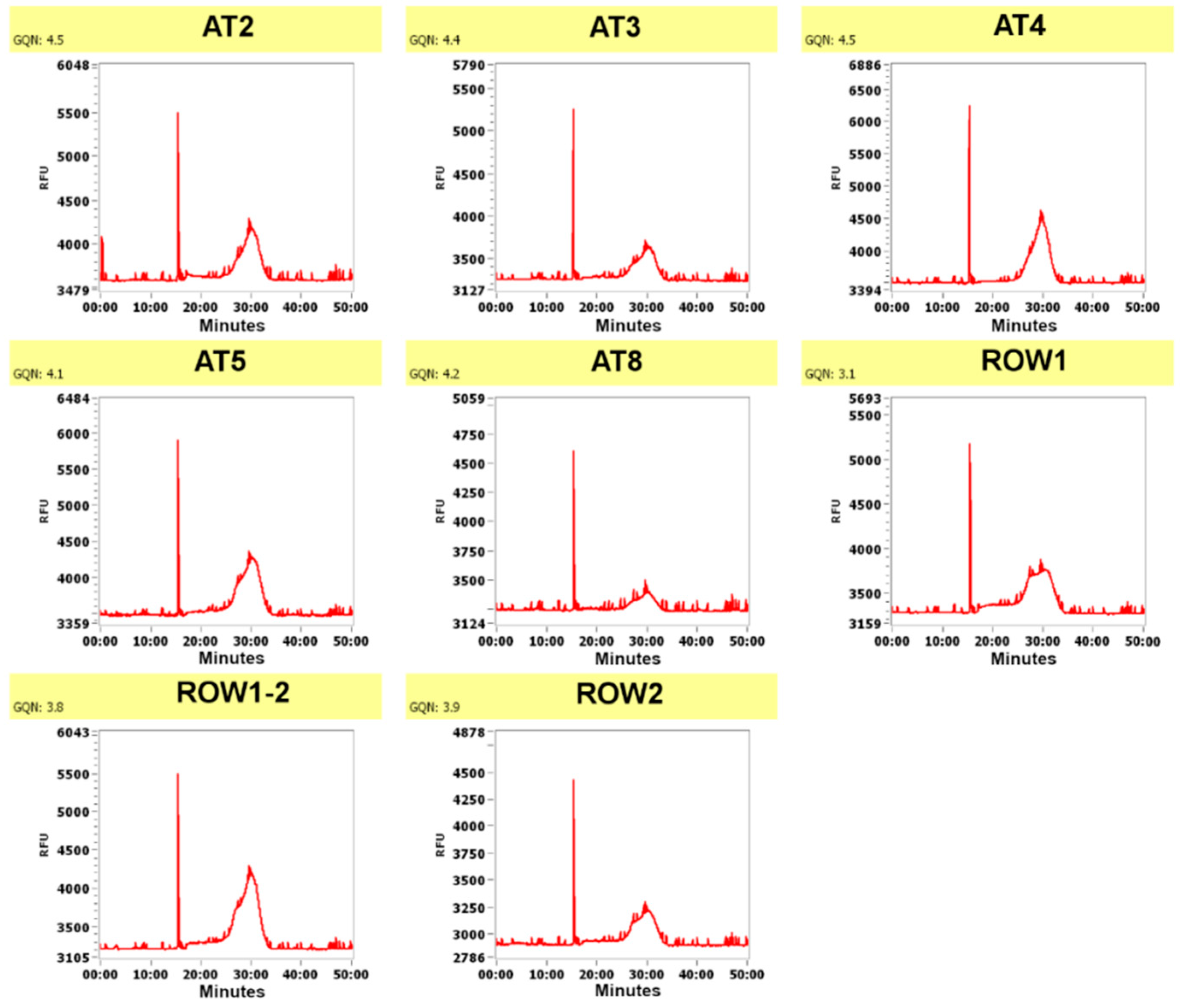{kind=link}
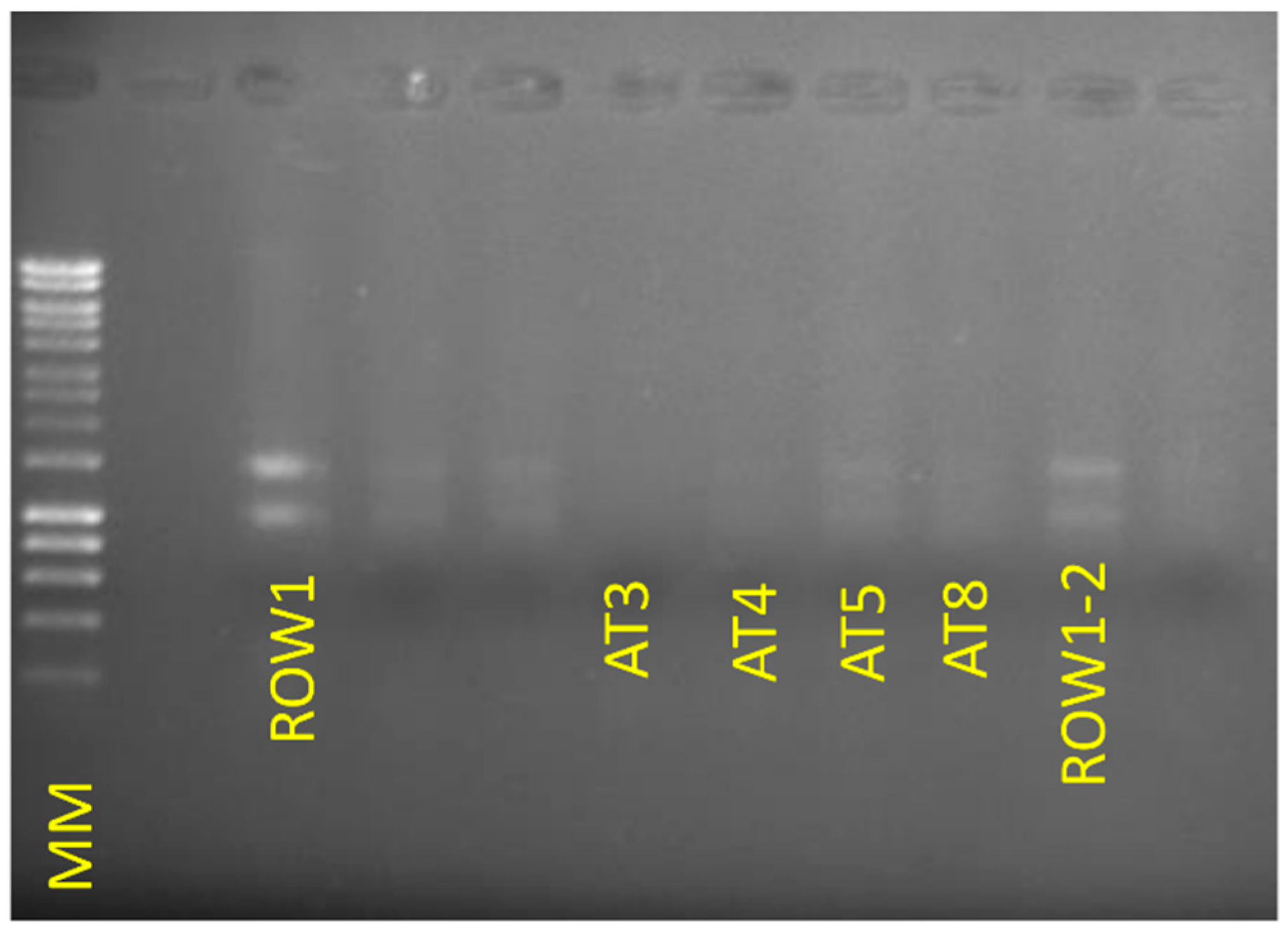{kind=link}
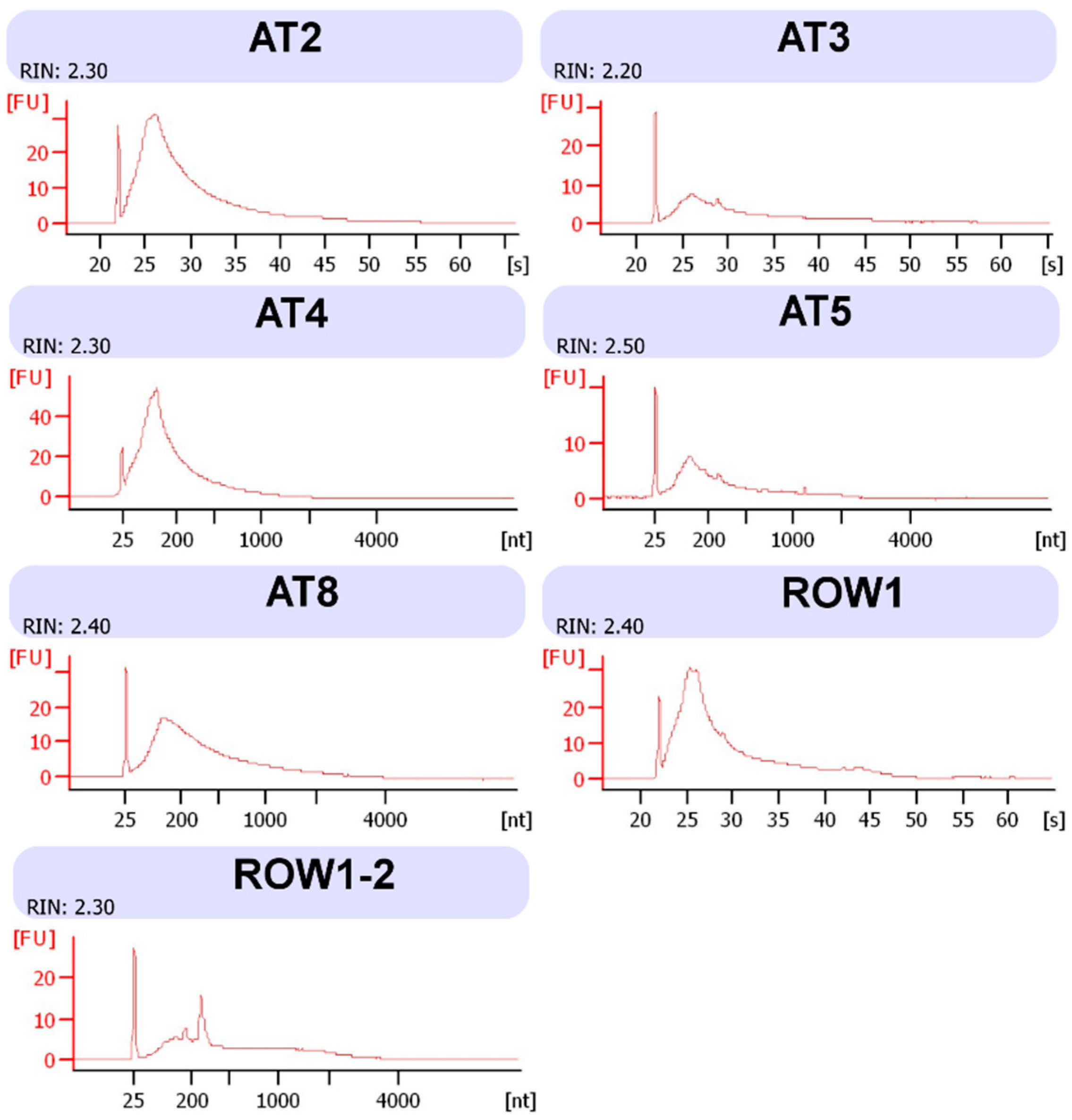{kind=link}
| Sample Name | Total Amount (ng) DNA after Three Rounds of Extraction (Qubit dsDNA HS Assay) |
| AT2 | 355 |
| AT3 | 381 |
| AT4 | 277 |
| AT5-2 | 416 |
| AT8 | 448 |
| ROW1 | 293 |
| ROW1-2 | 1334 |
| ROW2 | 507 |
| Sample Name | Total Amount (ng) RNA after Two Rounds of Extraction (Qubit RNA HS Assay) |
| AT2 | 2236 |
| AT3 | 1725 |
| AT4 | 1786 |
| AT5-2 | 150 |
| AT8 | 1551 |
| ROW1 | 588 |
| ROW1-2 | 5687 |
| ROW2 | Not successful |
| Sample Name | Total Amount (ng) Protein (Qubit Protein Assay Kit) |
| AT2 | 194 |
| AT3 | 184 |
| AT4 | 167 |
| AT5-2 | 284 |
| AT8 | 187 |
| ROW1 | 212 |
| ROW1-2 | 814 |
| ROW2 | 248 |
| Sample | DNA Sequencing bp | RNA Sequencing bp |
|---|---|---|
| AT2 | 6.60 × 109 | 8.94 × 109 |
| AT3 | 5.57 × 109 | 6.07 × 109 |
| AT4 | 7.04 × 109 | 8.78 × 109 |
| AT5 | 6.66 × 109 | 5.60 × 109 |
| AT8 | 6.98 × 109 | 7.52 × 109 |
| ROW1 | 6.66 × 109 | 8.97 × 109 |
| ROW1-2 | 6.42 × 109 | 8.43 × 109 |
| ROW2 | 6.11 × 109 | Did not produce any |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Broberg, M.; McDonald, J.E. Extraction of Microbial and Host DNA, RNA, and Proteins from Oak Bark Tissue. Methods Protoc. 2019, 2, 15. https://doi.org/10.3390/mps2010015
Broberg M, McDonald JE. Extraction of Microbial and Host DNA, RNA, and Proteins from Oak Bark Tissue. Methods and Protocols. 2019; 2(1):15. https://doi.org/10.3390/mps2010015
Chicago/Turabian StyleBroberg, Martin, and James E. McDonald. 2019. "Extraction of Microbial and Host DNA, RNA, and Proteins from Oak Bark Tissue" Methods and Protocols 2, no. 1: 15. https://doi.org/10.3390/mps2010015
APA StyleBroberg, M., & McDonald, J. E. (2019). Extraction of Microbial and Host DNA, RNA, and Proteins from Oak Bark Tissue. Methods and Protocols, 2(1), 15. https://doi.org/10.3390/mps2010015