Abstract
Introduction. Paroxysmal nocturnal hemoglobinuria (PNH) is an acquired non-malignant hematological disorder which affects the pluripotent hematopoietic stem cell. The cause of PNH development is the occurrence of somatic mutations in the phosphatidylinositol glycan-A gene which encodes a protein necessary for the biosynthesis of glycosylphosphatidylinositol anchors. The diagnosis of PNH requires the presence of signs of intravascular hemolysis, thrombosis, and (or) bone marrow failure. Case Report. We report the case of a 42-year-old female, diagnosed with PNH at the age of 27, whose evolution was initially characterized predominantly by hemolytic attacks and whose disease pattern evolved towards thromboembolic episodes with the advancement of age. Conclusions. Establishing the diagnosis of PNH is a difficult task and its management requires teamwork. During the evolution of the disease, a PNH patient can acquire supplementary risk factors for thrombosis, in addition to the pro-coagulant potential of the disease itself. We reported this case to remind physicians that establishing the diagnosis of PNH is troublesome, and thus it is questionable whether PNH is a rare disease or just underdiagnosed. In this context, in the clinical practice of hematologists and other physicians as well, PNH remains a veritable Pandora's box.
Introduction
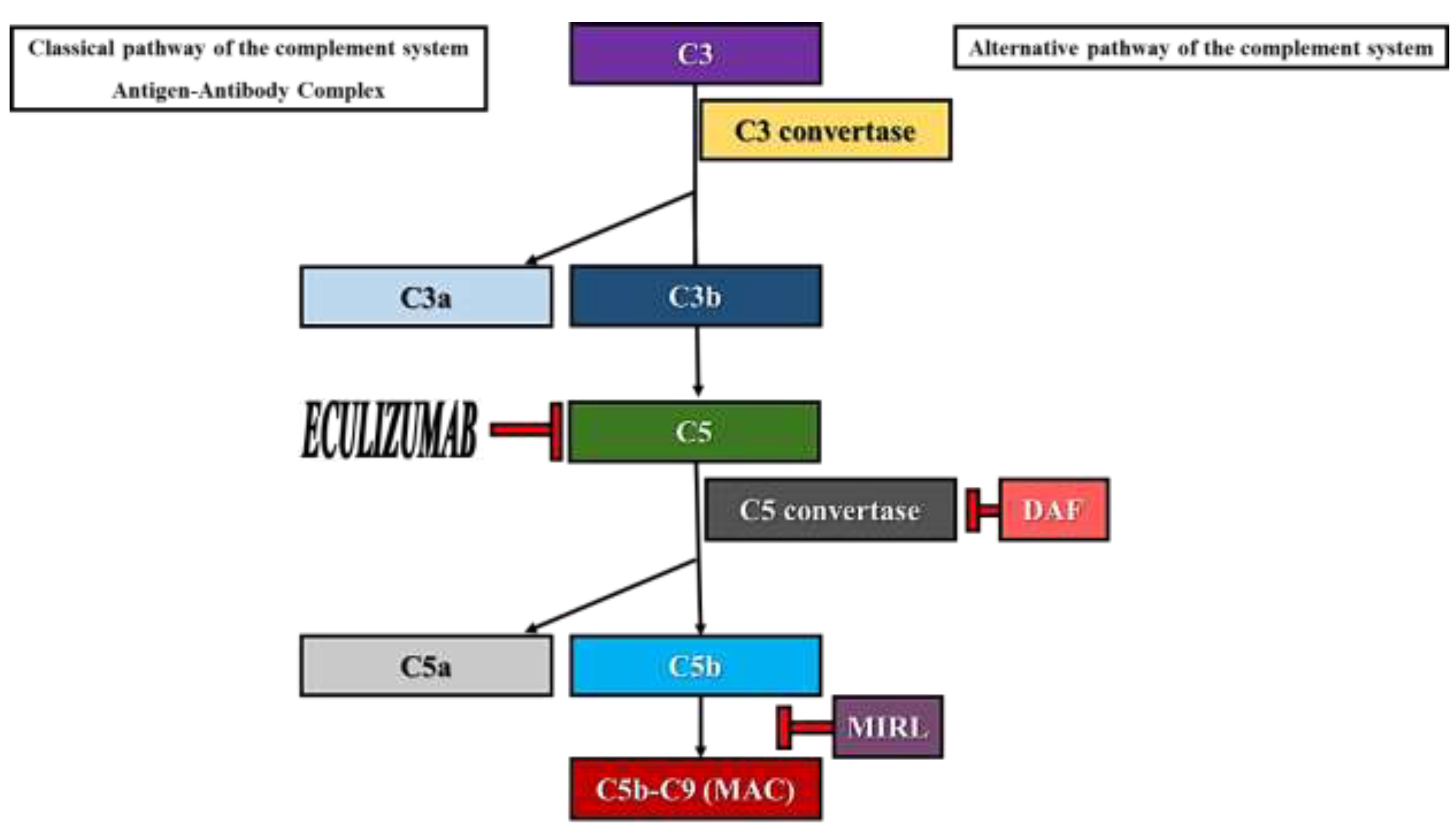
Paroxysmal nocturnal hemoglobinuria (PNH) is an acquired non-malignant hematological disorder which affects the pluripotent hematopoietic stem cell. The cause of PNH development is the occurrence of somatic mutations in the phosphatidylinositol glycan-A (PIGA) gene located on the X chromosome (Xp22.1) which encodes a protein necessary for the biosynthesis of glycosylphosphatidylinositol (GPI) anchors [1,2,3,4]. Consequently, in PNH there is a deficiency in proteins that employ GPI anchors to bind to the surface of blood cells, such as decay-accelerating factor (DAF or CD55) and membrane inhibitor of reactive lysis (MIRL or CD59). These molecules are involved in the inhibition of the membrane attack complex (MAC), regulating the action of the complement system activated via the alternative pathway (Figure 1) [1,2]. Although all cell lines derived from the affected stem cell are deficient in GPI-anchored proteins, erythrocytes are more likely to be destroyed by complement-mediated lysis since they lack a nucleus [2].
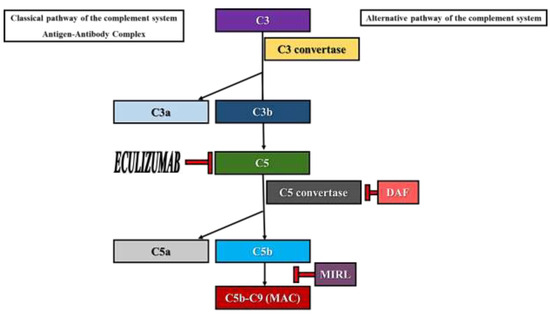
Figure 1.
Activation of the complement system via the alternative pathway. Role of DAF, MIRL and Eculizumab.
PNH is characterized by phenotypic mosaicism [4]:
- the sensitivity of type I clones to complement- mediated lysis is normal. The expression of MIRL (CD59) and DAF (CD55) on the surface of erythrocytes is normal.
- the sensitivity of type II clones to complement- mediated lysis is 2-4 times higher than normal. These cells have a partial deficiency in MIRL and DAF. The clinical course of PNH patients whose erythrocytes’ phenotype corresponds to type II clones is benign, suffering from hemolytic crises only when supervening events (infections, trauma etc.) lead to the brisk activation of the complement system.
- the sensitivity of type III clones to complement- mediated lysis is 15-25 times higher than normal. These cells are totally deficient in MIRL and DAF and, thus, a higher representation of this clone will lead to a severe clinical PNH course.
Although PNH is regarded as a rare disease in clinical practice, with an estimated incidence of 1-1.5/1.000.000, this hematological disease might be underdiagnosed, mainly due to its myriad of presenting conditions: anemia and related-symptoms, thrombosis, smooth muscle dystonia, fatigue, hemoglobinuria due to intravascular hemolysis, renal impairment, or pulmonary hypertension [2]. Herein, we present the case of a female patient with long-lasting PNH, with special emphasis on the onset, evolution, and management of this great mimicking disease.
Case Presentation
We report the case of a 42-year-old female, initially diagnosed at 15 years of age in a pediatric unit with Evans syndrome, based on the presence of hemolytic anemia (Coombs test slightly positive) and thrombocytopenia. The patient was treated with prednisone and cyclophosphamide pulse-therapy without any response and was subjected to splenectomy 12 months after the first hospitalization. At the age of 18, the patient was admitted to an Internal Medicine unit with abdominal discomfort in the upper right quadrant and jaundice. Abdominal ultrasound revealed the partial thrombosis of a branch of the portal vein and the presence of an accessory spleen. In this context, anticoagulation therapy with LMWH and thereafter acenocoumarol was prescribed.
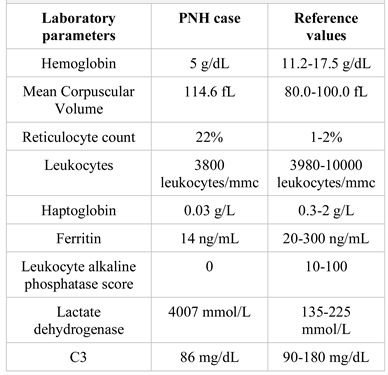
The patient was lost for follow-up for nearly 7 years and was admitted at the age of 27 to the Hematology Clinic, Fundeni Clinical Institute, Bucharest, Romania, with fever, jaundice, severe macrocytic anemia with a pattern of hemolysis, slight leukopenia, low haptoglobin, dark urine (hemoglobinuria), elevated LDH, low C3 values, negative Coombs test, low ferritin and leukocyte alkaline phosphatase levels, negative anti-DNAds, anti-Ro/SSA and anti-LA/SSB antibodies, positive Ham (Figure 2) and sucrose tests, and a positive test for hemosiderinuria (the main blood tests results are reported in Table 1). The final diagnosis of PNH was established before any blood transfusions, based on peripheral blood immunophenotyping by flow-cytometry. The flow- cytometric analysis showed a dominant PNH clone within the granulocytes (98% type II), monocytes (98% type II) and a minor PNH clone in the red blood cells (7.9% type II and 0.8% type III, decreased expression of CD59). Concomitantly, the patient was diagnosed with hepatitis C and blood tests indicated signs of autoimmunity (positive rheumatoid factor and positive antinuclear antibodies).

Figure 2.
Representation of the Ham test in the workup of PNH.

Table 1.
Laboratory tests results in our patient at the time of the PNH diagnosis.
Until 2019, the patient was treated conservatively (blood transfusions and anticoagulants), since eculizumab (recombinant humanized monoclonal antibody targeted against the complement protein C5) could not be prescribed to manage any hemolytic crisis as the drug was not available in Romania. The evolution of the patient was characterized by hemolytic crises triggered by common infections and (or) menses. In June 2019, the patient presented to the Neurology Clinic of our hospital with dysarthria, paresthesia, and loss of function of the upper left limb as signs of a stroke. She had also developed obesity and hypertension.
Computed tomography revealed:
- spontaneous cortico-subcortical hypodensity with loss of grey-white matter differentiation located in the right frontal area (compatible with a subacute ischemic stroke).
- small spontaneous hypodense lesion similar in density to the cerebrospinal fluid depicted as a craniocaudal band in the frontal white matter on the right side (possible sequelae lesion).
- minimal calcified atheromata of the parasellar segment of the internal carotid artery and left vertebral artery.
The patient received LMWH, mannitol, and cerebrolysin, followed by co-administration of acenocoumarol and LMWH, and was ultimately discharged with the recommendation to continue acenocoumarol treatment indefinitely. The patient had admitted the administration of herbal supplements for weight loss and she was advised not to consume such products due to a possible interaction with acenocoumarol. Also, Doppler echocardiography revealed the presence of an atrial septal defect (possible ostium secundum) and distal occlusion of the internal carotid artery.
In July 2019, the patient was hospitalized in our clinic for abdominal discomfort and nausea, with normal hemoglobin, no clinical or biological signs of a hemolytic attack, elevated liver enzymes and elevated amylase and lipase. Based on the abdominal ultrasound, the diagnosis of chronic cholecystitis with possible involvement of the pancreas was established. The patient was treated with antibiotics, antispasmodics, and antisecretory medication and was discharged with the recommendation of laparoscopic cholecystectomy if the symptoms persisted. In January 2020, due to the development of pericholecystic adhesions, open cholecystectomy was performed and the patient was discharged seven days after the surgical procedure.
Soon after the intervention, the patient was admitted to the Neurology Clinic of our hospital with left central facial paresis, left hemiplegia (0/5 on the Medical Research Council scale), and left hemihypesthesia. The examination by carotid ultrasound imaging underlined the occlusion of the internal right carotid artery. Also, echocardiographic examination revealed the presence of a patent foramen ovale with a right-to-left shunt. Blood tests revealed the presence of lupus anticoagulant and a positive cryoglobulin test (++). Screening for thrombophilia was negative. Computed tomography was performed, showing an area of early sub(acute) ischemia located in the frontoparietal region, indicative of a re-infarction in the right frontal area, and no signs of hemorrhagic transformation. Magnetic resonance imaging also confirmed the presence of bilateral supratentorial acute ischemic stroke, especially in the right fronto-parietal region, and multiple acute lacunar lesions which suggested a possible embolic etiology.
Discussions
We have presented the case of a patient with long- lasting PNH. Although the evolution of PNH in her case was typical, the definitive diagnosis was established only around the age of 27. The onset of the clinical signs was precocious, during adolescence, but was misinterpreted as Evans syndrome, i.e., autoimmune hemolytic anemia and autoimmune thrombocytopenia, frequently associated with systemic lupus erythematosus [5]. Thus, the definitive diagnosis of PNH was delayed. However, the development of thrombotic complications in unusual sites, namely splanchnic thrombosis, should have oriented the clinical reasoning towards the correct diagnosis. According to Fan et al. (2019), PNH is a risk factor for noncirrhotic nonmalignant portal vein thrombosis and Budd-Chiari syndrome. However, myeloproliferative neoplasms and mutations in thrombophilia-related genes (factor V Leiden G1691A, prothrombin G20210A) comprise the most frequent genetic association with the two aforementioned conditions [6]. Interestingly, splanchnic thrombosis at a young age is not uncommon in PNH, with Ain et al. (2018) reporting the occurrence of splanchnic thrombosis in a PNH female patient of just 14 years of age, who presented to the hospital with abdominal pain, palpitations and dyspnea. In her case, blood tests revealed the presence of hemolytic anemia and a negative Coombs test, and the final diagnosis of PNH was confirmed by flow-cytometry (the erythrocytes had lost the expression of CD59) [7]. The diagnosis of PNH requires the presence of signs of intravascular hemolysis, thrombosis, and (or) bone marrow failure. In our case report, the definitive diagnosis was established with great difficulty because the presentation of these signs was sequential, spanning a time frame of approximately 12 years.
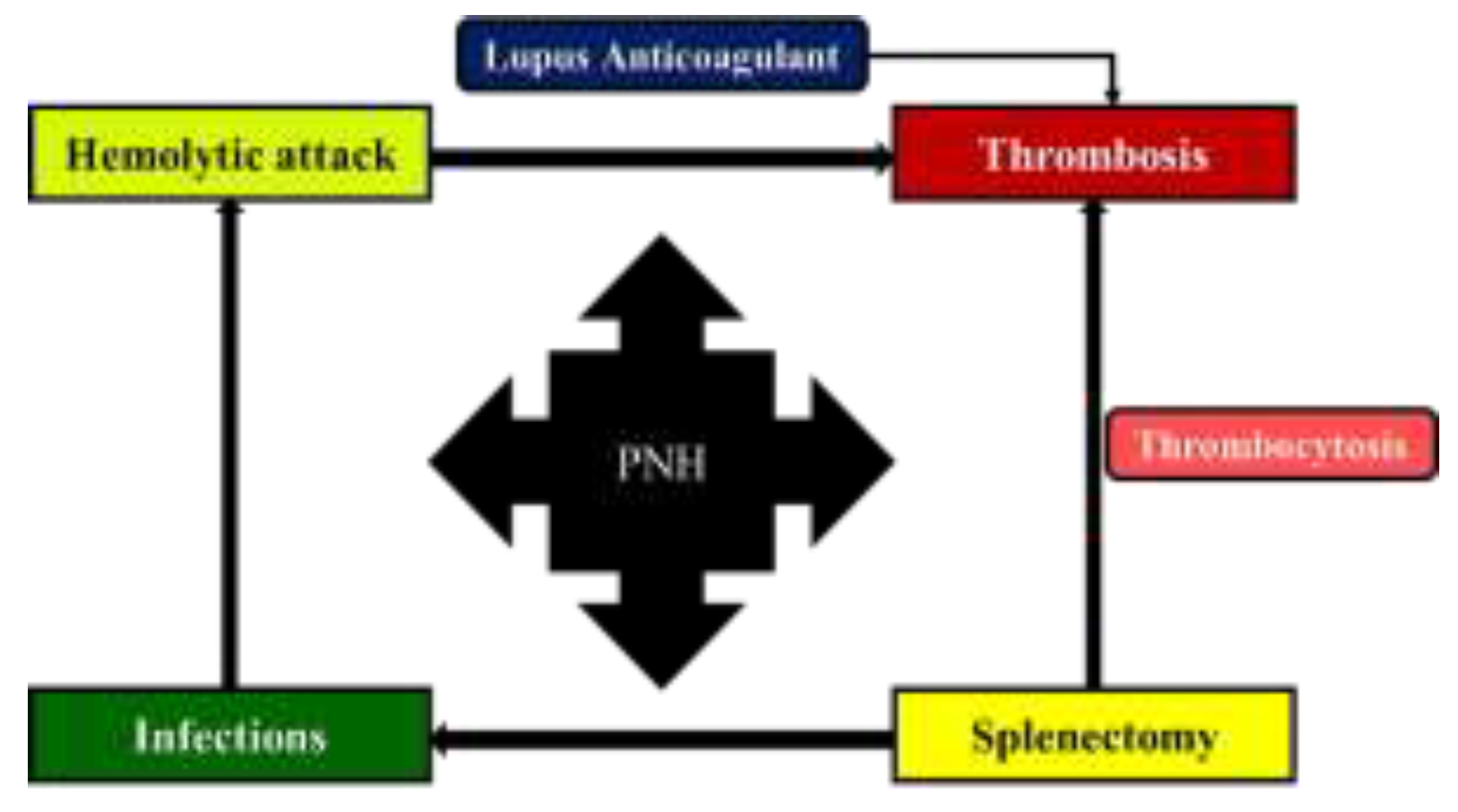
Interestingly, in our case, the pattern of disease changed over time. Up to the age of 42, the patient experienced numerous hemolytic attacks triggered by supervening events (infections, menses etc.) and only one thrombotic episode, namely the visceral splanchnic thrombosis involving the portal vein. Thereafter, the PNH pattern changed from predominantly hemolytic to predominantly thrombotic (with recurrent ischemic stroke involving the frontal region of the cortex), probably in direct relationship with the presence of the patent foramen ovale, occlusion of the internal carotid artery, additional cardiovascular risk factors (e.g. hypertension, obesity), the positive lupus anticoagulant and cryoglobulin test, as well as to the fact that the patient had previously undergone splenectomy. Patent foramen ovale is a possible cause of cryptogenic stroke in young adults, and recurrent ischemic stroke due to patent foramen ovale has already been reported in PNH patients [8,9,10]. Moreover, patients who underwent splenectomy seem to have a higher incidence of thromboembolic events, with portal-splenic mesenteric vein thrombosis being the most common finding [11]. Our patient associated additional risk factors for thrombosis (obesity, hypertension, lupus anticoagulant, cryoglobulins), although the screening for thrombophilia yielded negative results. Thus, as a take-home message, close monitoring of the patient diagnosed with PNH is of utmost importance, since ageing also leads to the acquisition of new disorders. Also, PNH patients who experience thrombosis should be prescribed anticoagulation therapy indefinitely [12,13,14]. The possible pathophysiological links explaining the evolution of PNH in this case report are depicted in Figure 3.
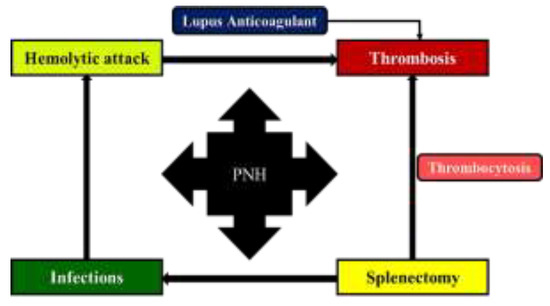
Figure 3.
Possible pathophysiological links explaining the evolution of PNH in our patient.
The patient would have been an ideal candidate for eculizumab, a recombinant humanized monoclonal antibody targeted against the complement protein C5, for multiple reasons: young age, intermediary-high risk, dominant major PNH major clone (type II PNH clone >80% within the granulocytes), frequent hemolytic attacks, and thrombotic events. However, the drug was unavailable in Romania at the time.
Highlights
- ✔
- Paroxysmal nocturnal hemoglobinuria (PNH) is an acquired non-malignant hematological disorder which affects the pluripotent hematopoietic stem cell.
- ✔
- PNH is characterized by intravascular hemolysis, thrombosis, and (or) bone marrow failure.
- ✔
- Although the clinical picture in PNH is dominated by the presence of hemolytic attacks, sometimes the pattern of the disease can change towards the predominance of thromboembolic episodes.
Conclusions
Establishing the diagnosis of PNH is a difficult task and its management requires teamwork. During the evolution of the disease, a PNH patient can acquire supplementary risk factors for thrombosis (obesity, hypertension, hypercholesterolemia), in addition to the pro-coagulant potential of the disease itself. Thus, the patient should be closely monitored. We reported this case to remind physicians that establishing the diagnosis of PNH is troublesome, and thus it is questionable whether PNH is a rare disease or just underdiagnosed. In this context, in the clinical practice of hematologists as well as other physicians, PNH remains a veritable Pandora's box.
Author Contributions
M.A.G. and I.U. wrote the paper. D.C. critically revised the paper for scientific content. M.A.G. and I.U. equally contributed to this paper and share first authorship. All authors read and approved the final version of the manuscript.
Compliance with ethical standards
Any aspect of the work covered in this manuscript has been conducted with the ethical approval of all relevant bodies and that such approvals are acknowledged within the manuscript. Written informed consent was obtained from the patient for the presentation of this case report.
Acknowledgments
We express our gratitude to all the physicians and allied health personnel who contributed to the management of this case.
Conflicts of interest
There are no known conflicts of interest in the publication of this article. The manuscript was read and approved by all authors.
Abbreviations
- Anti-DNAds antibodies, anti-double-stranded deoxyribonucleic acid antibodies.
- Anti-Ro/SSA antibodies, anti-Sjögren’s-syndrome- related antigen A antibodies.
- Anti-LA/SSB antibodies, anti-Sjögren’s-syndrome- related antigen B antibodies.
- CD, cluster of differentiation.
- DAF, decay-accelerating factor.
- GPI, glycosylphosphatidylinositol.
- LMWH, low molecular weight heparin.
- MAC, membrane attack complex.
- MIRL, membrane inhibitor of reactive lysis.
- PIGA, phosphatidylinositol glycan-A.
- PNH, paroxysmal nocturnal hemoglobinuria.
References
- Patriquin, C.J.; Kiss, T.; Caplan, S.; et al. How we treat paroxysmal nocturnal hemoglobinuria: A consensus statement of the Canadian PNH Network and review of the national registry. Eur J Haematol 2019, 102, 36–52. [Google Scholar] [CrossRef] [PubMed]
- Hill, A.; DeZern, A.E.; Kinoshita, T.; et al. Paroxysmal nocturnal haemoglobinuria. Nat Rev Dis Primers. 2017, 3, 17028. [Google Scholar] [CrossRef] [PubMed]
- Devalet, B.; Mullier, F.; Chatelain, B.; et al. Pathophysiology, diagnosis, and treatment of paroxysmal nocturnal hemoglobinuria: A review. Eur J Haematol 2015, 95, 190–198. [Google Scholar] [CrossRef] [PubMed]
- Parker, C.J. Update on the diagnosis and management of paroxysmal nocturnal hemoglobinuria. Hematology Am Soc Hematol Educ Program 2016, 2016, 208–216. [Google Scholar] [CrossRef] [PubMed]
- Jaime-Pérez, J.C.; Aguilar-Calderón, P.E.; Salazar-Cavazos, L.; et al. Evans syndrome: Clinical perspectives, biological insights and treatment modalities. J Blood Med 2018, 9, 171–184. [Google Scholar] [CrossRef] [PubMed]
- Fan, J.; Wang, Q.; Luo, B.; et al. Prevalence of prothrombotic factors in patients with Budd-Chiari syndrome or non-cirrhotic nonmalignant portal vein thrombosis: A hospital-based observational study. J Gastroenterol Hepatol 2020, 35, 1215–1222. [Google Scholar] [CrossRef] [PubMed]
- Ain, Q.U.; Saleem, H.; Iqbal, S.; Ghayas, R. A Case of Thrombosis Due To Paroxysmal Nocturnal Haemoglobinuria Presenting at An Early Age. J Ayub Med Coll Abbottabad 2018, 30, 138–139. [Google Scholar] [PubMed]
- Gad, M.M.; Ya’qoub, L.; Mahmoud, A.N.; et al. Echocardiography, Transcranial Doppler, and Oximetry for Imaging and Quantification of PFO-Mediated. In Patent Foramen Ovale Closure for Stroke, Myocardial Infarction, Peripheral Embolism, Migraine, and Hypoxemia; Academic Press: Cambridge, MA, USA, 2020; pp. 15–28. [Google Scholar] [CrossRef]
- Miranda, B.; Fonseca, A.C.; Ferro, J.M. Patent foramen ovale and stroke. J Neurol 2018, 265, 1943–1949. [Google Scholar] [CrossRef] [PubMed]
- Tiu, R.; Yacoub, H.; Maciejewski, J.; et al. Recurrent ischemic stroke in paroxysmal nocturnal hemoglobinuria: Paroxysmal nocturnal hemoglobinuria or missed patent foramen ovale? J Stroke Cerebrovasc Dis 2009, 18, 409–410. [Google Scholar] [CrossRef] [PubMed]
- Rottenstreich, A.; Kleinstern, G.; Spectre, G.; et al. Thromboembolic Events Following Splenectomy: Risk Factors, Prevention, Management and Outcomes. World J Surg 2018, 42, 675–681. [Google Scholar] [CrossRef] [PubMed]
- Ahsan, M.J.; Ishtiaq, R.; Ishtiaq, D. Ischemic Stroke Presenting as the First Symptom in a Setting of Paroxysmal Nocturnal Hemoglobinuria. Cureus 2017, 9, e1439. [Google Scholar] [CrossRef] [PubMed]
- Laslo, C.L.; Stoian, A.P.; Socea, B.; et al. New oral anticoagulants and their reversal agents. J Mind Med Sci. 2018, 5, 195–201. [Google Scholar] [CrossRef]
- Gaman, M.A.; Epingeac, M.E.; Gaman, A.M. The relationship between oxidative stress markers, age, neutrophil-to-lymphocyte ration and obesity. HemaSphere 2019, 3 (Suppl. S1). [Google Scholar] [CrossRef]

© 2020 by the author. 2020 Mihnea-Alexandru Găman, Iulia Ursuleac, Daniel Coriu