
Epigenetics and Congenital Heart Diseases



Abstract
1. Introduction
2. Morphogenesis, Embryology and Disease Spectrum
3. Epigenetic Alterations in Heart Disease
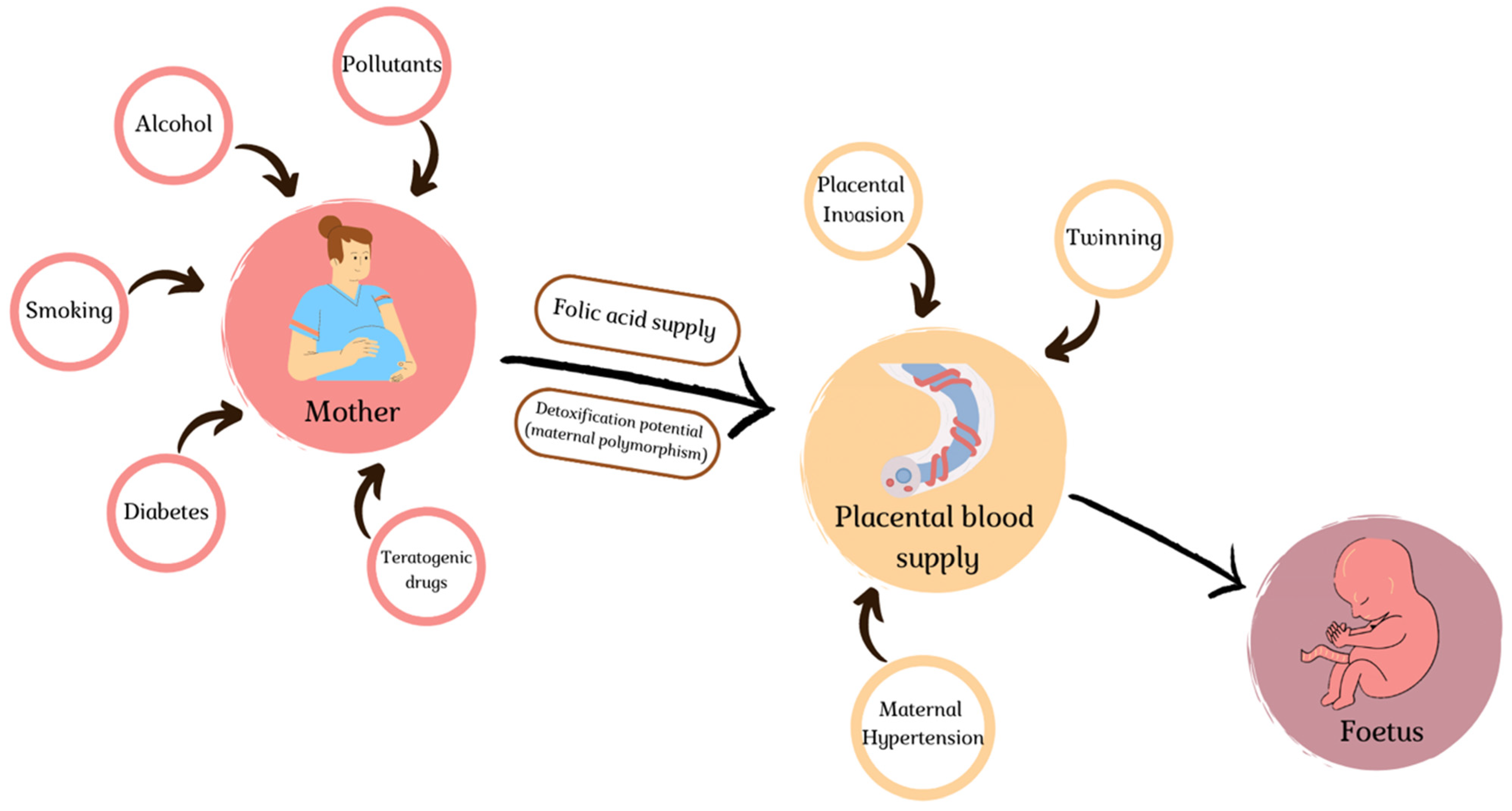
4. Environmental Slights
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- van der Linde, D.; Konings, E.E.M.; Slager, M.A.; Witsenburg, M.; Helbing, W.A.; Takkenberg, J.J.M.; Roos-Hesselink, J.W. Birth Prevalence of Congenital Heart Disease Worldwide: A Systematic Review and Meta-Analysis. J. Am. Coll. Cardiol. 2011, 58, 2241–2247. [Google Scholar] [CrossRef] [PubMed]
- Thiene, G.; Frescura, C. Anatomical and Pathophysiological Classification of Congenital Heart Disease. Cardiovasc. Pathol. 2010, 19, 259–274. [Google Scholar] [CrossRef] [PubMed]
- Bajolle, F.; Zaffran, S.; Bonnet, D. Genetics and Embryological Mechanisms of Congenital Heart Diseases. Arch. Cardiovasc. Dis. 2009, 102, 59–63. [Google Scholar] [CrossRef] [PubMed]
- Lim, T.B.; Foo, S.Y.R.; Chen, C.K. The Role of Epigenetics in Congenital Heart Disease. Genes 2021, 12, 390. [Google Scholar] [CrossRef]
- Fahed, A.C.; Gelb, B.D.; Seidman, J.G.; Seidman, C.E. Genetics of Congenital Heart Disease: The Glass Half Empty. Circ. Res. 2013, 112, 707–720. [Google Scholar] [CrossRef]
- Zaidi, S.; Choi, M.; Wakimoto, H.; Ma, L.; Jiang, J.; Overton, J.D.; Romano-Adesman, A.; Bjornson, R.D.; Breitbart, R.E.; Brown, K.K.; et al. De Novo Mutations in Histone-Modifying Genes in Congenital Heart Disease. Nature 2013, 498, 220–223. [Google Scholar] [CrossRef]
- Imany-Shakibai, H.; Yin, O.; Russell, M.R.; Sklansky, M.; Satou, G.; Afshar, Y. Discordant Congenital Heart Defects in Monochorionic Twins: Risk Factors and Proposed Pathophysiology. PLoS ONE 2021, 16, e0251160. [Google Scholar] [CrossRef]
- AlRais, F.; Feldstein, V.A.; Srivastava, D.; Gosnell, K.; Moon-Grady, A.J. Monochorionic Twins Discordant for Congenital Heart Disease: A Referral Center’s Experience and Possible Pathophysiologic Mechanisms. Prenat. Diagn. 2011, 31, 978–984. [Google Scholar] [CrossRef]
- Sedmera, D. Function and Form in the Developing Cardiovascular System. Cardiovasc. Res. 2011, 91, 252–259. [Google Scholar] [CrossRef]
- Asp, M.; Giacomello, S.; Larsson, L.; Wu, C.; Fürth, D.; Qian, X.; Wärdell, E.; Custodio, J.; Reimegård, J.; Salmén, F.; et al. A Spatiotemporal Organ-Wide Gene Expression and Cell Atlas of the Developing Human Heart. Cell 2019, 179, 1647–1660.e19. [Google Scholar] [CrossRef]
- Srivastava, D. Making or Breaking the Heart: From Lineage Determination to Morphogenesis. Cell 2006, 126, 1037–1048. [Google Scholar] [CrossRef] [PubMed]
- Buckingham, M.; Meilhac, S.; Zaffran, S. Building the Mammalian Heart from Two Sources of Myocardial Cells. Nat. Rev. Genet. 2005, 6, 826–835. [Google Scholar] [CrossRef] [PubMed]
- Restivo, A.; Piacentini, G.; Placidi, S.; Saffirio, C.; Marino, B. Cardiac Outflow Tract: A Review of Some Embryogenetic Aspects of the Conotruncal Region of the Heart. Anat. Rec. A Discov. Mol. Cell. Evol. Biol. 2006, 288, 936–943. [Google Scholar] [CrossRef] [PubMed]
- Schleich, J.-M.; Abdulla, T.; Summers, R.; Houyel, L. An Overview of Cardiac Morphogenesis. Arch. Cardiovasc. Dis. 2013, 106, 612–623. [Google Scholar] [CrossRef]
- Sadler, T.W. Establishing the Embryonic Axes: Prime Time for Teratogenic Insults. J. Cardiovasc. Dev. Dis. 2017, 4, 15. [Google Scholar] [CrossRef]
- Gittenberger-de Groot, A.C.; Calkoen, E.E.; Poelmann, R.E.; Bartelings, M.M.; Jongbloed, M.R.M. Morphogenesis and Molecular Considerations on Congenital Cardiac Septal Defects. Ann. Med. 2014, 46, 640–652. [Google Scholar] [CrossRef]
- Azhar, M.; Ware, S.M. Genetic and Developmental Basis of Cardiovascular Malformations. Clin. Perinatol. 2016, 43, 39–53. [Google Scholar] [CrossRef]
- Muntean, I.; Togănel, R.; Benedek, T. Genetics of Congenital Heart Disease: Past and Present. Biochem. Genet. 2017, 55, 105–123. [Google Scholar] [CrossRef]
- VanOudenhove, J.; Yankee, T.N.; Wilderman, A.; Cotney, J. Epigenomic and Transcriptomic Dynamics During Human Heart Organogenesis. Circ. Res. 2020, 127, e184–e209. [Google Scholar] [CrossRef]
- Acemel, R.D.; Maeso, I.; Gómez-Skarmeta, J.L. Topologically Associated Domains: A Successful Scaffold for the Evolution of Gene Regulation in Animals. Wiley Interdiscip. Rev. Dev. Biol. 2017, 6, e265. [Google Scholar] [CrossRef]
- George, R.M.; Firulli, A.B. Epigenetics and Heart Development. Front. Cell Dev. Biol. 2021, 9, 637996. [Google Scholar] [CrossRef] [PubMed]
- Piché, J.; Van Vliet, P.P.; Pucéat, M.; Andelfinger, G. The Expanding Phenotypes of Cohesinopathies: One Ring to Rule Them All! Cell Cycle Georget. Tex 2019, 18, 2828–2848. [Google Scholar] [CrossRef] [PubMed]
- Tessarz, P.; Kouzarides, T. Histone Core Modifications Regulating Nucleosome Structure and Dynamics. Nat. Rev. Mol. Cell Biol. 2014, 15, 703–708. [Google Scholar] [CrossRef]
- Miller, S.A.; Weinmann, A.S. An Essential Interaction between T-Box Proteins and Histone-Modifying Enzymes. Epigenetics 2009, 4, 85–88. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Fulcoli, F.G.; Franzese, M.; Liu, X.; Zhang, Z.; Angelini, C.; Baldini, A. Rebalancing Gene Haploinsufficiency in Vivo by Targeting Chromatin. Nat. Commun. 2016, 7, 11688. [Google Scholar] [CrossRef] [PubMed]
- Hou, Y.-S.; Wang, J.-Z.; Shi, S.; Han, Y.; Zhang, Y.; Zhi, J.-X.; Xu, C.; Li, F.-F.; Wang, G.-Y.; Liu, S.-L. Identification of Epigenetic Factor KAT2B Gene Variants for Possible Roles in Congenital Heart Diseases. Biosci. Rep. 2020, 40, BSR20191779. [Google Scholar] [CrossRef]
- Li, N.; Subrahmanyan, L.; Smith, E.; Yu, X.; Zaidi, S.; Choi, M.; Mane, S.; Nelson-Williams, C.; Behjati, M.; Kazemi, M.; et al. Mutations in the Histone Modifier PRDM6 Are Associated with Isolated Nonsyndromic Patent Ductus Arteriosus. Am. J. Hum. Genet. 2016, 98, 1082–1091. [Google Scholar] [CrossRef]
- Zhou, J.; Xiong, Y.; Dong, X.; Wang, H.; Qian, Y.; Ma, D.; Li, X. Genome-Wide Methylation Analysis Reveals Differentially Methylated CpG Sites and Altered Expression of Heart Development-Associated Genes in Fetuses with Cardiac Defects. Exp. Ther. Med. 2021, 22, 1032. [Google Scholar] [CrossRef]
- Chang, S.; Wang, Y.; Xin, Y.; Wang, S.; Luo, Y.; Wang, L.; Zhang, H.; Li, J. DNA Methylation Abnormalities of Imprinted Genes in Congenital Heart Disease: A Pilot Study. BMC Med. Genom. 2021, 14, 4. [Google Scholar] [CrossRef]
- Bahado-Singh, R.O.; Vishweswaraiah, S.; Aydas, B.; Yilmaz, A.; Saiyed, N.M.; Mishra, N.K.; Guda, C.; Radhakrishna, U. Precision Cardiovascular Medicine: Artificial Intelligence and Epigenetics for the Pathogenesis and Prediction of Coarctation in Neonates. J. Matern.-Fetal Neonatal Med. 2022, 35, 457–464. [Google Scholar] [CrossRef]
- Grunert, M.; Appelt, S.; Grossfeld, P.; Sperling, S.R. The Needle in the Haystack-Searching for Genetic and Epigenetic Differences in Monozygotic Twins Discordant for Tetralogy of Fallot. J. Cardiovasc. Dev. Dis. 2020, 7, 55. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Ye, M.; Xu, H.; Gu, R.; Ma, X.; Chen, M.; Li, X.; Sheng, W.; Huang, G. Methylation Status of CpG Sites in the NOTCH4 Promoter Region Regulates NOTCH4 Expression in Patients with Tetralogy of Fallot. Mol. Med. Rep. 2020, 22, 4412–4422. [Google Scholar] [CrossRef] [PubMed]
- Xie, P.; Zang, L.-Q.; Li, X.-K.; Shu, Q. An Epigenetic View of Developmental Diseases: New Targets, New Therapies. World J. Pediatr. WJP 2016, 12, 291–297. [Google Scholar] [CrossRef] [PubMed]
- Lan, Y.; Banks, K.M.; Pan, H.; Verma, N.; Dixon, G.R.; Zhou, T.; Ding, B.; Elemento, O.; Chen, S.; Huangfu, D.; et al. Stage-Specific Regulation of DNA Methylation by TET Enzymes during Human Cardiac Differentiation. Cell Rep. 2021, 37, 110095. [Google Scholar] [CrossRef] [PubMed]
- George, M.R.; Duan, Q.; Nagle, A.; Kathiriya, I.S.; Huang, Y.; Rao, K.; Haldar, S.M.; Bruneau, B.G. Minimal in Vivo Requirements for Developmentally Regulated Cardiac Long Intergenic Non-Coding RNAs. Dev. Camb. Engl. 2019, 146, dev185314. [Google Scholar] [CrossRef]
- Haunschild, J.; Schellinger, I.N.; Barnard, S.J.; von Aspern, K.; Davierwala, P.; Misfeld, M.; Petroff, D.; Borger, M.A.; Etz, C.D. Bicuspid Aortic Valve Patients Show Specific Epigenetic Tissue Signature Increasing Extracellular Matrix Destruction. Interact. Cardiovasc. Thorac. Surg. 2019, 29, 937–943. [Google Scholar] [CrossRef]
- Patterson, D. Molecular Genetic Analysis of Down Syndrome. Hum. Genet. 2009, 126, 195–214. [Google Scholar] [CrossRef]
- Vilardell, M.; Rasche, A.; Thormann, A.; Maschke-Dutz, E.; Pérez-Jurado, L.A.; Lehrach, H.; Herwig, R. Meta-Analysis of Heterogeneous Down Syndrome Data Reveals Consistent Genome-Wide Dosage Effects Related to Neurological Processes. BMC Genom. 2011, 12, 229. [Google Scholar] [CrossRef]
- Huang, A.C.; Olson, S.B.; Maslen, C.L. A Review of Recent Developments in Turner Syndrome Research. J. Cardiovasc. Dev. Dis. 2021, 8, 138. [Google Scholar] [CrossRef]
- Yan, S.; Lu, J.; Jiao, K. Epigenetic Regulation of Cardiac Neural Crest Cells. Front. Cell Dev. Biol. 2021, 9, 678954. [Google Scholar] [CrossRef]
- Rufaihah, A.J.; Chen, C.K.; Yap, C.H.; Mattar, C.N.Z. Mending a Broken Heart: In Vitro, in Vivo and in Silico Models of Congenital Heart Disease. Dis. Model. Mech. 2021, 14, dmm047522. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Liu, S.; Gao, H.; Han, L.; Chu, X.; Sheng, Y.; Shou, W.; Wang, Y.; Liu, Y.; Wan, J.; et al. Genome-Wide Studies Reveal the Essential and Opposite Roles of ARID1A in Controlling Human Cardiogenesis and Neurogenesis from Pluripotent Stem Cells. Genome Biol. 2020, 21, 169. [Google Scholar] [CrossRef] [PubMed]
- Moore-Morris, T.; van Vliet, P.P.; Andelfinger, G.; Puceat, M. Role of Epigenetics in Cardiac Development and Congenital Diseases. Physiol. Rev. 2018, 98, 2453–2475. [Google Scholar] [CrossRef] [PubMed]
- Weiss, K.; Lazar, H.P.; Kurolap, A.; Martinez, A.F.; Paperna, T.; Cohen, L.; Smeland, M.F.; Whalen, S.; Heide, S.; Keren, B.; et al. The CHD4-Related Syndrome: A Comprehensive Investigation of the Clinical Spectrum, Genotype-Phenotype Correlations, and Molecular Basis. Genet. Med. 2020, 22, 389–397. [Google Scholar] [CrossRef] [PubMed]
- Lavery, W.J.; Barski, A.; Wiley, S.; Schorry, E.K.; Lindsley, A.W. KMT2C/D COMPASS Complex-Associated Diseases [KCDCOM-ADs]: An Emerging Class of Congenital Regulopathies. Clin. Epigenetics 2020, 12, 10. [Google Scholar] [CrossRef]
- Froimchuk, E.; Jang, Y.; Ge, K. Histone H3 Lysine 4 Methyltransferase KMT2D. Gene 2017, 627, 337–342. [Google Scholar] [CrossRef]
- Blackburn, P.R.; Tischer, A.; Zimmermann, M.T.; Kemppainen, J.L.; Sastry, S.; Knight Johnson, A.E.; Cousin, M.A.; Boczek, N.J.; Oliver, G.; Misra, V.K.; et al. A Novel Kleefstra Syndrome-Associated Variant That Affects the Conserved TPLX Motif within the Ankyrin Repeat of EHMT1 Leads to Abnormal Protein Folding. J. Biol. Chem. 2017, 292, 3866–3876. [Google Scholar] [CrossRef]
- Fan, Z.; Yamaza, T.; Lee, J.S.; Yu, J.; Wang, S.; Fan, G.; Shi, S.; Wang, C.-Y. BCOR Regulates Mesenchymal Stem Cell Function by Epigenetic Mechanisms. Nat. Cell Biol. 2009, 11, 1002–1009. [Google Scholar] [CrossRef]
- Davoody, A.; Chen, I.-P.; Nanda, R.; Uribe, F.; Reichenberger, E.J. Oculofaciocardiodental Syndrome: A Rare Case and Review of the Literature. Cleft Palate-Craniofac. J. 2012, 49, e55–e60. [Google Scholar] [CrossRef]
- Graham, J.M.; Schwartz, C.E. MED12 Related Disorders. Am. J. Med. Genet. A. 2013, 161A, 2734–2740. [Google Scholar] [CrossRef]
- Schwartz, C.E.; Tarpey, P.S.; Lubs, H.A.; Verloes, A.; May, M.M.; Risheg, H.; Friez, M.J.; Futreal, P.A.; Edkins, S.; Teague, J.; et al. The Original Lujan Syndrome Family Has a Novel Missense Mutation (p.N1007S) in the MED12 Gene. J. Med. Genet. 2007, 44, 472–477. [Google Scholar] [CrossRef] [PubMed]
- Herskind, A.M.; Almind Pedersen, D.; Christensen, K. Increased Prevalence of Congenital Heart Defects in Monozygotic and Dizygotic Twins. Circulation 2013, 128, 1182–1188. [Google Scholar] [CrossRef] [PubMed]
- Bahtiyar, M.O.; Dulay, A.T.; Weeks, B.P.; Friedman, A.H.; Copel, J.A. Prevalence of Congenital Heart Defects in Monochorionic/Diamniotic Twin Gestations: A Systematic Literature Review. J. Ultrasound Med. 2007, 26, 1491–1498. [Google Scholar] [CrossRef] [PubMed]
- Karatza, A.A.; Wolfenden, J.L.; Taylor, M.J.O.; Wee, L.; Fisk, N.M.; Gardiner, H.M. Influence of Twin-Twin Transfusion Syndrome on Fetal Cardiovascular Structure and Function: Prospective Case-Control Study of 136 Monochorionic Twin Pregnancies. Heart Br. Card. Soc. 2002, 88, 271–277. [Google Scholar] [CrossRef]
- Hidaka, N.; Tsukimori, K.; Chiba, Y.; Hara, T.; Wake, N. Monochorionic Twins in Which at Least One Fetus Has a Congenital Heart Disease with or without Twin-Twin Transfusion Syndrome. J. Perinat. Med. 2007, 35, 425–430. [Google Scholar] [CrossRef] [PubMed]
- Manning, N. The Influence of Twinning on Cardiac Development. Early Hum. Dev. 2008, 84, 173–179. [Google Scholar] [CrossRef] [PubMed]
- Pruetz, J.D.; Sklansky, M.; Detterich, J.; Korst, L.M.; Llanes, A.; Chmait, R.H. Twin-Twin Transfusion Syndrome Treated with Laser Surgery: Postnatal Prevalence of Congenital Heart Disease in Surviving Recipients and Donors. Prenat. Diagn. 2011, 31, 973–977. [Google Scholar] [CrossRef] [PubMed]
- Hove, J.R.; Köster, R.W.; Forouhar, A.S.; Acevedo-Bolton, G.; Fraser, S.E.; Gharib, M. Intracardiac Fluid Forces Are an Essential Epigenetic Factor for Embryonic Cardiogenesis. Nature 2003, 421, 172–177. [Google Scholar] [CrossRef]
- Johnson, B.; Bark, D.; Van Herck, I.; Garrity, D.; Dasi, L.P. Altered Mechanical State in the Embryonic Heart Results in Time-Dependent Decreases in Cardiac Function. Biomech. Model. Mechanobiol. 2015, 14, 1379–1389. [Google Scholar] [CrossRef]
- Rugonyi, S. Genetic and Flow Anomalies in Congenital Heart Disease. AIMS Genet. 2016, 3, 157–166. [Google Scholar] [CrossRef]
- Santhanakrishnan, A.; Miller, L.A. Fluid Dynamics of Heart Development. Cell Biochem. Biophys. 2011, 61, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Jarrell, D.K.; Lennon, M.L.; Jacot, J.G. Epigenetics and Mechanobiology in Heart Development and Congenital Heart Disease. Diseases 2019, 7, 52. [Google Scholar] [CrossRef] [PubMed]
- Manning, N.; Archer, N. A Study to Determine the Incidence of Structural Congenital Heart Disease in Monochorionic Twins. Prenat. Diagn. 2006, 26, 1062–1064. [Google Scholar] [CrossRef] [PubMed]
- Hajdu, J.; Beke, A.; Marton, T.; Hruby, E.; Pete, B.; Papp, Z. Congenital Heart Diseases in Twin Pregnancies. Fetal Diagn. Ther. 2006, 21, 198–203. [Google Scholar] [CrossRef]
- Yuan, S.; Zaidi, S.; Brueckner, M. Congenital Heart Disease: Emerging Themes Linking Genetics and Development. Curr. Opin. Genet. Dev. 2013, 23, 352–359. [Google Scholar] [CrossRef]
- Giorgione, V.; Fesslova, V.; Boveri, S.; Candiani, M.; Khalil, A.; Cavoretto, P. Adverse Perinatal Outcome and Placental Abnormalities in Pregnancies with Major Fetal Congenital Heart Defects: A Retrospective Case-Control Study. Prenat. Diagn. 2020, 40, 1390–1397. [Google Scholar] [CrossRef]
- Maslen, C.L. Recent Advances in Placenta-Heart Interactions. Front. Physiol. 2018, 9, 735. [Google Scholar] [CrossRef]
- Andescavage, N.N.; Limperopoulos, C. Placental Abnormalities in Congenital Heart Disease. Transl. Pediatr. 2021, 10, 2148–2156. [Google Scholar] [CrossRef]
- Ozcan, T.; Kikano, S.; Plummer, S.; Strainic, J.; Ravishankar, S. The Association of Fetal Congenital Cardiac Defects and Placental Vascular Malperfusion. Pediatr. Dev. Pathol. 2021, 24, 187–192. [Google Scholar] [CrossRef]
- Courtney, J.A.; Cnota, J.F.; Jones, H.N. The Role of Abnormal Placentation in Congenital Heart Disease; Cause, Correlate, or Consequence? Front. Physiol. 2018, 9, 1045. [Google Scholar] [CrossRef]
- Bateman, B.T.; Huybrechts, K.F.; Fischer, M.A.; Seely, E.W.; Ecker, J.L.; Oberg, A.S.; Franklin, J.M.; Mogun, H.; Hernandez-Diaz, S. Chronic Hypertension in Pregnancy and the Risk of Congenital Malformations: A Cohort Study. Am. J. Obstet. Gynecol. 2015, 212, 337.E1–337.E14. [Google Scholar] [CrossRef] [PubMed]
- Ramakrishnan, A.; Lee, L.J.; Mitchell, L.E.; Agopian, A.J. Maternal Hypertension During Pregnancy and the Risk of Congenital Heart Defects in Offspring: A Systematic Review and Meta-Analysis. Pediatr. Cardiol. 2015, 36, 1442–1451. [Google Scholar] [CrossRef] [PubMed]
- Leirgul, E.; Brodwall, K.; Greve, G.; Vollset, S.E.; Holmstrøm, H.; Tell, G.S.; Øyen, N. Maternal Diabetes, Birth Weight, and Neonatal Risk of Congenital Heart Defects in Norway, 1994–2009. Obstet. Gynecol. 2016, 128, 1116–1125. [Google Scholar] [CrossRef] [PubMed]
- Lisowski, L.A.; Verheijen, P.M.; Copel, J.A.; Kleinman, C.S.; Wassink, S.; Visser, G.H.A.; Meijboom, E.-J. Congenital Heart Disease in Pregnancies Complicated by Maternal Diabetes Mellitus. An International Clinical Collaboration, Literature Review, and Meta-Analysis. Herz 2010, 35, 19–26. [Google Scholar] [CrossRef]
- Ding, Z.; Zhou, H.; McCauley, N.; Ko, G.; Zhang, K.K.; Xie, L. In Ovo Hyperglycemia Causes Congenital Limb Defects in Chicken Embryos via Disruption of Cell Proliferation and Apoptosis. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165955. [Google Scholar] [CrossRef]
- Gabbay-Benziv, R.; Reece, E.A.; Wang, F.; Yang, P. Birth Defects in Pregestational Diabetes: Defect Range, Glycemic Threshold and Pathogenesis. World J. Diabetes 2015, 6, 481–488. [Google Scholar] [CrossRef]
- Wang, F.; Wu, Y.; Quon, M.J.; Li, X.; Yang, P. ASK1 Mediates the Teratogenicity of Diabetes in the Developing Heart by Inducing ER Stress and Inhibiting Critical Factors Essential for Cardiac Development. Am. J. Physiol. Endocrinol. Metab. 2015, 309, E487–E499. [Google Scholar] [CrossRef]
- Persson, M.; Razaz, N.; Edstedt Bonamy, A.-K.; Villamor, E.; Cnattingius, S. Maternal Overweight and Obesity and Risk of Congenital Heart Defects. J. Am. Coll. Cardiol. 2019, 73, 44–53. [Google Scholar] [CrossRef]
- Helle, E.; Priest, J.R. Maternal Obesity and Diabetes Mellitus as Risk Factors for Congenital Heart Disease in the Offspring. J. Am. Heart Assoc. 2020, 9, e011541. [Google Scholar] [CrossRef]
- Linask, K.K.; Huhta, J. Folate Protection from Congenital Heart Defects Linked with Canonical Wnt Signaling and Epigenetics. Curr. Opin. Pediatr. 2010, 22, 561–566. [Google Scholar] [CrossRef]
- Padmanabhan, N.; Jia, D.; Geary-Joo, C.; Wu, X.; Ferguson-Smith, A.C.; Fung, E.; Bieda, M.C.; Snyder, F.F.; Gravel, R.A.; Cross, J.C.; et al. Mutation in Folate Metabolism Causes Epigenetic Instability and Transgenerational Effects on Development. Cell 2013, 155, 81–93. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Li, J.; Wei, C.; Lu, Q.; Tang, X.; Erickson, S.W.; MacLeod, S.L.; Hobbs, C.A. A Three-Way Interaction among Maternal and Fetal Variants Contributing to Congenital Heart Defects. Ann. Hum. Genet. 2016, 80, 20–31. [Google Scholar] [CrossRef] [PubMed]
- McKay, J.A.; Groom, A.; Potter, C.; Coneyworth, L.J.; Ford, D.; Mathers, J.C.; Relton, C.L. Genetic and Non-Genetic Influences during Pregnancy on Infant Global and Site Specific DNA Methylation: Role for Folate Gene Variants and Vitamin B12. PLoS ONE 2012, 7, e33290. [Google Scholar] [CrossRef] [PubMed]
- Joshi, R.O.; Chellappan, S.; Kukshal, P. Exploring the Role of Maternal Nutritional Epigenetics in Congenital Heart Disease. Curr. Dev. Nutr. 2020, 4, nzaa166. [Google Scholar] [CrossRef]
- Ionescu-Ittu, R.; Marelli, A.J.; Mackie, A.S.; Pilote, L. Prevalence of Severe Congenital Heart Disease after Folic Acid Fortification of Grain Products: Time Trend Analysis in Quebec, Canada. BMJ 2009, 338, b1673. [Google Scholar] [CrossRef]
- Eckmann-Scholz, C.; von Kaisenberg, C.S.; Alkatout, I.; Jonat, W.; Rajabi-Wieckhorst, A. Pathologic Ultrasound Findings and Risk for Congenital Anomalies in Teenage Pregnancies. J. Matern.-Fetal Neonatal Med. 2012, 25, 1950–1952. [Google Scholar] [CrossRef]
- Khalil, A.; Tanos, R.; El-Hachem, N.; Kurban, M.; Bouvagnet, P.; Bitar, F.; Nemer, G. A HAND to TBX5 Explains the Link Between Thalidomide and Cardiac Diseases. Sci. Rep. 2017, 7, 1416. [Google Scholar] [CrossRef]
- Gutgesell, H. Lithium Use in Pregnancy and the Risk of Cardiac Malformations. N. Engl. J. Med. 2017, 377, 893–894. [Google Scholar] [CrossRef]
- Han, M.; Serrano, M.C.; Lastra-Vicente, R.; Brinez, P.; Acharya, G.; Huhta, J.C.; Chen, R.; Linask, K.K. Folate Rescues Lithium-, Homocysteine- and Wnt3A-Induced Vertebrate Cardiac Anomalies. Dis. Model. Mech. 2009, 2, 467–478. [Google Scholar] [CrossRef]
- Tanoshima, M.; Kobayashi, T.; Tanoshima, R.; Beyene, J.; Koren, G.; Ito, S. Risks of Congenital Malformations in Offspring Exposed to Valproic Acid in Utero: A Systematic Review and Cumulative Meta-Analysis. Clin. Pharmacol. Ther. 2015, 98, 417–441. [Google Scholar] [CrossRef]
- Duan, H.-Y.; Zhou, K.-Y.; Wang, T.; Zhang, Y.; Li, Y.-F.; Hua, Y.-M.; Wang, C. Disruption of Planar Cell Polarity Pathway Attributable to Valproic Acid-Induced Congenital Heart Disease through Hdac3 Participation in Mice. Chin. Med. J. 2018, 131, 2080–2088. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.-H.; Ho, Y.-L.; Huang, P.-T.; Chu, S.-L.; Tsai, H.-J.; Liou, H.-H. The Phosphorylation State of GSK3β Serine 9 Correlated to the Development of Valproic Acid-Associated Fetal Cardiac Teratogenicity, Fetal VPA Syndrome, Rescued by Folic Acid Administration. Cardiovasc. Toxicol. 2016, 16, 34–45. [Google Scholar] [CrossRef] [PubMed]
- Caldwell, P.T.; Manziello, A.; Howard, J.; Palbykin, B.; Runyan, R.B.; Selmin, O. Gene Expression Profiling in the Fetal Cardiac Tissue after Folate and Low-Dose Trichloroethylene Exposure. Birt. Defects Res. A Clin. Mol. Teratol. 2010, 88, 111–127. [Google Scholar] [CrossRef] [PubMed]
- Nicoll, R. Environmental Contaminants and Congenital Heart Defects: A Re-Evaluation of the Evidence. Int. J. Environ. Res. Public. Health 2018, 15, 2096. [Google Scholar] [CrossRef]
- Kuehl, K.S.; Loffredo, C.A. A Cluster of Hypoplastic Left Heart Malformation in Baltimore, Maryland. Pediatr. Cardiol. 2006, 27, 25–31. [Google Scholar] [CrossRef]
- de Gannes, M.; Ko, C.-I.; Zhang, X.; Biesiada, J.; Niu, L.; Koch, S.E.; Medvedovic, M.; Rubinstein, J.; Puga, A. Dioxin Disrupts Dynamic DNA Methylation Patterns in Genes That Govern Cardiomyocyte Maturation. Toxicol. Sci. 2020, 178, 325–337. [Google Scholar] [CrossRef]
- Vecoli, C.; Pulignani, S.; Andreassi, M.G. Genetic and Epigenetic Mechanisms Linking Air Pollution and Congenital Heart Disease. J. Cardiovasc. Dev. Dis. 2016, 3, 32. [Google Scholar] [CrossRef]
- Cheng, W.; Zhou, R.; Feng, Y.; Wang, Y. Mainstream Smoke and Sidestream Smoke Affect the Cardiac Differentiation of Mouse Embryonic Stem Cells Discriminately. Toxicology 2016, 357–358, 1–10. [Google Scholar] [CrossRef]
- Hoyt, A.T.; Canfield, M.A.; Romitti, P.A.; Botto, L.D.; Anderka, M.T.; Krikov, S.V.; Tarpey, M.K.; Feldkamp, M.L. Associations between Maternal Periconceptional Exposure to Secondhand Tobacco Smoke and Major Birth Defects. Am. J. Obstet. Gynecol. 2016, 215, 613.e1–613.e11. [Google Scholar] [CrossRef]
- Gianicolo, E.A.L.; Cresci, M.; Ait-Ali, L.; Foffa, I.; Andreassi, M.G. Smoking and Congenital Heart Disease: The Epidemiological and Biological Link. Curr. Pharm. Des. 2010, 16, 2572–2577. [Google Scholar] [CrossRef]
- Zhang, S.; Wang, L.; Yang, T.; Chen, L.; Zhao, L.; Wang, T.; Chen, L.; Ye, Z.; Zheng, Z.; Qin, J. Parental Alcohol Consumption and the Risk of Congenital Heart Diseases in Offspring: An Updated Systematic Review and Meta-Analysis. Eur. J. Prev. Cardiol. 2020, 27, 410–421. [Google Scholar] [CrossRef] [PubMed]
- Jawaid, S.; Strainic, J.P.; Kim, J.; Ford, M.R.; Thrane, L.; Karunamuni, G.H.; Sheehan, M.M.; Chowdhury, A.; Gillespie, C.A.; Rollins, A.M.; et al. Glutathione Protects the Developing Heart from Defects and Global DNA Hypomethylation Induced by Prenatal Alcohol Exposure. Alcohol. Clin. Exp. Res. 2021, 45, 69–78. [Google Scholar] [CrossRef] [PubMed]
- Pan, B.; Zhu, J.; Lv, T.; Sun, H.; Huang, X.; Tian, J. Alcohol Consumption during Gestation Causes Histone3 Lysine9 Hyperacetylation and an Alternation of Expression of Heart Development-Related Genes in Mice. Alcohol. Clin. Exp. Res. 2014, 38, 2396–2402. [Google Scholar] [CrossRef]
- Chen, Z.; Li, S.; Guo, L.; Peng, X.; Liu, Y. Prenatal Alcohol Exposure Induced Congenital Heart Diseases: From Bench to Bedside. Birth Defects Res. 2021, 113, 521–534. [Google Scholar] [CrossRef]
- Tararbit, K.; Houyel, L.; Bonnet, D.; De Vigan, C.; Lelong, N.; Goffinet, F.; Khoshnood, B. Risk of Congenital Heart Defects Associated with Assisted Reproductive Technologies: A Population-Based Evaluation. Eur. Heart J. 2011, 32, 500–508. [Google Scholar] [CrossRef] [PubMed]
- Giorgione, V.; Parazzini, F.; Fesslova, V.; Cipriani, S.; Candiani, M.; Inversetti, A.; Sigismondi, C.; Tiberio, F.; Cavoretto, P. Congenital Heart Defects in IVF/ICSI Pregnancy: Systematic Review and Meta-Analysis. Ultrasound Obstet. Gynecol. 2018, 51, 33–42. [Google Scholar] [CrossRef] [PubMed]
- Panagiotopoulou, O.; Fouzas, S.; Sinopidis, X.; Mantagos, S.P.; Dimitriou, G.; Karatza, A.A. Congenital Heart Disease in Twins: The Contribution of Type of Conception and Chorionicity. Int. J. Cardiol. 2016, 218, 144–149. [Google Scholar] [CrossRef] [PubMed]
- Tenorio, J.; Romanelli, V.; Martin-Trujillo, A.; Fernández, G.-M.; Segovia, M.; Perandones, C.; Pérez Jurado, L.A.; Esteller, M.; Fraga, M.; Arias, P.; et al. Clinical and Molecular Analyses of Beckwith-Wiedemann Syndrome: Comparison between Spontaneous Conception and Assisted Reproduction Techniques. Am. J. Med. Genet. A. 2016, 170, 2740–2749. [Google Scholar] [CrossRef]
- Benincasa, G.; Marfella, R.; Della Mura, N.; Schiano, C.; Napoli, C. Strengths and Opportunities of Network Medicine in Cardiovascular Diseases. Circ. J. 2020, 84, 144–152. [Google Scholar] [CrossRef]
- Napoli, C.; Benincasa, G.; Donatelli, F.; Ambrosio, G. Precision Medicine in Distinct Heart Failure Phenotypes: Focus on Clinical Epigenetics. Am. Heart J. 2020, 224, 113–128. [Google Scholar] [CrossRef]
- Cheng, F.; Zhao, J.; Wang, Y.; Lu, W.; Liu, Z.; Zhou, Y.; Martin, W.R.; Wang, R.; Huang, J.; Hao, T.; et al. Comprehensive Characterization of Protein–Protein Interactions Perturbed by Disease Mutations. Nat. Genet. 2021, 53, 342–353. [Google Scholar] [CrossRef] [PubMed]
- Morris, J.K.; Springett, A.L.; Greenlees, R.; Loane, M.; Addor, M.-C.; Arriola, L.; Barisic, I.; Bergman, J.E.H.; Csaky-Szunyogh, M.; Dias, C.; et al. Trends in Congenital Anomalies in Europe from 1980 to 2012. PLoS ONE 2018, 13, e0194986. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Epigenetic Function | Syndrome Affected Gene | Cardiac Manifestation | Reference |
|---|---|---|---|
| ATP-dependent chromatin modifiers | Coffin–Siris syndrome ARID1A/B, SMARCB1, SMARCA4, SAMRCE1 | ASD VSD TOF PDA | [40,41,42] |
| CHARGE syndrome CHD7 | TOF DORV VSD AVSD PDA PS IAA | [40,43] | |
| Sifrim-Hitz-Weiss syndrome CHD4 | ASD VSD PS PDA TOF MV anomalies | [44] | |
| Williams syndrome WSTF | AS PS | [40] | |
| Histone modifiers | Kabuki syndrome KMT2D, KDM6A, WDR5 | Coarctation ASD VSD | [40,45,46] |
| Kleefstra syndrome EHMT1 | ASD VSD TOF Coarctation BAV PS | [40,45,47] | |
| Wolf–Hirschhorn syndrome WHSC1/2, LETM1 | ASD | [40] | |
| Rubinstein–Taybi CREBBP, EP300 | PDA ASD VSD HLHS BAV | [41,45] | |
| KAT2B | ASD VSD PDA TOF PS | [26] | |
| PRDM6 | PDA | [27] | |
| Oculofaciocardiodental syndrome BCOR | ASD VSD MV anomalies | [48,49] | |
| Cohesinopathies | Cornelia de Lange NIPBL, HDAC8, SMC1, SMC3, RAD21, BRD4, ANKRD11 | TOF ASD VSD PDA PS | [22,41] |
| Robert’s syndrome ESCO2 | VSD ASD PDA | [22] | |
| Warsaw breakage syndrome DDX11 | TOF VSD | [22] | |
| ARTX syndrome ATRX | VSD ASD TOF PDA PS/AS | [22] | |
| CHOPS syndrome AFF4 | VSD PDA | [22] | |
| STAG2-related X-linked Intellectual Deficiency STAG2 | VSD | [22] | |
| CAID syndrome SGO1 | PS/AS VSD | [22] | |
| Mediatorpathies | Opitz–Kaveggia syndrome MED12 | TOF | [43,50] |
| Lujan–Fryns syndrome MED12 | TOF | [43,51] | |
| Ohdo syndrome MED13L | TOF | [43] | |
| DNA methylation modulators | ICF syndrome DMNT3B | ASD VSD | [40] |
| Chromatin-modifier regulators | DiGeorge syndrome TBX1 | IAA Truncus arteriosus TOF TGA VSD | [21,43] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Linglart, L.; Bonnet, D. Epigenetics and Congenital Heart Diseases. J. Cardiovasc. Dev. Dis. 2022, 9, 185. https://doi.org/10.3390/jcdd9060185
Linglart L, Bonnet D. Epigenetics and Congenital Heart Diseases. Journal of Cardiovascular Development and Disease. 2022; 9(6):185. https://doi.org/10.3390/jcdd9060185
Chicago/Turabian StyleLinglart, Léa, and Damien Bonnet. 2022. "Epigenetics and Congenital Heart Diseases" Journal of Cardiovascular Development and Disease 9, no. 6: 185. https://doi.org/10.3390/jcdd9060185
APA StyleLinglart, L., & Bonnet, D. (2022). Epigenetics and Congenital Heart Diseases. Journal of Cardiovascular Development and Disease, 9(6), 185. https://doi.org/10.3390/jcdd9060185