
PDE-Mediated Cyclic Nucleotide Compartmentation in Vascular Smooth Muscle Cells: From Basic to a Clinical Perspective




Abstract
1. Introduction
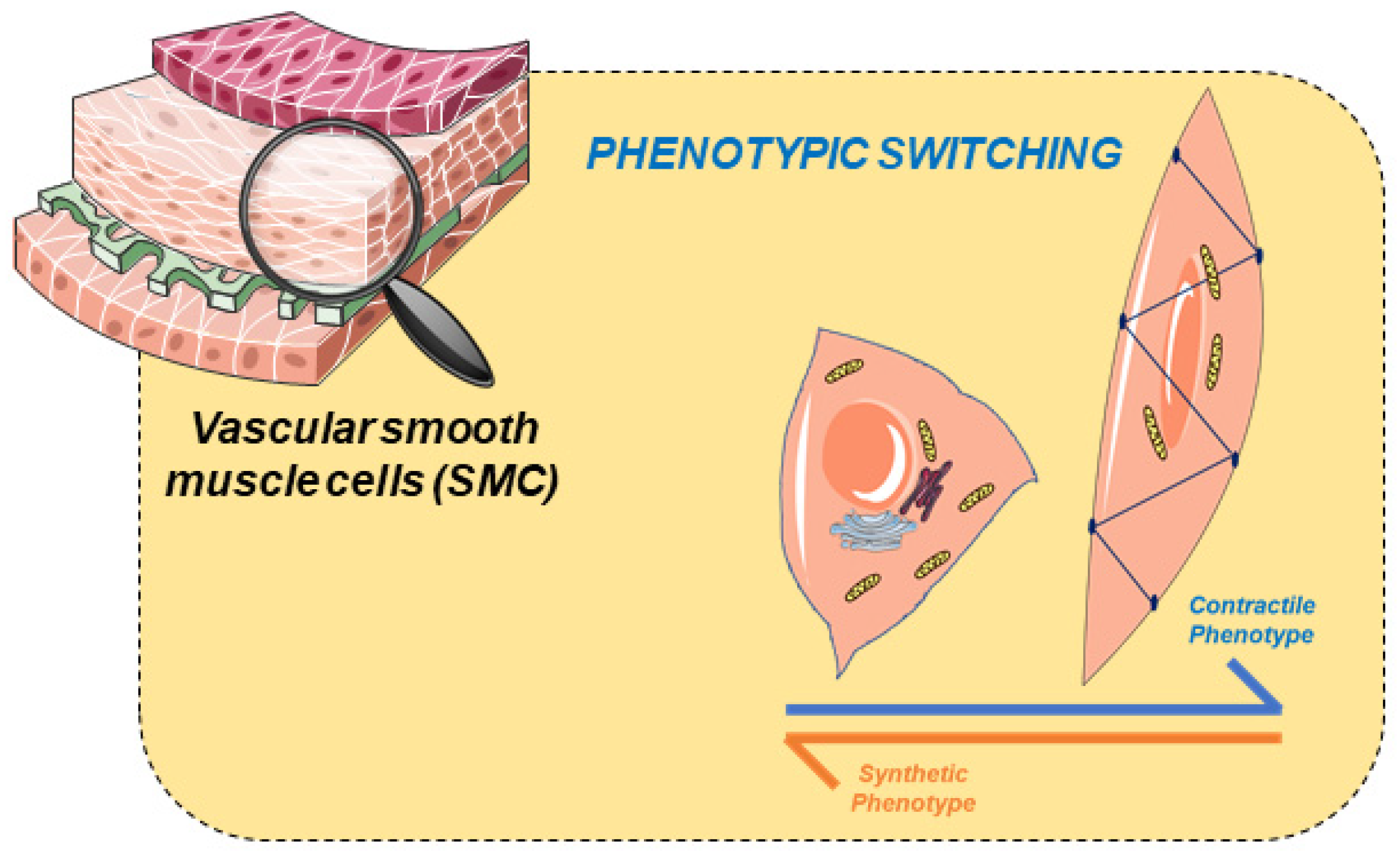
2. Vascular Smooth Muscle
3. Vascular Cyclic Nucleotides
4. Vascular Function of Cyclic Nucleotide Phosphodiesterases
5. How Do Cyclic Nucleotides Regulate Vascular Tone?
5.1. cAMP Effector Proteins
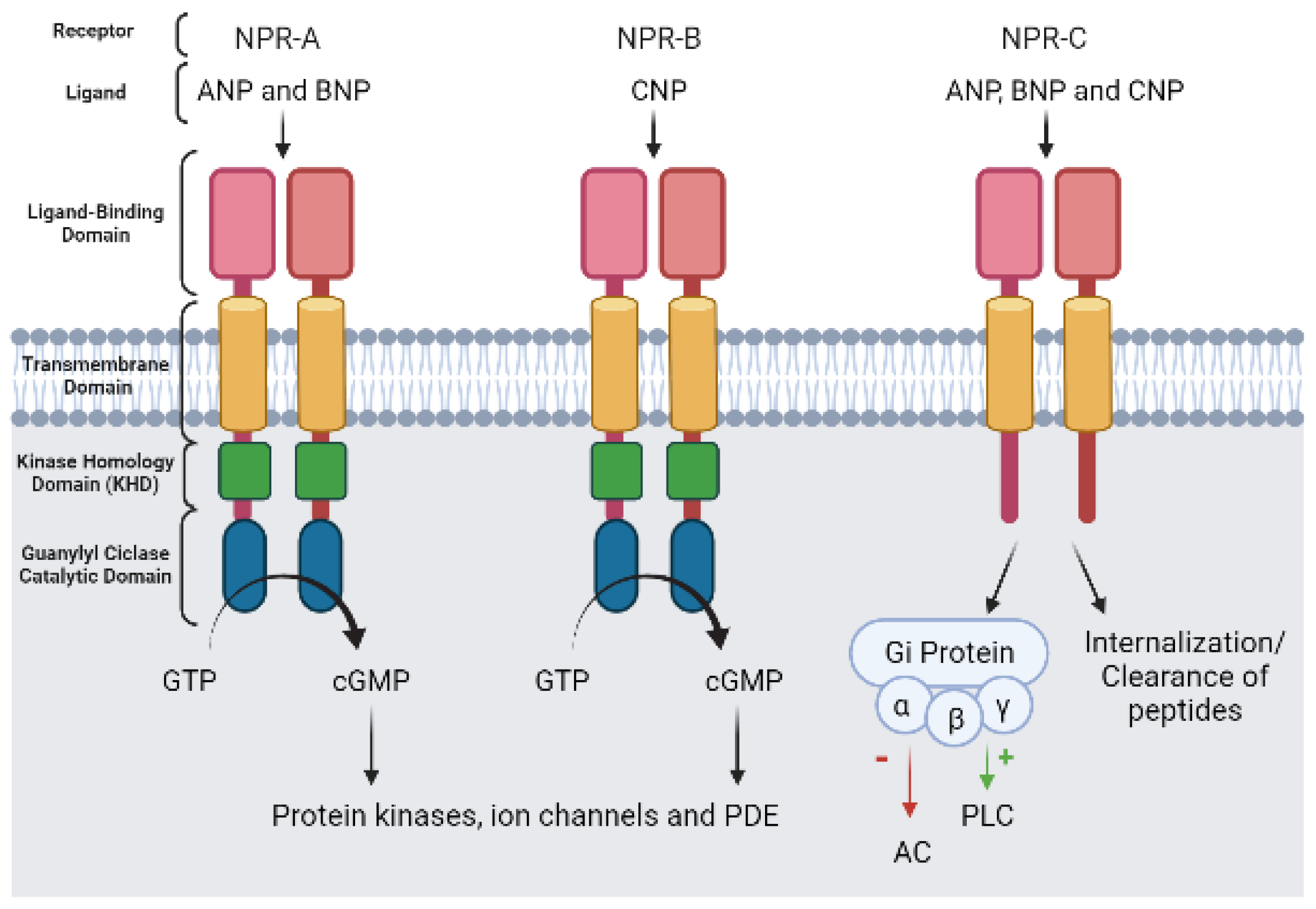
5.2. cGMP Effector Proteins
6. Compartmentation of Cyclic Nucleotides Signaling
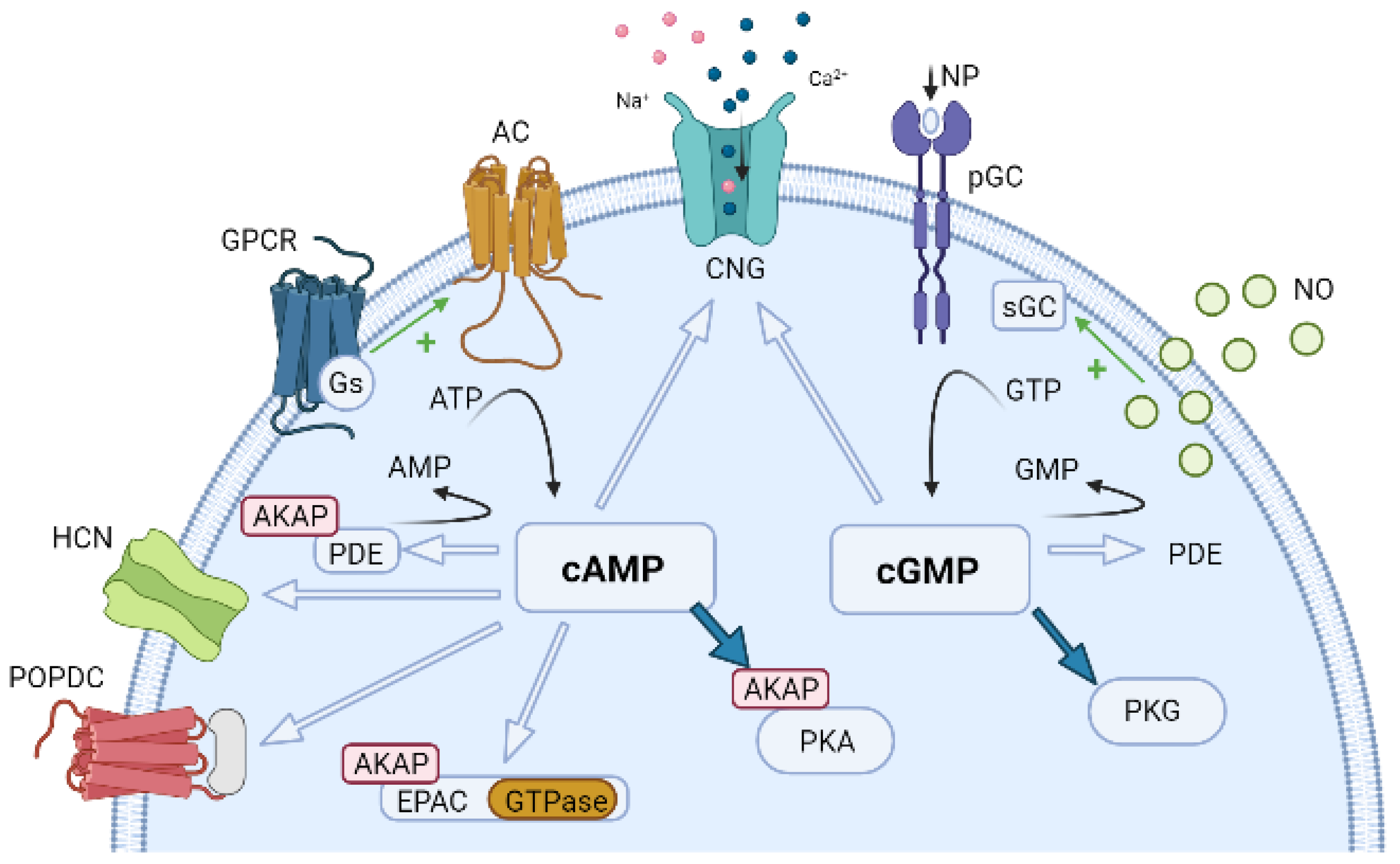
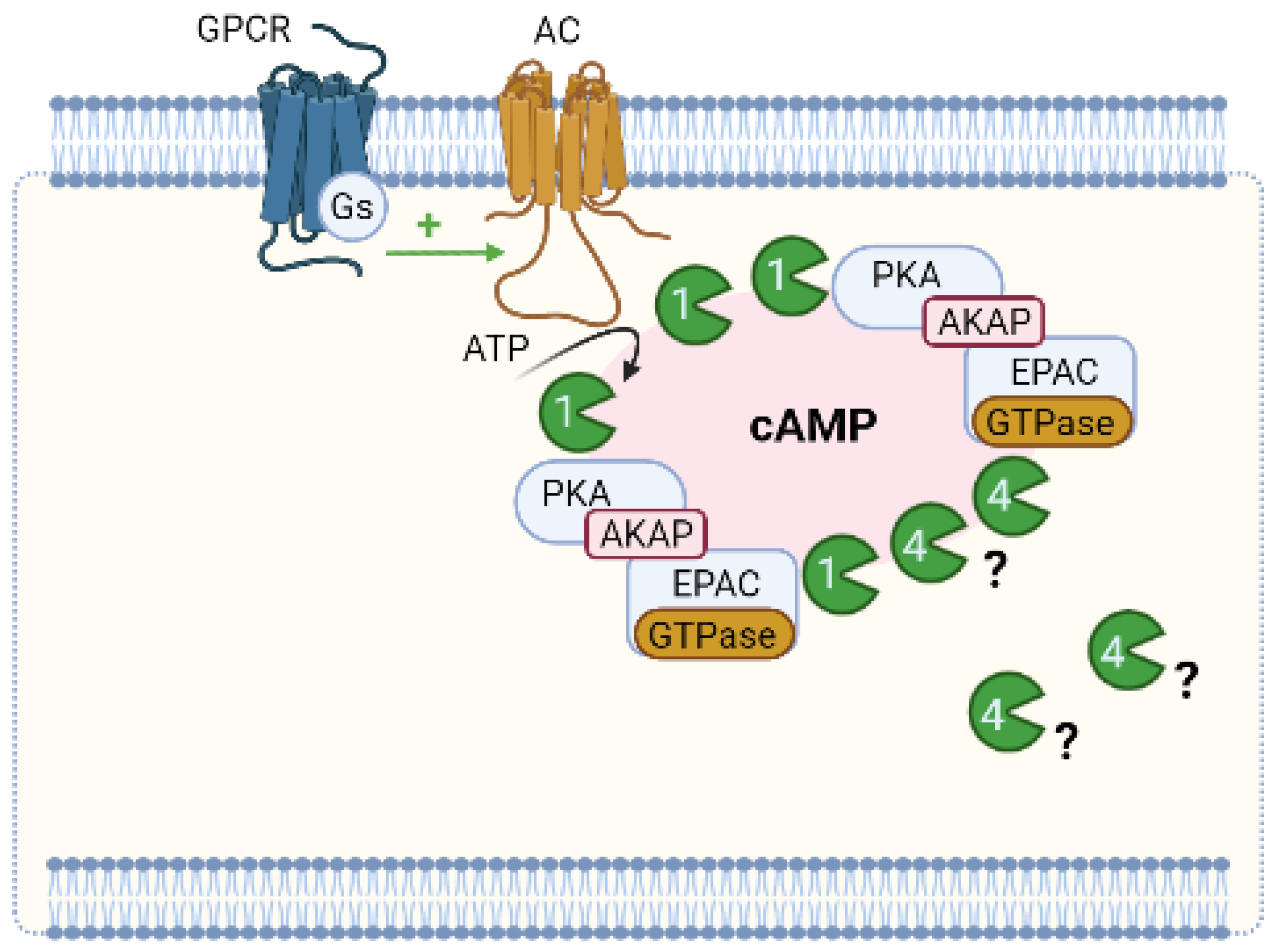
6.1. Compartmentation of cAMP Signalling
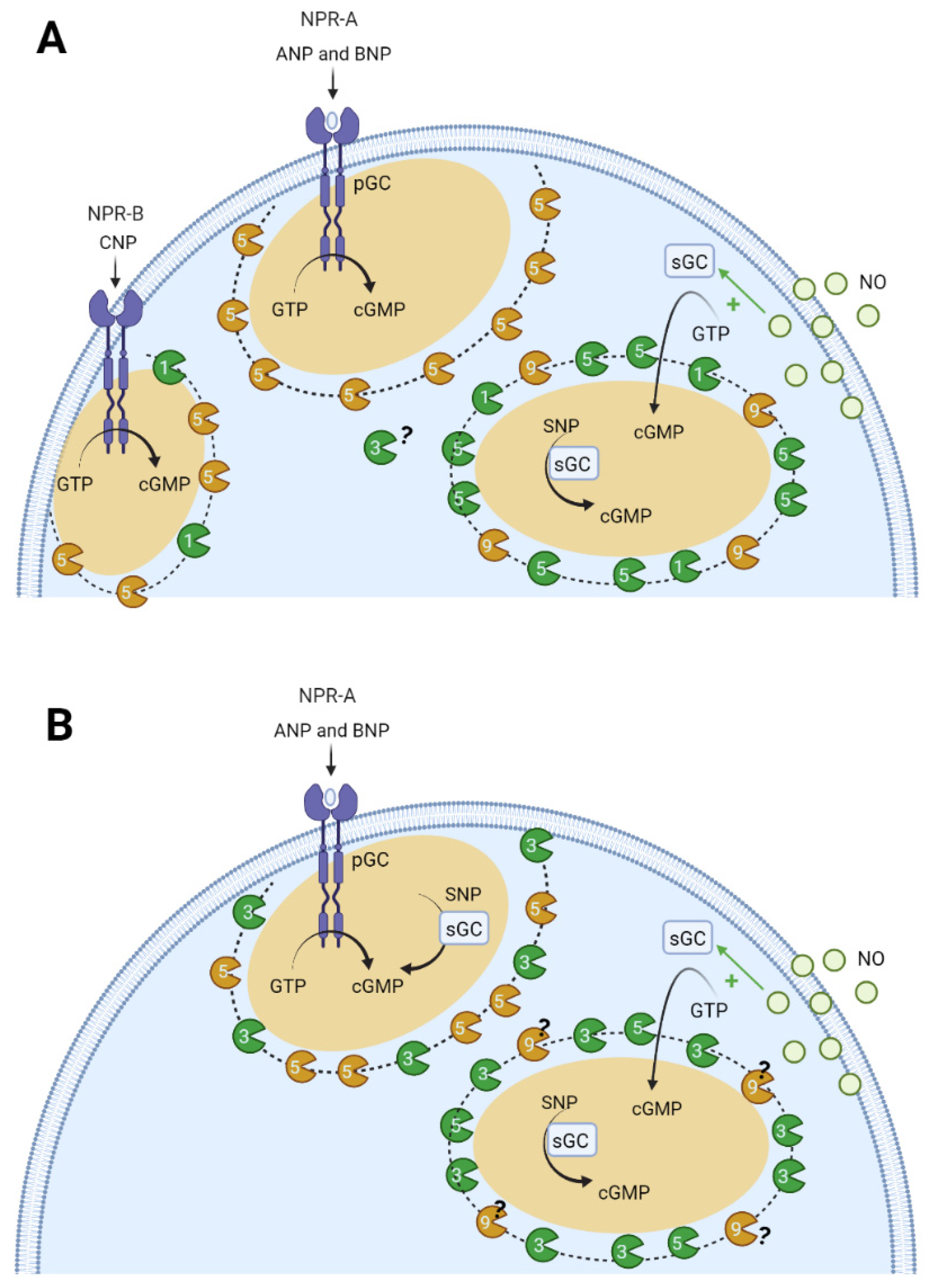
6.2. Compartmentation of cGMP Signalling
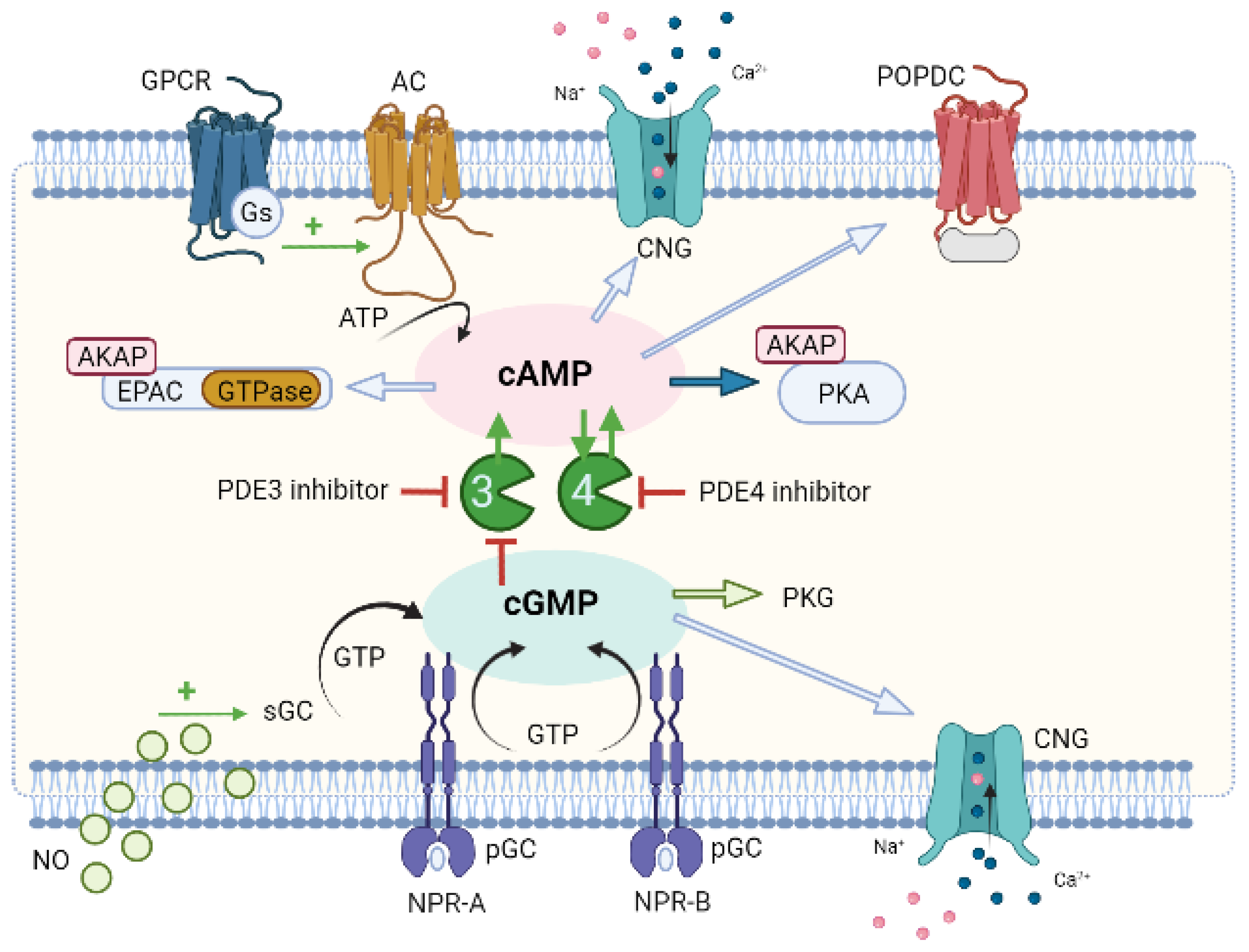
7. Compartmentation as a Potential Therapeutic Approach for CVD
8. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
Abbreviations
References
- Herrmann, J.; Babic, M.; Tölle, M.; Van Der Giet, M.; Schuchardt, M. Research Models for Studying Vascular Calcification. Int. J. Mol. Sci. 2020, 21, 2204. [Google Scholar] [CrossRef] [PubMed]
- Dejea, H.; Garcia-Canadilla, P.; Cook, A.C.; Guasch, E.; Zamora, M.; Crispi, F.; Stampanoni, M.; Bijnens, B.; Bonnin, A. Comprehensive Analysis of Animal Models of Cardiovascular Disease using Multiscale X-Ray Phase Contrast Tomography. Sci. Rep. 2019, 9, 1–12. [Google Scholar] [CrossRef]
- Lorigo, M.; Mariana, M.; Feiteiro, J.; Cairrao, E. How is the human umbilical artery regulated? J. Obstet. Gynaecol. Res. 2018, 44, 1193–1201. [Google Scholar] [CrossRef] [PubMed]
- Mangana, C.; Lorigo, M.; Cairrao, E. Implications of Endothelial Cell-Mediated Dysfunctions in Vasomotor Tone Regulation. Biologics 2021, 1, 231–251. [Google Scholar] [CrossRef]
- Morgado, M.; Cairrão, E.; Santos-Silva, A.J.; Verde, I. Cyclic nucleotide-dependent relaxation pathways in vascular smooth muscle. Experientia 2011, 69, 247–266. [Google Scholar] [CrossRef]
- Santos-Silva, A.J.; Cairrão, E.; Morgado, M.; Álvarez, E.; Verde, I. PDE4 and PDE5 regulate cyclic nucleotides relaxing effects in human umbilical arteries. Eur. J. Pharmacol. 2008, 582, 102–109. [Google Scholar] [CrossRef] [PubMed]
- Manoury, B.; Idres, S.; Leblais, V.; Fischmeister, R. Ion channels as effectors of cyclic nucleotide pathways: Functional relevance for arterial tone regulation. Pharmacol. Ther. 2020, 209, 107499. [Google Scholar] [CrossRef] [PubMed]
- Holland, N.A.; Francisco, J.T.; Johnson, S.C.; Morgan, J.S.; Dennis, T.J.; Gadireddy, N.R.; Tulis, D.A. Cyclic Nucleotide-Directed Protein Kinases in Cardiovascular Inflammation and Growth. J. Cardiovasc. Dev. Dis. 2018, 5, 6. [Google Scholar] [CrossRef]
- Bork, N.I.; Nikolaev, V.O. cGMP Signaling in the Cardiovascular System—The Role of Compartmentation and Its Live Cell Imaging. Int. J. Mol. Sci. 2018, 19, 801. [Google Scholar] [CrossRef] [PubMed]
- Feiteiro, J.; Verde, I.; Cairrão, E. Cyclic guanosine monophosphate compartmentation in human vascular smooth muscle cells. Cell. Signal. 2015, 28, 109–116. [Google Scholar] [CrossRef] [PubMed]
- Maurice, D.H.; Ke, H.; Ahmad, F.; Wang, Y.; Chung, J.; Manganiello, V.C. Advances in targeting cyclic nucleotide phosphodiesterases. Nat. Rev. Drug Discov. 2014, 13, 290–314. [Google Scholar] [CrossRef]
- Owens, G.K.; Kumar, M.S.; Wamhoff, B.R. Molecular Regulation of Vascular Smooth Muscle Cell Differentiation in Development and Disease. Physiol. Rev. 2004, 84, 767–801. [Google Scholar] [CrossRef] [PubMed]
- Ashraf, J.V.; Zen, A.A.H. Role of Vascular Smooth Muscle Cell Phenotype Switching in Arteriogenesis. Int. J. Mol. Sci. 2021, 22, 10585. [Google Scholar] [CrossRef] [PubMed]
- Brozovich, F.; Nicholson, C.; Degen, C.; Gao, Y.Z.; Aggarwal, M.; Morgan, K. Mechanisms of Vascular Smooth Muscle Contraction and the Basis for Pharmacologic Treatment of Smooth Muscle Disorders. Pharmacol. Rev. 2016, 68, 476–532. [Google Scholar] [CrossRef]
- Van Der Heijden, O.W.H.; Essers, Y.P.G.; Simkens, L.H.J.; Teunissen, Q.G.A.; Peeters, L.L.H.; De Mey, J.G.R.; Van Eys, G.J.J.M. Aging Blunts Remodeling of the Uterine Artery during Murine Pregnancy. J. Soc. Gynecol. Investig. 2004, 11, 304–310. [Google Scholar] [CrossRef]
- Pagé, E.L.; Robitaille, G.A.; Pouysségur, J.; Richard, D. Induction of Hypoxia-inducible Factor-1α by Transcriptional and Translational Mechanisms. J. Biol. Chem. 2002, 277, 48403–48409. [Google Scholar] [CrossRef]
- Lorigo, M.; Cairrao, E. Fetoplacental vasculature as a model to study human cardiovascular endocrine disruption. Mol. Asp. Med. 2021, 101054. [Google Scholar] [CrossRef]
- Zaccolo, M. Spatial control of cAMP signalling in health and disease. Curr. Opin. Pharmacol. 2011, 11, 649–655. [Google Scholar] [CrossRef] [PubMed]
- Lugnier, C. Cyclic nucleotide phosphodiesterase (PDE) superfamily: A new target for the development of specific therapeutic agents. Pharmacol. Ther. 2006, 109, 366–398. [Google Scholar] [CrossRef]
- Tschaikner, P.; Enzler, F.; Torres-Quesada, O.; Aanstad, P.; Stefan, E. Hedgehog and Gpr161: Regulating cAMP Signaling in the Primary Cilium. Cells 2020, 9, 118. [Google Scholar] [CrossRef]
- Owens, G.K. Molecular Control of Vascular Smooth Muscle Cell Differentiation and Phenotypic Plasticity. In Novartis Foundation Symposium; John Wiley: Chichester, UK; New York, NY, USA, 1999; Volume 283, pp. 174–193. [Google Scholar] [CrossRef]
- McCarron, J.G.; Craig, J.W.; Bradley, K.N.; Muir, T.C. Agonist-induced phasic and tonic responses in smooth muscle are mediated by InsP3. J. Cell Sci. 2002, 115, 2207–2218. [Google Scholar] [CrossRef]
- Hilgers, R.H.P.; Webb, R.C. Molecular Aspects of Arterial Smooth Muscle Contraction: Focus on Rho. Exp. Biol. Med. 2005, 230, 829–835. [Google Scholar] [CrossRef]
- Somara, S.; Bitar, K.N. Phosphorylated HSP27 modulates the association of phosphorylated caldesmon with tropomyosin in colonic smooth muscle. Am. J. Physiol. Liver Physiol. 2006, 291, G630–G639. [Google Scholar] [CrossRef]
- Porras-González, C.; Ordóñez, A.; Castellano, A.; Ureña, J. Regulation of RhoA/ROCK and sustained arterial contraction by low cytosolic Ca 2+ levels during prolonged depolarization of arterial smooth muscle. Vasc. Pharmacol. 2017, 93–95, 33–41. [Google Scholar] [CrossRef] [PubMed]
- Owens, G.K. Regulation of differentiation of vascular smooth muscle cells. Physiol. Rev. 1995, 75, 487–517. [Google Scholar] [CrossRef]
- Owens, G.K. Role of Mechanical Strain in Regulation of Differentiation of Vascular Smooth Muscle Cells. Circ. Res. 1996, 79, 1054–1055. [Google Scholar] [CrossRef] [PubMed]
- Rensen, S.S.M.; Doevendans, P.A.F.M.; van Eys, G.J.J.M. Regulation and characteristics of vascular smooth muscle cell phenotypic diversity. Neth. Hear. J. 2007, 15, 100–108. [Google Scholar] [CrossRef]
- Margarida Lorigo, M.; Melissa Mariana, M.D.; Joana Feiteiro, M.D.; Cairrao, E. Human Umbilical Artery Smooth Muscle Cells: Vascular Function and Clinical Importance. In Horizons in World Cardiovascular Research; Bennington, E.H., Ed.; Nova Science Publisher: New York, NY, USA, 2019; Volume 16. [Google Scholar]
- Salmon, M.; Gomez, D.; Greene, E.; Shankman, L.; Owens, G.K. Cooperative Binding of KLF4, pELK-1, and HDAC2 to a G/C Repressor Element in the SM22α Promoter Mediates Transcriptional Silencing During SMC Phenotypic Switching In Vivo. Circ. Res. 2012, 111, 685–696. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.-H.; Ciou, J.-S.; Chen, S.-T.; Kok, V.C.; Chung, Y.; Tsai, J.J.P.; Kurubanjerdjit, N.; Huang, C.-Y.F.; Ng, K.-L. Identify potential drugs for cardiovascular diseases caused by stress-induced genes in vascular smooth muscle cells. PeerJ 2016, 4, e2478. [Google Scholar] [CrossRef]
- Jensen, L.F.; Bentzon, J.F.; Albarrán-Juárez, J. The Phenotypic Responses of Vascular Smooth Muscle Cells Exposed to Mechanical Cues. Cells 2021, 10, 2209. [Google Scholar] [CrossRef]
- Shi, N.; Chen, S.-Y. Mechanisms simultaneously regulate smooth muscle proliferation and differentiation. J. Biomed. Res. 2014, 28, 40–46. [Google Scholar] [CrossRef]
- Cairrão, E.; Santos-Silva, A.J.; Alvarez, E.; Correia, I.; Verde, I. Isolation and culture of human umbilical artery smooth muscle cells expressing functional calcium channels. Vitr. Cell. Dev. Biol.-Anim. 2009, 45, 175–184. [Google Scholar] [CrossRef] [PubMed]
- Worth, N.F.; Rolfe, B.E.; Song, J.; Campbell, G.R. Vascular smooth muscle cell phenotypic modulation in culture is associated with reorganisation of contractile and cytoskeletal proteins. Cell Motil. Cytoskelet. 2001, 49, 130–145. [Google Scholar] [CrossRef]
- Tulis, D.A. Novel Therapies for Cyclic GMP Control of Vascular Smooth Muscle Growth. Am. J. Ther. 2008, 15, 551–564. [Google Scholar] [CrossRef]
- Hanoune, J.; Defer, N. Regulation and role of adenylyl cyclase isoforms. Annu. Rev. Pharmacol. Toxicol. 2001, 41, 145–174. [Google Scholar] [CrossRef]
- Jaggupilli, A.; Dhanaraj, P.; Pritchard, A.; Dakshinamurti, S.; Chelikani, P.; Sorensen, J.L. Study of adenylyl cyclase-GαS interactions and identification of novel AC ligands. Mol. Cell. Biochem. 2018, 446, 63–72. [Google Scholar] [CrossRef]
- Dessauer, C.W.; Watts, V.J.; Ostrom, R.S.; Conti, M.; Dove, S.; Seifert, R. International Union of Basic and Clinical Pharmacology. CI. Structures and Small Molecule Modulators of Mammalian Adenylyl Cyclases. Pharmacol. Rev. 2017, 69, 93–139. [Google Scholar] [CrossRef] [PubMed]
- Halls, M.L.; Cooper, D.M.F. Adenylyl cyclase signalling complexes—Pharmacological challenges and opportunities. Pharmacol. Ther. 2017, 172, 171–180. [Google Scholar] [CrossRef]
- Khannpnavar, B.; Mehta, V.; Qi, C.; Korkhov, V. Structure and function of adenylyl cyclases, key enzymes in cellular signaling. Curr. Opin. Struct. Biol. 2020, 63, 34–41. [Google Scholar] [CrossRef]
- Bassler, J.; Schultz, J.E.; Lupas, A.N. Adenylate cyclases: Receivers, transducers, and generators of signals. Cell. Signal. 2018, 46, 135–144. [Google Scholar] [CrossRef]
- Dove, S. Mammalian Nucleotidyl Cyclases and Their Nucleotide Binding Sites. Non-Canonical Cycl. Nucleotides 2015, 238, 49–66. [Google Scholar] [CrossRef]
- Tesmer, J.J.; Sprang, S.R. The structure, catalytic mechanism and regulation of adenylyl cyclase. Curr. Opin. Struct. Biol. 1998, 8, 713–719. [Google Scholar] [CrossRef]
- Willoughby, D.; Cooper, D.M.F. Organization and Ca2+Regulation of Adenylyl Cyclases in cAMP Microdomains. Physiol. Rev. 2007, 87, 965–1010. [Google Scholar] [CrossRef]
- Patel, T.B.; Du, Z.; Pierre, S.; Cartin, L.; Scholich, K. Molecular biological approaches to unravel adenylyl cyclase signaling and function. Gene 2001, 269, 13–25. [Google Scholar] [CrossRef]
- Rokolya, A.; Singer, H.A. Inhibition of CaM kinase II activation and force maintenance by KN-93 in arterial smooth muscle. Am. J. Physiol. Physiol. 2000, 278, C537–C545. [Google Scholar] [CrossRef]
- Marganski, W.A.; Gangopadhyay, S.S.; Je, H.-D.; Gallant, C.; Morgan, K.G. Targeting of a Novel Ca+ 2/Calmodulin-Dependent Protein Kinase II Is Essential for Extracellular Signal-Regulated Kinase–Mediated Signaling in Differentiated Smooth Muscle Cells. Circ. Res. 2005, 97, 541–549. [Google Scholar] [CrossRef]
- Wong, S.T.; Baker, L.P.; Trinh, K.; Hetman, M.; Suzuki, L.A.; Storm, D.R.; Bornfeldt, K.E. Adenylyl Cyclase 3 Mediates Prostaglandin E2-induced Growth Inhibition in Arterial Smooth Muscle Cells. J. Biol. Chem. 2001, 276, 34206–34212. [Google Scholar] [CrossRef] [PubMed]
- Cary, S.P.; Winger, J.; Derbyshire, E.R.; Marletta, M.A. Nitric oxide signaling: No longer simply on or off. Trends Biochem. Sci. 2006, 31, 231–239. [Google Scholar] [CrossRef]
- Carvajal, J.A.; Germain, A.M.; Huidobro-Toro, J.P.; Weiner, C.P. Molecular mechanism of cGMP-mediated smooth muscle relaxation. J. Cell. Physiol. 2000, 184, 409–420. [Google Scholar] [CrossRef]
- Silva, B.R.; Paula, T.D.; Paulo, M.; Bendhack, L.M. Nitric Oxide Signaling and the Cross Talk with Prostanoids Pathways in Vascular System. Med. Chem. 2017, 13, 319–333. [Google Scholar] [CrossRef]
- Potter, L.R.; Abbey-Hosch, S.; Dickey, D.M. Natriuretic Peptides, Their Receptors, and Cyclic Guanosine Monophosphate-Dependent Signaling Functions. Endocr. Rev. 2005, 27, 47–72. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, M. Molecular Physiology of Membrane Guanylyl Cyclase Receptors. Physiol. Rev. 2016, 96, 751–804. [Google Scholar] [CrossRef]
- Montfort, W.R.; Wales, J.A.; Weichsel, A. Structure and Activation of Soluble Guanylyl Cyclase, the Nitric Oxide Sensor. Antioxid. Redox Signal. 2017, 26, 107–121. [Google Scholar] [CrossRef]
- Russwurm, M.; Wittau, N.; Koesling, D. Guanylyl Cyclase/PSD-95 Interaction: Targeting of the nitric oxide-sensitive alpha2beta1 guanylyl cyclase to synaptic membranes. The Journal of biological chemistry. J. Biol. Chem. 2001, 276, 44647–44652. [Google Scholar] [CrossRef]
- Cerra, M.; Pellegrino, M.C.C.A.D. Cardiovascular cGMP-Generating Systems in Physiological and Pathological Conditions. Curr. Med. Chem. 2007, 14, 585–599. [Google Scholar] [CrossRef]
- Hofmann, F. The cGMP system: Components and function. Biol. Chem. 2019, 401, 447–469. [Google Scholar] [CrossRef] [PubMed]
- Rahmutula, D.; Nakayama, T.; Soma, M.; Kosuge, K.; Aoi, N.; Izumi, Y.; Kanmatsuse, K.; Ozawa, Y. Structure and Polymorphisms of the Human Natriuretic Peptide Receptor C Gene. Endocrine 2002, 17, 085–090. [Google Scholar] [CrossRef]
- Suga, S.; Nakao, K.; Kishimoto, I.; Hosoda, K.; Mukoyama, M.; Arai, H.; Shirakami, G.; Ogawa, Y.; Komatsu, Y.; Nakagawa, O. Phenotype-related alteration in expression of natriuretic peptide receptors in aortic smooth muscle cells. Circ. Res. 1992, 71, 34–39. [Google Scholar] [CrossRef]
- Rehemudula, D.; Nakayama, T.; Soma, M.; Takahashi, Y.; Uwabo, J.; Sato, M.; Izumi, Y.; Kanmatsuse, K.; Ozawa, Y. Structure of the type B human natriuretic peptide receptor gene and association of a novel microsatellite polymorphism with essential hypertension. Circ. Res. 1999, 84, 605–610. [Google Scholar] [CrossRef]
- Fedele, E.; Ricciarelli, R. Memory Enhancers for Alzheimer’s Dementia: Focus on cGMP. Pharmaceuticals 2021, 14, 61. [Google Scholar] [CrossRef] [PubMed]
- He, X.-L.; Dukkipati, A.; Garcia, K.C. Structural Determinants of Natriuretic Peptide Receptor Specificity and Degeneracy. J. Mol. Biol. 2006, 361, 698–714. [Google Scholar] [CrossRef]
- Schulz, S.; Chinkers, M.; Garbers, D.L. The guanylate cyclase/receptor family of proteins. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 1989, 3, 2026–2035. [Google Scholar] [CrossRef]
- Lumsden, N.G.; Khambata, R.S.; Hobbs, A.J. C-type Natriuretic Peptide (CNP): Cardiovascular Roles and Potential as a Therapeutic Target. Curr. Pharm. Des. 2010, 16, 4080–4088. [Google Scholar] [CrossRef]
- Mergia, E.; Stegbauer, J. Role of Phosphodiesterase 5 and Cyclic GMP in Hypertension. Curr. Hypertens. Rep. 2016, 18, 1–8. [Google Scholar] [CrossRef]
- Koesling, D.; Mergia, E.; Russwurm, M. Physiological Functions of NO-Sensitive Guanylyl Cyclase Isoforms. Curr. Med. Chem. 2016, 23, 2653–2665. [Google Scholar] [CrossRef] [PubMed]
- Mergia, E.; Russwurm, M.; Zoidl, G.; Koesling, D. Major occurrence of the new α2β1 isoform of NO-sensitive guanylyl cyclase in brain. Cell. Signal. 2003, 15, 189–195. [Google Scholar] [CrossRef]
- Foerster, J.; Harteneck, C.; Malkewitz, J.; Schultz, G.; Koesling, D. A Functional Heme-Binding Site of Soluble Guanylyl Cyclase Requires Intact N-Termini of alpha1 and beta1 Subunits. JBIC J. Biol. Inorg. Chem. 1996, 240, 380–386. [Google Scholar] [CrossRef]
- Wobst, J.; Schunkert, H.; Kessler, T. Genetic alterations in the NO-cGMP pathway and cardiovascular risk. Nitric Oxide 2018, 76, 105–112. [Google Scholar] [CrossRef]
- Underbakke, E.; Iavarone, A.T.; Chalmers, M.J.; Pascal, B.D.; Novick, S.; Griffin, P.R.; Marletta, M.A. Nitric Oxide-Induced Conformational Changes in Soluble Guanylate Cyclase. Structure 2014, 22, 602–611. [Google Scholar] [CrossRef] [PubMed]
- Horst, B.G.; Yokom, A.L.; Rosenberg, D.J.; Morris, K.L.; Hammel, M.; Hurley, J.H.; Marletta, M.A. Allosteric activation of the nitric oxide receptor soluble guanylate cyclase mapped by cryo-electron microscopy. eLife 2019, 8, e50634. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.; Liu, R.; Wu, J.-X.; Chen, L. Structural insights into the mechanism of human soluble guanylate cyclase. Nature 2019, 574, 206–210. [Google Scholar] [CrossRef]
- Zhou, Z.; Martin, E.; Sharina, I.; Esposito, I.; Szabo, C.; Bucci, M.; Cirino, G.; Papapetropoulos, A. Regulation of soluble guanylyl cyclase redox state by hydrogen sulfide. Pharmacol. Res. 2016, 111, 556–562. [Google Scholar] [CrossRef]
- Krumenacker, J.S.; Hanafy, K.A.; Murad, F. Regulation of nitric oxide and soluble guanylyl cyclase. Brain Res. Bull. 2004, 62, 505–515. [Google Scholar] [CrossRef]
- Shaul, P.W. Regulation of Endothelial Nitric Oxide Synthase: Location, Location, Location. Annu. Rev. Physiol. 2002, 64, 749–774. [Google Scholar] [CrossRef] [PubMed]
- Costa, E.D.; Rezende, B.A.; Cortes, S.F.; Lemos, V.S. Neuronal Nitric Oxide Synthase in Vascular Physiology and Diseases. Front. Physiol. 2016, 7, 206. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.I.; Sessa, W.C. Vascular Endothelial Growth Factor Signaling to Endothelial Nitric Oxide Synthase. Circ. Res. 2006, 99, 666–668. [Google Scholar] [CrossRef]
- Ginnan, R.; Guikema, B.J.; Halligan, K.E.; Singer, H.A.; Jourd’Heuil, D. Regulation of smooth muscle by inducible nitric oxide synthase and NADPH oxidase in vascular proliferative diseases. Free Radic. Biol. Med. 2008, 44, 1232–1245. [Google Scholar] [CrossRef]
- Zhao, Y.; Vanhoutte, P.M.; Leung, S.W. Vascular nitric oxide: Beyond eNOS. J. Pharmacol. Sci. 2015, 129, 83–94. [Google Scholar] [CrossRef]
- Derbyshire, E.R.; Marletta, M.A. Structure and Regulation of Soluble Guanylate Cyclase. Annu. Rev. Biochem. 2012, 81, 533–559. [Google Scholar] [CrossRef]
- Ding, X.; Ren, F. Leucine-rich Repeat Kinase 2 Inhibitors: A Patent Review (2014-present). Expert Opin. Ther. Pat. 2020, 30, 275–286. [Google Scholar] [CrossRef] [PubMed]
- Evgenov, O.V.; Pacher, P.; Schmidt, P.; Haskó, G.; Schmidt, H.H.H.W.; Stasch, J.-P. NO-independent stimulators and activators of soluble guanylate cyclase: Discovery and therapeutic potential. Nat. Rev. Drug Discov. 2006, 5, 755–768. [Google Scholar] [CrossRef]
- Omori, K.; Kotera, J. Overview of PDEs and Their Regulation. Circ. Res. 2007, 100, 309–327. [Google Scholar] [CrossRef]
- Rybalkin, S.D.; Yan, C.; Bornfeldt, K.E.; Beavo, J.A. Cyclic GMP Phosphodiesterases and Regulation of Smooth Muscle Function. Circ. Res. 2003, 93, 280–291. [Google Scholar] [CrossRef] [PubMed]
- Lugnier, C.; Keravis, T.; Eckly-Michel, A. Cross talk between NO and cyclic nucleotide phosphodiesterases in the modulation of signal transduction in blood vessel. J. Physiol. Pharmacol. Off. J. Pol. Physiol. Soc. 1999, 50, 639–652. [Google Scholar]
- Ercu, M.; Klussmann, E. Roles of A-Kinase Anchoring Proteins and Phosphodiesterases in the Cardiovascular System. J. Cardiovasc. Dev. Dis. 2018, 5, 14. [Google Scholar] [CrossRef]
- Wennogle, L.P.; Hoxie, H.; Peng, Y.; Hendrick, J.P. Phosphodiesterase 1: A Unique Drug Target for Degenerative Diseases and Cognitive Dysfunction. Phosphodiesterases CNS Funct. Dis. 2017, 17, 349–384. [Google Scholar] [CrossRef]
- Larré, A.B.; Parisotto, A.; Rockenbach, B.F.; Pasin, D.M.; Capellari, C.; Escouto, D.C.; da Costa, B.E.P.; Poli-De-Figueiredo, C.E. Phosphodiesterases and preeclampsia. Med. Hypotheses 2017, 108, 94–100. [Google Scholar] [CrossRef]
- Azevedo, M.F.; Faucz, F.R.; Bimpaki, E.; Horvath, A.; Levy, I.; De Alexandre, R.B.; Ahmad, F.; Manganiello, V.; Stratakis, C.A. Clinical and Molecular Genetics of the Phosphodiesterases (PDEs). Endocr. Rev. 2013, 35, 195–233. [Google Scholar] [CrossRef] [PubMed]
- Beavo, J.A. Cyclic nucleotide phosphodiesterases: Functional implications of multiple isoforms. Physiol. Rev. 1995, 75, 725–748. [Google Scholar] [CrossRef]
- Bender, A.T.; Beavo, J.A. Cyclic Nucleotide Phosphodiesterases: Molecular Regulation to Clinical Use. Pharmacol. Rev. 2006, 58, 488–520. [Google Scholar] [CrossRef]
- Lugnier, C.; Schoeffter, P.; Le Bec, A.; Strouthou, E.; Stoclet, J. Selective inhibition of cyclic nucleotide phosphodiesterases of human, bovine and rat aorta. Biochem. Pharmacol. 1986, 35, 1743–1751. [Google Scholar] [CrossRef]
- Matsumoto, T.; Kobayashi, T.; Kamata, K. Phosphodiesterases in the Vascular System. J. Smooth Muscle Res. 2003, 39, 67–86. [Google Scholar] [CrossRef] [PubMed]
- Pagani, E.D.; Buchholz, R.A.; Silver, P.J. Cardiovascular cyclic nucleotide phosphodiesterases and their role in regulating cardiovascular function. In Cellular and Molecular Alterations in the Failing Human Heart; Steinkopff: Heidelberg, Germany, 1992; pp. 73–86. [Google Scholar] [CrossRef]
- Chan, S.; Yan, C. PDE1 isozymes, key regulators of pathological vascular remodeling. Curr. Opin. Pharmacol. 2011, 11, 720–724. [Google Scholar] [CrossRef]
- Rybalkin, S.D.; Bornfeldt, K.E.; Sonnenburg, W.K.; Rybalkina, I.G.; Kwak, K.S.; Hanson, K.; Krebs, E.G.; Beavo, J.A. Calmodulin-stimulated cyclic nucleotide phosphodiesterase (PDE1C) is induced in human arterial smooth muscle cells of the synthetic, proliferative phenotype. J. Clin. Investig. 1997, 100, 2611–2621. [Google Scholar] [CrossRef] [PubMed]
- Murray, F.; Patel, H.H.; Suda, R.Y.S.; Zhang, S.; Thistlethwaite, P.A.; Yuan, J.X.-J.; Insel, P.A. Expression and activity of cAMP phosphodiesterase isoforms in pulmonary artery smooth muscle cells from patients with pulmonary hypertension: Role for PDE1. Am. J. Physiol. Cell. Mol. Physiol. 2007, 292, L294–L303. [Google Scholar] [CrossRef]
- Nan, Y.; Zeng, X.; Jin, Z.; Li, N.; Chen, Z.; Chen, J.; Wang, D.; Wang, Y.; Lin, Z.; Ying, L. PDE1 or PDE5 inhibition augments NO-dependent hypoxic constriction of porcine coronary artery via elevating inosine 3′,5′-cyclic monophosphate level. J. Cell. Mol. Med. 2020, 24, 14514–14524. [Google Scholar] [CrossRef]
- Lugnier, C.; Schini, V.B. Characterization of cyclic nucleotide phosphodiesterases from cultured bovine aortic endothelial cells. Biochem. Pharmacol. 1990, 39, 75–84. [Google Scholar] [CrossRef]
- Favot, L.; Keravis, T.; Holl, V.; Le Bec, A.; Lugnier, C. VEGF-induced HUVEC migration and proliferation are decreased by PDE2 and PDE4 inhibitors. Thromb. Haemost. 2003, 90, 334–343. [Google Scholar] [CrossRef]
- Bubb, K.J.; Trinder, S.L.; Baliga, R.S.; Patel, J.; Clapp, L.H.; MacAllister, R.J.; Hobbs, A.J. Inhibition of Phosphodiesterase 2 Augments cGMP and cAMP Signaling to Ameliorate Pulmonary Hypertension. Circulation 2014, 130, 496–507. [Google Scholar] [CrossRef]
- Murray, F.; MacLean, M.; Pyne, N. Increased expression of the cGMP-inhibited cAMP-specific (PDE3) and cGMP binding cGMP-specific (PDE5) phosphodiesterases in models of pulmonary hypertension. Br. J. Pharmacol. 2002, 137, 1187–1194. [Google Scholar] [CrossRef]
- Dunkerley, H.A.; Tilley, D.; Palmer, D.; Liu, H.; Jimmo, S.L.; Maurice, D.H. Reduced phosphodiesterase 3 activity and phosphodiesterase 3A level in synthetic vascular smooth muscle cells: Implications for use of phosphodiesterase 3 inhibitors in cardiovascular tissues. Mol. Pharmacol. 2002, 61, 1033–1040. [Google Scholar] [CrossRef]
- Hubert, F.; Belacel-Ouari, M.; Manoury, B.; Zhai, K.; Domergue-Dupont, V.; Mateo, P.; Joubert, F.; Fischmeister, R.; Leblais, V. Alteration of vascular reactivity in heart failure: Role of phosphodiesterases 3 and 4. Br. J. Pharmacol. 2014, 171, 5361–5375. [Google Scholar] [CrossRef]
- Movsesian, M. Novel approaches to targeting PDE3 in cardiovascular disease. Pharmacol. Ther. 2016, 163, 74–81. [Google Scholar] [CrossRef]
- Ercu, M.; Markó, L.; Schächterle, C.; Tsvetkov, D.; Cui, Y.; Maghsodi, S.; Bartolomaeus, T.U.; Maass, P.G.; Zühlke, K.; Gregersen, N.; et al. Phosphodiesterase 3A and Arterial Hypertension. Circulation 2020, 142, 133–149. [Google Scholar] [CrossRef]
- Fertig, B.A.; Baillie, G.S. PDE4-Mediated cAMP Signalling. J. Cardiovasc. Dev. Dis. 2018, 5, 8. [Google Scholar] [CrossRef] [PubMed]
- Peng, T.; Qi, B.; He, J.; Ke, H.; Shi, J. Advances in the Development of Phosphodiesterase-4 Inhibitors. J. Med. Chem. 2020, 63, 10594–10617. [Google Scholar] [CrossRef] [PubMed]
- Lugnier, C.; Meyer, A.; Talha, S.; Geny, B. Cyclic nucleotide phosphodiesterases: New targets in the metabolic syndrome? Pharmacol. Ther. 2020, 208, 107475. [Google Scholar] [CrossRef] [PubMed]
- Houslay, M.D. Underpinning compartmentalised cAMP signalling through targeted cAMP breakdown. Trends Biochem. Sci. 2010, 35, 91–100. [Google Scholar] [CrossRef]
- Mika, D.; Conti, M. PDE4D phosphorylation: A coincidence detector integrating multiple signaling pathways. Cell. Signal. 2016, 28, 719–724. [Google Scholar] [CrossRef]
- Saeki, T.; Saito, I. Isolation of cyclic nucleotide phosphodiesterase isoenzymes from pig aorta. Biochem. Pharmacol. 1993, 46, 833–839. [Google Scholar] [CrossRef]
- Komas, N.; Lugnier, C.; Andriantsitohaina, R.; Stoclet, J.-C. Characterisation of cyclic nucleotide phosphodiesterases from rat mesenteric artery. Eur. J. Pharmacol. Mol. Pharmacol. 1991, 208, 85–87. [Google Scholar] [CrossRef]
- Rabe, K.F.; Tenor, H.; Dent, G.; Schudt, C.; Nakashima, M.; Magnussen, H. Identification of PDE isozymes in human pulmonary artery and effect of selective PDE inhibitors. Am. J. Physiol. Content 1994, 266, L536–L543. [Google Scholar] [CrossRef]
- Willette, R.N.; Shiloh, A.O.; Sauermelch, C.F.; Sulpizio, A.; Michell, M.P.; Cieslinski, L.B.; Torphy, T.J.; Ohlstein, E.H. Identification, Characterization, and Functional Role of Phosphodiesterase Type IV in Cerebral Vessels: Effects of Selective Phosphodiesterase Inhibitors. J. Cereb. Blood Flow Metab. 1997, 17, 210–219. [Google Scholar] [CrossRef]
- Houslay, M.D.; Baillie, G.; Maurice, D.H. cAMP-Specific Phosphodiesterase-4 Enzymes in the Cardiovascular System: A molecular toolbox for generating compartmentalized cAMP signaling. Circulation research. Circ. Res. 2007, 100, 950–966. [Google Scholar] [CrossRef]
- Tilley, D.; Maurice, D.H. Vascular Smooth Muscle Cell Phenotype-Dependent Phosphodiesterase 4D Short Form Expression: Role of Differential Histone Acetylation on cAMP-Regulated Function. Mol. Pharmacol. 2005, 68, 596–605. [Google Scholar] [CrossRef]
- Kotera, J.; Fujishige, K.; Akatsuka, H.; Imai, Y.; Yanaka, N.; Omori, K. Novel alternative splice variants of cGMP-binding cGMP-specific phosphodiesterase. J. Biol. Chem. 1998, 273, 26982–26990. [Google Scholar] [CrossRef]
- Loughney, K.; Hill, T.R.; Florio, V.A.; Uher, L.; Rosman, G.J.; Wolda, S.L.; Jones, B.A.; Howard, M.L.; McAllister-Lucas, L.M.; Sonnenburg, W.K.; et al. Isolation and characterization of cDNAs encoding PDE5A, a human cGMP-binding, cGMP-specific 3′,5′-cyclic nucleotide phosphodiesterase. Gene 1998, 216, 139–147. [Google Scholar] [CrossRef]
- Kotera, J.; Grimes, K.A.; Corbin, J.D.; Francis, S.H. cGMP-dependent protein kinase protects cGMP from hydrolysis by phosphodiesterase-5. Biochem. J. 2003, 372, 419–426. [Google Scholar] [CrossRef]
- Tcheudji, J.F.; Lebeau, L.; Virmaux, N.; Maftei, C.G.; Cote, R.; Lugnier, C.; Schultz, P. Molecular organization of bovine rod cGMP-phosphodiesterase 6. J. Mol. Biol. 2001, 310, 781–791. [Google Scholar] [CrossRef]
- Ahmed, W.S.; Geethakumari, A.M.; Biswas, K.H. Phosphodiesterase 5 (PDE5): Structure-function regulation and therapeutic applications of inhibitors. Biomed. Pharmacother. 2020, 134, 111128. [Google Scholar] [CrossRef]
- Maurice, D.H. Cardiovascular implications in the use of PDE5 inhibitor therapy. Int. J. Impot. Res. 2004, 16, S20–S23. [Google Scholar] [CrossRef][Green Version]
- Pauvert, O.; Lugnier, C.; Keravis, T.; Marthan, R.; Rousseau, E.; Savineau, J.P. Effect of sildenafil on cyclic nucleotide phosphodiesterase activity, vascular tone and calcium signaling in rat pulmonary artery. Br. J. Pharmacol. 2003, 139, 513–522. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Bouadjel, K.; Manoury, B.; Vandecasteele, G.; Fischmeister, R.; Leblais, V. Cyclic nucleotide signalling compartmentation by PDEs in cultured vascular smooth muscle cells. Br. J. Pharmacol. 2019, 176, 1780–1792. [Google Scholar] [CrossRef]
- Smith, S.J.; Brookes-Fazakerley, S.; Donnelly, L.; Barnes, P.J.; Barnette, M.S.; Giembycz, M.A. Ubiquitous expression of phosphodiesterase 7A in human proinflammatory and immune cells. Am. J. Physiol. Cell. Mol. Physiol. 2003, 284, L279–L289. [Google Scholar] [CrossRef]
- Phillips, P.G.; Long, L.; Wilkins, M.R.; Morrell, N.W. cAMP phosphodiesterase inhibitors potentiate effects of prostacyclin analogs in hypoxic pulmonary vascular remodeling. Am. J. Physiol. Cell. Mol. Physiol. 2005, 288, L103–L115. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Zhai, K.; Hubert, F.; Nicolas, V.; Ji, G.; Fischmeister, R.; Leblais, V. β-Adrenergic cAMP Signals Are Predominantly Regulated by Phosphodiesterase Type 4 in Cultured Adult Rat Aortic Smooth Muscle Cells. PLoS ONE 2012, 7, e47826. [Google Scholar] [CrossRef] [PubMed]
- Belacel-Ouari, M.; Zhang, L.; Hubert, F.; Assaly, R.; Gerbier, R.; Jockers, R.; Dauphin, F.; Lechêne, P.; Fischmeister, R.; Manoury, B.; et al. Influence of cell confluence on the cAMP signalling pathway in vascular smooth muscle cells. Cell. Signal. 2017, 35, 118–128. [Google Scholar] [CrossRef] [PubMed]
- Soderling, S.H.; Bayuga, S.J.; Beavo, J.A. Cloning and characterization of a cAMP-specific cyclic nucleotide phosphodiesterase. Proc. Natl. Acad. Sci. USA 1998, 95, 8991–8996. [Google Scholar] [CrossRef]
- Wunder, F.; Tersteegen, A.; Rebmann, A.; Erb, C.; Fahrig, T.; Hendrix, M. Characterization of the First Potent and Selective PDE9 Inhibitor Using a cGMP Reporter Cell Line. Mol. Pharmacol. 2005, 68, 1775–1781. [Google Scholar] [CrossRef]
- Rentero, C.; Monfort, A.; Puigdomènech, P. Identification and distribution of different mRNA variants produced by differential splicing in the human phosphodiesterase 9A gene. Biochem. Biophys. Res. Commun. 2003, 301, 686–692. [Google Scholar] [CrossRef]
- Gross-Langenhoff, M.; Hofbauer, K.; Weber, J.; Schultz, A.; Schultz, J.E. cAMP Is a Ligand for the Tandem GAF Domain of Human Phosphodiesterase 10 and cGMP for the Tandem GAF Domain of Phosphodiesterase 11. J. Biol. Chem. 2006, 281, 2841–2846. [Google Scholar] [CrossRef]
- Tian, X.; Vroom, C.; Ghofrani, H.A.; Weissmann, N.; Bieniek, E.; Grimminger, F.; Seeger, W.; Schermuly, R.T.; Pullamsetti, S.S. Phosphodiesterase 10A Upregulation Contributes to Pulmonary Vascular Remodeling. PLoS ONE 2011, 6, e18136. [Google Scholar] [CrossRef] [PubMed]
- Amunjela, J.N.; Swan, A.H.; Brand, T. The Role of the Popeye Domain Containing Gene Family in Organ Homeostasis. Cells 2019, 8, 1594. [Google Scholar] [CrossRef] [PubMed]
- Walsh, D.A.; Perkins, J.P.; Krebs, E.G. An adenosine 3’,5’-monophosphate-dependant protein kinase from rabbit skeletal muscle. J. Biol. Chem. 1968, 243, 3763–3765. [Google Scholar] [CrossRef]
- Smith, F.D.; Esseltine, J.L.; Nygren, P.J.; Veesler, D.; Byrne, D.P.; Vonderach, M.; Strashnov, I.; Eyers, C.E.; Eyers, P.A.; Langeberg, L.K.; et al. Local protein kinase A action proceeds through intact holoenzymes. Science 2017, 356, 1288–1293. [Google Scholar] [CrossRef] [PubMed]
- Colledge, M.; Scott, J.D. AKAPs: From structure to function. Trends Cell Biol. 1999, 9, 216–221. [Google Scholar] [CrossRef]
- Dostmann, W.R.; Taylor, S.S.; Genieser, H.G.; Jastorff, B.; Døskeland, S.O.; Ogreid, D. Probing the cyclic nucleotide binding sites of cAMP-dependent protein kinases I and II with analogs of adenosine 3′,5′-cyclic phosphorothioates. J. Biol. Chem. 1990, 265, 10484–10491. [Google Scholar] [CrossRef]
- Cummings, D.E.; Brandon, E.P.; Planas, J.V.; Motamed, K.; Idzerda, R.L.; McKnight, G.S. Genetically lean mice result from targeted disruption of the RIIβ subunit of protein kinase A. Nature 1996, 382, 622–626. [Google Scholar] [CrossRef] [PubMed]
- Taskén, K.; Sklhegg, B.S.; Austlid, T.K.; Solberg, R.; Knutsen, H.K.; Levy, F.O.; Sandberg, M.; Ørstavik, S.; Larsen, T.; Johansen, A.K.; et al. Structure, function, and regulation of human cAMP-dependent protein kinases. Adv. Second. Messenger Phosphoprot. Res. 1997, 31, 191–204. [Google Scholar] [CrossRef]
- Søberg, K.; Jahnsen, T.; Rognes, T.; Skålhegg, B.S.; Laerdahl, J.K. Evolutionary Paths of the cAMP-Dependent Protein Kinase (PKA) Catalytic Subunits. PLoS ONE 2013, 8, e60935. [Google Scholar] [CrossRef]
- Wang, L.; Sunahara, R.K.; Krumins, A.; Perkins, G.; Crochiere, M.L.; Mackey, M.; Bell, S.; Ellisman, M.H.; Taylor, S.S. Cloning and mitochondrial localization of full-length D-AKAP2, a protein kinase A anchoring protein. Proc. Natl. Acad. Sci. USA 2001, 98, 3220–3225. [Google Scholar] [CrossRef] [PubMed]
- Taskén, K.; Aandahl, E.M. Localized Effects of cAMP Mediated by Distinct Routes of Protein Kinase A. Physiol. Rev. 2004, 84, 137–167. [Google Scholar] [CrossRef]
- Jarnæss, E.; Tasken, K. Spatiotemporal control of cAMP signalling processes by anchored signalling complexes. Biochem. Soc. Trans. 2007, 35, 931–937. [Google Scholar] [CrossRef]
- Carr, D.W.; Stofko-Hahn, R.E.; Fraser, I.D.; Bishop, S.M.; Acott, T.S.; Brennan, R.G.; Scott, J.D. Interaction of the regulatory subunit (RII) of cAMP-dependent protein kinase with RII-anchoring proteins occurs through an amphipathic helix binding motif. J. Biol. Chem. 1991, 266, 14188–14192. [Google Scholar] [CrossRef]
- Scott, J.D.; Stofko, R.E.; McDonald, J.R.; Comer, J.D.; Vitalis, E.A.; Mangili, J.A. Type II regulatory subunit dimerization determines the subcellular localization of the cAMP-dependent protein kinase. J. Biol. Chem. 1990, 265, 21561–21566. [Google Scholar] [CrossRef]
- Turnham, R.E.; Scott, J.D. Protein kinase A catalytic subunit isoform PRKACA.; History, function and physiology. Gene 2015, 577, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Bauman, A.L.; Soughayer, J.; Nguyen, B.T.; Willoughby, D.; Carnegie, G.K.; Wong, W.; Hoshi, N.; Langeberg, L.K.; Cooper, D.M.; Dessauer, C.; et al. Dynamic Regulation of cAMP Synthesis through Anchored PKA-Adenylyl Cyclase V/VI Complexes. Mol. Cell 2006, 23, 925–931. [Google Scholar] [CrossRef] [PubMed]
- Kirschner, L.S.; Carney, J.A.; Pack, S.; Taymans, S.E.; Giatzakis, C.; Cho, Y.S.; Cho-Chung, Y.S.; Stratakis, C.A. Mutations of the gene encoding the protein kinase A type I-α regulatory subunit in patients with the Carney complex. Nat. Genet. 2000, 26, 89–92. [Google Scholar] [CrossRef]
- Beuschlein, F.; Fassnacht, M.; Assié, G.; Calebiro, D.; Stratakis, C.A.; Osswald, A.; Ronchi, C.; Wieland, T.; Sbiera, S.; Faucz, F.R.; et al. Constitutive Activation of PKA Catalytic Subunit in Adrenal Cushing’s Syndrome. N. Engl. J. Med. 2014, 370, 1019–1028. [Google Scholar] [CrossRef]
- Gold, M.G.; Gonen, T.; Scott, J.D. Local cAMP signaling in disease at a glance. J. Cell Sci. 2013, 126, 4537–4543. [Google Scholar] [CrossRef]
- Krebs, E.G.; Beavo, J.A. Phosphorylation-Dephosphorylation of Enzymes. Annu. Rev. Biochem. 1979, 48, 923–959. [Google Scholar] [CrossRef] [PubMed]
- Bjorn, S.S.; Skalhegg, B.S.; Tasken, K. Specificity in the cAMP/PKA signaling pathway. differential expression, regulation, and subcellular localization of subunits of PKA. Front. Biosci. 2000, 5, d678–d693. [Google Scholar] [CrossRef]
- Kim, C.; Xuong, N.-H.; Taylor, S.S. Crystal Structure of a Complex Between the Catalytic and Regulatory (RIα) Subunits of PKA. Science 2005, 307, 690–696. [Google Scholar] [CrossRef]
- Sjoberg, T.J.; Kornev, A.P.; Taylor, S.S. Dissecting the cAMP-inducible allosteric switch in protein kinase A RIα. Protein Sci. 2010, 19, 1213–1221. [Google Scholar] [CrossRef]
- Anand, G.S.; Krishnamurthy, S.; Bishnoi, T.; Kornev, A.; Taylor, S.S.; Johnson, D.A. Cyclic AMP- and (Rp)-cAMPS-induced Conformational Changes in a Complex of the Catalytic and Regulatory (RIα) Subunits of Cyclic AMP-dependent Protein Kinase. Mol. Cell. Proteom. 2010, 9, 2225–2237. [Google Scholar] [CrossRef]
- Wong, W.; Scott, J.D. AKAP signalling complexes: Focal points in space and time. Nat. Rev. Mol. Cell Biol. 2004, 5, 959–970. [Google Scholar] [CrossRef]
- Taylor, S.S.; Buechler, J.A.; Yonemoto, W. cAMP-DEPENDENT PROTEIN KINASE: FRAMEWORK FOR A DIVERSE FAMILY OF REGULATORY ENZYMES. Annu. Rev. Biochem. 1990, 59, 971–1005. [Google Scholar] [CrossRef] [PubMed]
- Herberg, F.W.; Taylor, S.S.; Dostmann, W.R. Active Site Mutations Define the Pathway for the Cooperative Activation of cAMP-Dependent Protein Kinase. Biochemistry 1996, 35, 2934–2942. [Google Scholar] [CrossRef]
- Yang, S.; Fletcher, W.H.; Johnson, D.A. Regulation of cAMP-dependent protein kinase: Enzyme activation without dissociation. Biochemistry 1995, 34, 6267–6271. [Google Scholar] [CrossRef]
- Bittinger, M.A.; McWhinnie, E.; Meltzer, J.; Iourgenko, V.; Latario, B.; Liu, X.; Chen, C.H.; Song, C.; Garza, D.; Labow, M. Activation of cAMP Response Element-Mediated Gene Expression by Regulated Nuclear Transport of TORC Proteins. Curr. Biol. 2004, 14, 2156–2161. [Google Scholar] [CrossRef]
- Smith, F.D.; Reichow, S.; Esseltine, J.L.; Shi, D.; Langeberg, L.K.; Scott, J.D.; Gonen, T. Intrinsic disorder within an AKAP-protein kinase A complex guides local substrate phosphorylation. eLife 2013, 2, e01319. [Google Scholar] [CrossRef]
- Hogarth, D.K.; Sandbo, N.; Taurin, S.; Kolenko, V.; Miano, J.M.; Dulin, N.O. Dual role of PKA in phenotypic modulation of vascular smooth muscle cells by extracellular ATP. Am. J. Physiol. Physiol. 2004, 287, C449–C456. [Google Scholar] [CrossRef] [PubMed]
- Lincoln, T.M.; Cornwell, T.L.; Taylor, A.E. cGMP-dependent protein kinase mediates the reduction of Ca2+ by cAMP in vascular smooth muscle cells. Am. J. Physiol. Physiol. 1990, 258, C399–C407. [Google Scholar] [CrossRef] [PubMed]
- White, R.E.; Kryman, J.P.; El-Mowafy, A.M.; Han, G.; Carrier, G.O. cAMP-Dependent Vasodilators Cross-Activate the cGMP-Dependent Protein Kinase to Stimulate BK Ca Channel Activity in Coronary Artery Smooth Muscle Cells. Circ. Res. 2000, 86, 897–905. [Google Scholar] [CrossRef]
- Wu, Y.-J.; Bond, M.; Sala-Newby, G.B.; Newby, A.C. Altered S-Phase Kinase-Associated Protein-2 Levels Are a Major Mediator of Cyclic Nucleotide–Induced Inhibition of Vascular Smooth Muscle Cell Proliferation. Circ. Res. 2006, 98, 1141–1150. [Google Scholar] [CrossRef]
- VanSchouwen, B.; Selvaratnam, R.; Giri, R.; Lorenz, R.; Herberg, F.W.; Kim, C.; Melacini, G. Mechanism of cAMP Partial Agonism in Protein Kinase G (PKG). J. Biol. Chem. 2015, 290, 28631–28641. [Google Scholar] [CrossRef] [PubMed]
- Eckly-Michel, A.; Martin, V.; Lugnier, C. Involvement of cyclic nucleotide-dependent protein kinases in cyclic AMP-mediated vasorelaxation. Br. J. Pharmacol. 1997, 122, 158–164. [Google Scholar] [CrossRef]
- Wehbe, N.; Nasser, S.A.; Al-Dhaheri, Y.; Iratni, R.; Bitto, A.; El-Yazbi, A.F.; Badran, A.; Kobeissy, F.; Baydoun, E.; Eid, A.H. EPAC in Vascular Smooth Muscle Cells. Int. J. Mol. Sci. 2020, 21, 5160. [Google Scholar] [CrossRef]
- Kawasaki, H.; Springett, G.M.; Mochizuki, N.; Toki, S.; Nakaya, M.; Matsuda, M.; Housman, D.E.; Graybiel, A.M. A Family of cAMP-Binding Proteins That Directly Activate Rap1. Science 1998, 282, 2275–2279. [Google Scholar] [CrossRef]
- De Rooij, J.; Zwartkruis, F.J.T.; Verheijen, M.H.G.; Cool, R.H.; Nijman, S.M.B.; Wittinghofer, A.; Bos, J.L. Epac is a Rap1 guanine-nucleotide-exchange factor directly activated by cyclic AMP. Nature 1998, 396, 474–477. [Google Scholar] [CrossRef]
- Lezoualc’H, F.; Fazal, L.; Laudette, M.; Conte, C. Cyclic AMP Sensor EPAC Proteins and Their Role in Cardiovascular Function and Disease. Circ. Res. 2016, 118, 881–897. [Google Scholar] [CrossRef]
- Leung, Y.K.; Du, J.; Huang, Y.; Yao, X. Cyclic Nucleotide-Gated Channels Contribute to Thromboxane A2-Induced Contraction of Rat Small Mesenteric Arteries. PLoS ONE 2010, 5, e11098. [Google Scholar] [CrossRef]
- Yao, X.; Leung, P.-S.; Kwan, H.-Y.; Wong, T.-P.; Fong, M.-W. Rod-type cyclic nucleotide-gated cation channel is expressed in vascular endothelium and vascular smooth muscle cells. Cardiovasc. Res. 1999, 41, 282–290. [Google Scholar] [CrossRef][Green Version]
- Cheng, K.-T.; Chan, F.L.; Huang, Y.; Chan, W.-Y.; Yao, X. Expression of olfactory-type cyclic nucleotide-gated channel (CNGA2) in vascular tissues. Histochem. Cell Biol. 2003, 120, 475–481. [Google Scholar] [CrossRef] [PubMed]
- Shen, B.; Cheng, K.-T.; Leung, Y.-K.; Kwok, Y.-C.; Kwan, H.-Y.; Wong, C.-O.; Chen, Z.-Y.; Huang, Y.; Yao, X. Epinephrine-induced Ca2+ influx in vascular endothelial cells is mediated by CNGA2 channels. J. Mol. Cell. Cardiol. 2008, 45, 437–445. [Google Scholar] [CrossRef]
- Biel, M.; Michalakis, S. Cyclic nucleotide-gated channels. In Handbook of Experimental Pharmacology; Springer: Berlin/Heidelberg, Germany, 2009; pp. 111–136. [Google Scholar] [CrossRef]
- E.Reeseabc, D.; Zavaljevskib, M.; Streiff, N.L.; Baderabc, D. bves:A Novel Gene Expressed during Coronary Blood Vessel Development. Dev. Biol. 1999, 209, 159–171. [Google Scholar] [CrossRef] [PubMed]
- Wada, A.M.; Reese, D.E.; Bader, D.M. Bves: Prototype of a new class of cell adhesion molecules expressed during coronary artery development. Development 2001, 128, 2085–2093. [Google Scholar] [CrossRef]
- Walsh, D.A.; Van Patten, S.M. Multiple pathway signal transduction by the cAMP-dependent protein kinase. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 1994, 8, 1227–1236. [Google Scholar]
- Taylor, S.S.; Kim, C.; Vigil, D.; Haste, N.M.; Yang, J.; Wu, J.; Anand, G.S. Dynamics of signaling by PKA. Biochim. Biophys. Acta (BBA)-Proteins Proteom. 2005, 1754, 25–37. [Google Scholar] [CrossRef]
- Hofmann, F.; Feil, R.; Kleppisch, T.; Schlossmann, J. Function of cGMP-dependent protein kinases as revealed by gene deletion. Physiol. Rev. 2006, 86, 1–23. [Google Scholar] [CrossRef]
- Lincoln, T.M.; Dey, N.; Sellak, H. Invited review: cGMP-dependent protein kinase signaling mechanisms in smooth muscle: From the regulation of tone to gene expression. J. Appl. Physiol. 2001, 91, 1421–1430. [Google Scholar] [CrossRef]
- Lucas, K.A.; Pitari, G.M.; Kazerounian, S.; Ruiz-Stewart, I.; Park, J.; Schulz, S.; Chepenik, K.P.; Waldman, S.A. Guanylyl cyclases and signaling by cyclic GMP. Pharmacol. Rev. 2000, 52, 375–414. [Google Scholar]
- Geiselhöringer, A.; Werner, M.; Sigl, K.; Smital, P.; Wörner, R.; Acheo, L.; Stieber, J.; Weinmeister, P.; Feil, R.; Feil, S.; et al. IRAG is essential for relaxation of receptor-triggered smooth muscle contraction by cGMP kinase. EMBO J. 2004, 23, 4222–4231. [Google Scholar] [CrossRef]
- Hofmann, F. The Biology of Cyclic GMP-dependent Protein Kinases. J. Biol. Chem. 2005, 280, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Feil, R.; Hofmann, F. Alternative splicing of cGMP-dependent protein kinase I and nitrate tolerance. Circ. Res. 2003, 93, e143. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Cornwell, T.L.; Soff, G.A.; Traynor, A.E.; Lincoln, T.M. Regulation of the Expression of Cyclic GMP-Dependent Protein Kinase by Cell Density in Vascular Smooth Muscle Cells. J. Vasc. Res. 1994, 31, 330–337. [Google Scholar] [CrossRef]
- Boerth, N.J.; Dey, N.B.; Cornwell, T.L.; Lincoln, T.M. Cyclic GMP-Dependent Protein Kinase Regulates Vascular Smooth Muscle Cell Phenotype. J. Vasc. Res. 1997, 34, 245–259. [Google Scholar] [CrossRef]
- Kim, J.J.; Lorenz, R.; Arold, S.T.; Reger, A.S.; Sankaran, B.; Casteel, D.E.; Herberg, F.W.; Kim, C. Crystal Structure of PKG I:cGMP Complex Reveals a cGMP-Mediated Dimeric Interface that Facilitates cGMP-Induced Activation. Structure 2016, 24, 710–720. [Google Scholar] [CrossRef] [PubMed]
- Maryam, A.; Khalid, R.R.; Vedithi, S.C.; Ece, A.; Çınaroğlu, S.S.; Siddiqi, A.R.; Blundell, T.L. Exploring the structural basis of conformational heterogeneity and autoinhibition of human cGMP-specific protein kinase Iα through computational modelling and molecular dynamics simulations. Comput. Struct. Biotechnol. J. 2020, 18, 1625–1638. [Google Scholar] [CrossRef]
- Huang, G.Y.; Kim, J.J.; Reger, A.S.; Lorenz, R.; Moon, E.-W.; Zhao, C.; Casteel, D.E.; Bertinetti, D.; VanSchouwen, B.; Selvaratnam, R.; et al. Structural Basis for Cyclic-Nucleotide Selectivity and cGMP-Selective Activation of PKG I. Structure 2013, 22, 116–124. [Google Scholar] [CrossRef] [PubMed]
- Schlossmann, J.; Ammendola, A.; Ashman, K.; Zong, X.; Huber, A.; Neubauer, G.; Wang, G.-X.; Allescher, H.-D.; Korth, M.; Wilm, M.; et al. Regulation of intracellular calcium by a signalling complex of IRAG, IP3 receptor and cGMP kinase Iβ. Nature 2000, 404, 197–201. [Google Scholar] [CrossRef]
- Komalavilas, P.; Lincoln, T.M. Phosphorylation of the Inositol 1,4,5-Trisphosphate Receptor. J. Biol. Chem. 1996, 271, 21933–21938. [Google Scholar] [CrossRef] [PubMed]
- Osei-Owusu, P.; Sun, X.; Drenan, R.M.; Steinberg, T.H.; Blumer, K.J. Regulation of RGS2 and Second Messenger Signaling in Vascular Smooth Muscle Cells by cGMP-dependent Protein Kinase. J. Biol. Chem. 2007, 282, 31656–31665. [Google Scholar] [CrossRef]
- Sawada, N.; Itoh, H.; Yamashita, J.; Doi, K.; Inoue, M.; Masatsugu, K.; Fukunaga, Y.; Sakaguchi, S.; Sone, M.; Yamahara, K.-I.; et al. cGMP-Dependent Protein Kinase Phosphorylates and Inactivates RhoA. Biochem. Biophys. Res. Commun. 2001, 280, 798–805. [Google Scholar] [CrossRef] [PubMed]
- Sauzeau, V.; Le Jeune, H.; Cario-Toumaniantz, C.; Smolenski, A.; Lohmann, S.M.; Bertoglio, J.; Chardin, P.; Pacaud, P.; Loirand, G. Cyclic GMP-dependent Protein Kinase Signaling Pathway Inhibits RhoA-induced Ca2+ Sensitization of Contraction in Vascular Smooth Muscle. J. Biol. Chem. 2000, 275, 21722–21729. [Google Scholar] [CrossRef] [PubMed]
- Somlyo, A.V. Cyclic GMP Regulation of Myosin Phosphatase. Circ. Res. 2007, 101, 645–647. [Google Scholar] [CrossRef]
- Wooldridge, A.A.; MacDonald, J.A.; Erdodi, F.; Ma, C.; Borman, M.A.; Hartshorne, D.J.; Haystead, T.A.J. Smooth Muscle Phosphatase Is Regulated in Vivo by Exclusion of Phosphorylation of Threonine 696 of MYPT1 by Phosphorylation of Serine 695 in Response to Cyclic Nucleotides. J. Biol. Chem. 2004, 279, 34496–34504. [Google Scholar] [CrossRef]
- Beall, A.; Bagwell, D.; Woodrum, D.; Stoming, T.A.; Kato, K.; Suzuki, A.; Rasmussen, H.; Brophy, C.M. The Small Heat Shock-related Protein, HSP20, Is Phosphorylated on Serine 16 during Cyclic Nucleotide-dependent Relaxation. J. Biol. Chem. 1999, 274, 11344–11351. [Google Scholar] [CrossRef]
- Beall, A.C.; Kato, K.; Goldenring, J.R.; Rasmussen, H.; Brophy, C.M. Cyclic Nucleotide-dependent Vasorelaxation Is Associated with the Phosphorylation of a Small Heat Shock-related Protein. J. Biol. Chem. 1997, 272, 11283–11287. [Google Scholar] [CrossRef]
- Cairrão, E.; Santos-Silva, A.J.; Verde, I. PKG is involved in testosterone-induced vasorelaxation of human umbilical artery. Eur. J. Pharmacol. 2010, 640, 94–101. [Google Scholar] [CrossRef]
- Podda, M.V.; Grassi, C. New perspectives in cyclic nucleotide-mediated functions in the CNS: The emerging role of cyclic nucleotide-gated (CNG) channels. Pflügers Arch.-Eur. J. Physiol. 2013, 466, 1241–1257. [Google Scholar] [CrossRef]
- Feil, R.; Lehners, M.; Stehle, D.; Feil, S. Visualising and understanding cGMP signals in the cardiovascular system. Br. J. Pharmacol. 2021, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Rybalkin, S.D.; Rybalkina, I.G.; Shimizu-Albergine, M.; Tang, X.; Beavo, J.A. PDE5 is converted to an activated state upon cGMP binding to the GAF A domain. EMBO J. 2003, 22, 469–478. [Google Scholar] [CrossRef]
- Pani, B.; Singh, B.B. Lipid rafts/caveolae as microdomains of calcium signaling. Cell Calcium 2009, 45, 625–633. [Google Scholar] [CrossRef] [PubMed]
- Guellich, A.; Mehel, H.; Fischmeister, R. Cyclic AMP synthesis and hydrolysis in the normal and failing heart. Pflügers Arch.-Eur. J. Physiol. 2014, 466, 1163–1175. [Google Scholar] [CrossRef] [PubMed]
- Schrade, K.; Klussmann, E. Pharmacological Approaches for Delineating Functions of AKAP-Based Signalling Complexes and Finding Therapeutic Targets. In Microdomains in the Cardiovascular System; Springer: Cham, Switzerland, 2017; Volume 3, pp. 59–83. [Google Scholar] [CrossRef]
- Ghigo, A.; Pirozzi, F.; Li, M.; Hirsch, E. Chatting Second Messengers: PIP3 and cAMP. In Microdomains in the Cardiovascular System; Springer: Cham, Switzerland, 2017; Volume 3, pp. 85–95. [Google Scholar] [CrossRef]
- Francis, S.H.; Blount, M.A.; Corbin, J.D. Mammalian Cyclic Nucleotide Phosphodiesterases: Molecular Mechanisms and Physiological Functions. Physiol. Rev. 2011, 91, 651–690. [Google Scholar] [CrossRef] [PubMed]
- Hardin, C.D.; Vallejo, J. Caveolins in vascular smooth muscle: Form organizing function. Cardiovasc. Res. 2006, 69, 808–815. [Google Scholar] [CrossRef] [PubMed]
- Lefkimmiatis, K.; Zaccolo, M. cAMP signaling in subcellular compartments. Pharmacol. Ther. 2014, 143, 295–304. [Google Scholar] [CrossRef] [PubMed]
- Belleville-Rolland, T.; Sassi, Y.; Decouture, B.; Dreano, E.; Hulot, J.-S.; Gaussem, P.; Bachelot-Loza, C. MRP4 (ABCC4) as a potential pharmacologic target for cardiovascular disease. Pharmacol. Res. 2016, 107, 381–389. [Google Scholar] [CrossRef]
- Sassi, Y.; Lipskaia, L.; Vandecasteele, G.; Nikolaev, V.O.; Hatem, S.N.; Aubart, F.C.; Russel, F.G.; Mougenot, N.; Vrignaud, C.; Lechat, P.; et al. Multidrug resistance-associated protein 4 regulates cAMP-dependent signaling pathways and controls human and rat SMC proliferation. J. Clin. Investig. 2008, 118, 2747–2757. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, F.; Murata, T.; Shimizu, K.; Degerman, E.; Maurice, D.; Manganiello, V. Cyclic Nucleotide Phosphodiesterases: Important signaling modulators and therapeutic targets. Oral Dis. 2014, 21, e25–e50. [Google Scholar] [CrossRef] [PubMed]
- Dillard, J.; Meng, X.; Nelin, L.; Liu, Y.; Chen, B. Nitric oxide activates AMPK by modulating PDE3A in human pulmonary artery smooth muscle cells. Physiol. Rep. 2020, 8, e14559. [Google Scholar] [CrossRef]
- Sprenger, J.U.; Nikolaev, V.O. Biophysical Techniques for Detection of cAMP and cGMP in Living Cells. Int. J. Mol. Sci. 2013, 14, 8025–8046. [Google Scholar] [CrossRef] [PubMed]
- Thunemann, M.; Fomin, N.; Krawutschke, C.; Russwurm, M.; Feil, R. Visualization of cGMP with cGi Biosensors. In Guanylate Cyclase and Cyclic GMP; Humana Press: Totowa, NJ, USA, 2013; pp. 89–120. [Google Scholar] [CrossRef]
- Berisha, F.; Nikolaev, V.O. Cyclic nucleotide imaging and cardiovascular disease. Pharmacol. Ther. 2017, 175, 107–115. [Google Scholar] [CrossRef]
- Beavo, J.A.; Brunton, L.L. Cyclic nucleotide research—Still expanding after half a century. Nat. Rev. Mol. Cell Biol. 2002, 3, 710–717. [Google Scholar] [CrossRef]
- Kritzer, M.D.; Li, J.; Dodge-Kafka, K.; Kapiloff, M.S. AKAPs: The architectural underpinnings of local cAMP signaling. J. Mol. Cell. Cardiol. 2012, 52, 351–358. [Google Scholar] [CrossRef]
- Tröger, J.; Moutty, M.C.; Skroblin, P.; Klussmann, E. A-kinase anchoring proteins as potential drug targets. Br. J. Pharmacol. 2012, 166, 420–433. [Google Scholar] [CrossRef]
- Perino, A.; Ghigo, A.; Scott, J.D.; Hirsch, E. Anchoring Proteins as Regulators of Signaling Pathways. Circ. Res. 2012, 111, 482–492. [Google Scholar] [CrossRef]
- Scott, J.D.; Dessauer, C.W.; Taskén, K. Creating Order from Chaos: Cellular Regulation by Kinase Anchoring. Annu. Rev. Pharmacol. Toxicol. 2013, 53, 187–210. [Google Scholar] [CrossRef]
- Nystoriak, M.A.; Nieves-Cintrón, M.; Patriarchi, T.; Buonarati, O.R.; Prada, M.P.; Morotti, S.; Grandi, E.; Fernandes, J.D.S.; Forbush, K.; Hofmann, F.; et al. Ser 1928 phosphorylation by PKA stimulates the L-type Ca 2+ channel Ca V 1.2 and vasoconstriction during acute hyperglycemia and diabetes. Sci. Signal. 2017, 10, eaaf9647. [Google Scholar] [CrossRef] [PubMed]
- Navedo, M.F.; Nieves-Cintrón, M.; Amberg, G.C.; Yuan, C.; Votaw, V.S.; Lederer, W.J.; McKnight, G.S.; Santana, L.F. AKAP150 Is Required for Stuttering Persistent Ca2+Sparklets and Angiotensin II–Induced Hypertension. Circ. Res. 2008, 102, 1–11. [Google Scholar] [CrossRef]
- Baillie, G.; Houslay, M.D. Arrestin times for compartmentalised cAMP signalling and phosphodiesterase-4 enzymes. Curr. Opin. Cell Biol. 2005, 17, 129–134. [Google Scholar] [CrossRef] [PubMed]
- Zaccolo, M.; Di Benedetto, G.; Lissandron, V.; Mancuso, L.; Terrin, A.; Zamparo, I. Restricted diffusion of a freely diffusible second messenger: Mechanisms underlying compartmentalized cAMP signalling. Biochem. Soc. Trans. 2006, 34, 495–497. [Google Scholar] [CrossRef]
- McCormick, K.; Baillie, G.S. Compartmentalisation of second messenger signalling pathways. Curr. Opin. Genet. Dev. 2014, 27, 20–25. [Google Scholar] [CrossRef] [PubMed]
- Brescia, M.; Zaccolo, M. Modulation of Compartmentalised Cyclic Nucleotide Signalling via Local Inhibition of Phosphodiesterase Activity. Int. J. Mol. Sci. 2016, 17, 1672. [Google Scholar] [CrossRef]
- Richter, W.; Day, P.; Agrawal, R.; Bruss, M.D.; Granier, S.; Wang, Y.L.; Rasmussen, S.; Horner, K.; Wang, P.; Lei, T.; et al. Signaling from β1- and β2-adrenergic receptors is defined by differential interactions with PDE4. EMBO J. 2008, 27, 384–393. [Google Scholar] [CrossRef]
- Cai, Y.; Nagel, D.J.; Zhou, Q.; Cygnar, K.; Zhao, H.; Li, F.; Pi, X.; Knight, P.A.; Yan, C. Role of cAMP-phosphodiesterase 1C signaling in regulating growth factor receptor stability, vascular smooth muscle cell growth, migration, and neointimal hyperplasia. Circ. Res. 2015, 116, 1120–1132. [Google Scholar] [CrossRef]
- Kim, N.; Rybalkin, S.D.; Pi, X.; Wang, Y.; Zhang, C.; Munzel, T.; Beavo, J.A.; Berk, B.C.; Yan, C. Upregulation of phosphodiesterase 1A1 expression is associated with the development of nitrate tolerance. Circulation 2001, 104, 2338–2343. [Google Scholar] [CrossRef]
- Koschinski, A.; Zaccolo, M. Activation of PKA in cell requires higher concentration of cAMP than in vitro: Implications for compartmentalization of cAMP signalling. Sci. Rep. 2017, 7, 1–12. [Google Scholar] [CrossRef]
- Rich, T.C.; Xin, W.; Mehats, C.; Hassell, K.A.; Piggott, L.A.; Le, X.; Karpen, J.W.; Conti, M. Cellular mechanisms underlying prostaglandin-induced transient cAMP signals near the plasma membrane of HEK-293 cells. Am. J. Physiol. Physiol. 2007, 292, C319–C331. [Google Scholar] [CrossRef]
- Tsai, E.J.; Kass, D.A. Cyclic GMP signaling in cardiovascular pathophysiology and therapeutics. Pharmacol. Ther. 2009, 122, 216–238. [Google Scholar] [CrossRef] [PubMed]
- Zolle, O.; Lawrie, A.M.; Simpson, A.W. Activation of the Particulate and Not the Soluble Guanylate Cyclase Leads to the Inhibition of Ca2+ Extrusion through Localized Elevation of cGMP. J. Biol. Chem. 2000, 275, 25892–25899. [Google Scholar] [CrossRef]
- Rho, E.H.; Perkins, W.J.; Lorenz, R.R.; Warner, D.O.; Jones, K.A. Differential effects of soluble and particulate guanylyl cyclase on Ca(2+) sensitivity in airway smooth muscle. J. Appl. Physiol. 2002, 92, 257–263. [Google Scholar] [CrossRef]
- Subramanian, H.; Froese, A.; Jönsson, P.; Schmidt, H.; Gorelik, J.; Nikolaev, V.O. Distinct submembrane localisation compartmentalises cardiac NPR1 and NPR2 signalling to cGMP. Nat. Commun. 2018, 9, 2446. [Google Scholar] [CrossRef]
- Piggott, L.A.; Hassell, K.A.; Berkova, Z.; Morris, A.P.; Silberbach, M.; Rich, T.C. Natriuretic Peptides and Nitric Oxide Stimulate cGMP Synthesis in Different Cellular Compartments. J. Gen. Physiol. 2006, 128, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Cawley, S.M.; Sawyer, C.L.; Brunelle, K.F.; van der Vliet, A.; Dostmann, W.R. Nitric oxide-evoked transient kinetics of cyclic GMP in vascular smooth muscle cells. Cell. Signal. 2007, 19, 1023–1033. [Google Scholar] [CrossRef] [PubMed]
- Nausch, L.W.M.; Ledoux, J.; Bonev, A.D.; Nelson, M.T.; Dostmann, W.R. Differential patterning of cGMP in vascular smooth muscle cells revealed by single GFP-linked biosensors. Proc. Natl. Acad. Sci. USA 2007, 105, 365–370. [Google Scholar] [CrossRef] [PubMed]
- Wilson, L.S.; Guo, M.; Umana, M.B.; Maurice, D.H. Distinct phosphodiesterase 5A-containing compartments allow selective regulation of cGMP-dependent signalling in human arterial smooth muscle cells. Cell. Signal. 2017, 36, 204–211. [Google Scholar] [CrossRef] [PubMed]
- Ammon, H.P.T.; Müller, A.B. Forskolin: From an Ayurvedic Remedy to a Modern Agent. Planta Med. 1985, 51, 473–477. [Google Scholar] [CrossRef]
- DeNinno, M.P. Future directions in phosphodiesterase drug discovery. Bioorg. Med. Chem. Lett. 2012, 22, 6794–6800. [Google Scholar] [CrossRef]
- Martínez, A.; Gil, C. cAMP-specific phosphodiesterase inhibitors: Promising drugs for inflammatory and neurological diseases. Expert Opin. Ther. Patents 2014, 24, 1311–1321. [Google Scholar] [CrossRef]
- Keravis, T.; Lugnier, C. Cyclic nucleotide phosphodiesterase (PDE) isozymes as targets of the intracellular signalling network: Benefits of PDE inhibitors in various diseases and perspectives for future therapeutic developments. Br. J. Pharmacol. 2012, 165, 1288–1305. [Google Scholar] [CrossRef]
- Lugnier, C.; Meyer, A.; Charloux, A.; Andrès, E.; Gény, B.; Talha, S. The Endocrine Function of the Heart: Physiology and Involvements of Natriuretic Peptides and Cyclic Nucleotide Phosphodiesterases in Heart Failure. J. Clin. Med. 2019, 8, 1746. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.; Ji, Z.; Tsalkova, T.; Mei, F. Epac and PKA: A tale of two intracellular cAMP receptors. Acta Biochim. Biophys. Sin. 2008, 40, 651–662. [Google Scholar] [CrossRef]
- Parnell, E.; Palmer, T.M.; Yarwood, S.J. The future of EPAC-targeted therapies: Agonism versus antagonism. Trends Pharmacol. Sci. 2015, 36, 203–214. [Google Scholar] [CrossRef] [PubMed]
- Barker, G.; Parnell, E.; Van Basten, B.; Buist, H.; Adams, D.R.; Yarwood, S.J. The Potential of a Novel Class of EPAC-Selective Agonists to Combat Cardiovascular Inflammation. J. Cardiovasc. Dev. Dis. 2017, 4, 22. [Google Scholar] [CrossRef]
- Ahmed, A.; Boulton, S.; Shao, H.; Akimoto, M.; Natarajan, A.; Cheng, X.; Melacini, G. Recent Advances in EPAC-Targeted Therapies: A Biophysical Perspective. Cells 2019, 8, 1462. [Google Scholar] [CrossRef]
- Miller, M.R.; Megson, I.L. Recent developments in nitric oxide donor drugs. Br. J. Pharmacol. 2007, 151, 305–321. [Google Scholar] [CrossRef]
- Lima, B.; Forrester, M.T.; Hess, D.T.; Stamler, J.S. S -Nitrosylation in Cardiovascular Signaling. Circ. Res. 2010, 106, 633–646. [Google Scholar] [CrossRef] [PubMed]
- Petraina, A.; Nogales, C.; Krahn, T.; Mucke, H.; Lüscher, T.F.; Fischmeister, R.; Kass, D.A.; Burnett, J.C.; Hobbs, A.J.; Schmidt, H.H.H.W. Cyclic GMP modulating drugs in cardiovascular diseases: Mechanism-based network pharmacology. Cardiovasc. Res. 2021, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Alexander, S.P.; Striessnig, J.; Kelly, E.; Marrion, N.V.; Peters, J.A.; Faccenda, E.; Harding, S.D.; Pawson, A.J.; Sharman, J.L.; Southan, C.; et al. The concise guide to pharmacology 2017/18: Voltage-gated ion channels. Br. J. Pharmacol. 2017, 174, S160–S194. [Google Scholar] [CrossRef]
- McKie, P.M.; Cataliotti, A.; Huntley, B.K.; Martin, F.L.; Olson, T.M.; Burnett, J.C. A Human Atrial Natriuretic Peptide Gene Mutation Reveals a Novel Peptide with Enhanced Blood Pressure-Lowering, Renal-Enhancing, and Aldosterone-Suppressing Actions. J. Am. Coll. Cardiol. 2009, 54, 1024–1032. [Google Scholar] [CrossRef]
- McKie, P.M.; Iyer, S.R.; Scott, C.; Bailey, K.; Johnson, B.K.; Benike, S.L.; Chen, H.; Miller, W.L.; Volpi, R.; Cabassi, A.; et al. Abstract 15205: MANP: A Novel ANP Analog for Hypertension Associated With Obesity and Metabolic Syndrome. Circulation 2020, 142, A15205. [Google Scholar] [CrossRef]
- Cataliotti, A.; Costello-Boerrigter, L.C.; Chen, H.H.; Textor, S.C.; Burnett, J.C. Sustained Blood Pressure–Lowering Actions of Subcutaneous B-Type Natriuretic Peptide (Nesiritide) in a Patient With Uncontrolled Hypertension. Mayo Clin. Proc. 2012, 87, 413–415. [Google Scholar] [CrossRef] [PubMed]
- Conole, D.; Scott, L.J. Riociguat: First Global Approval. Drugs 2013, 73, 1967–1975. [Google Scholar] [CrossRef]
- Markham, A.; Duggan, S. Vericiguat: First Approval. Drugs 2021, 81, 721–726. [Google Scholar] [CrossRef] [PubMed]
- Hahn, M.G.; Lampe, T.; El Sheikh, S.; Griebenow, N.; Woltering, E.; Schlemmer, K.-H.; Dietz, L.; Gerisch, M.; Wunder, F.; Becker-Pelster, E.-M.; et al. Discovery of the Soluble Guanylate Cyclase Activator Runcaciguat (BAY 1101042). J. Med. Chem. 2021, 64, 5323–5344. [Google Scholar] [CrossRef]
- Sharma, U.; Cozier, G.E.; Sturrock, E.D.; Acharya, K.R. Molecular Basis for Omapatrilat and Sampatrilat Binding to Neprilysin—Implications for Dual Inhibitor Design with Angiotensin-Converting Enzyme. J. Med. Chem. 2020, 63, 5488–5500. [Google Scholar] [CrossRef] [PubMed]
- Shafiee-Nick, R.; Afshari, A.R.; Mousavi, S.H.; Rafighdoust, A.; Askari, V.R.; Mollazadeh, H.; Fanoudi, S.; Mohtashami, E.; Rahimi, V.B.; Mohebbi, M.; et al. A comprehensive review on the potential therapeutic benefits of phosphodiesterase inhibitors on cardiovascular diseases. Biomed. Pharmacother. 2017, 94, 541–556. [Google Scholar] [CrossRef]
- Karam, S.; Margaria, J.P.; Bourcier, A.; Mika, D.; Varin, A.; Bedioune, I.; Lindner, M.; Bouadjel, K.; Dessillons, M.; Gaudin, F.; et al. Cardiac Overexpression of PDE4B Blunts β-Adrenergic Response and Maladaptive Remodeling in Heart Failure. Circulation 2020, 142, 161–174. [Google Scholar] [CrossRef]
- Baillie, G.; Tejeda, G.S.; Kelly, M.P. Therapeutic targeting of 3′,5′-cyclic nucleotide phosphodiesterases: Inhibition and beyond. Nat. Rev. Drug Discov. 2019, 18, 770–796. [Google Scholar] [CrossRef] [PubMed]
- Real, J.; Serna, M.C.; Giner-Soriano, M.; Forés, R.; Pera, G.; Ribes, E.; Alzamora, M.; Marsal, J.R.; Heras, A.; Morros, R. Safety of cilostazol in peripheral artery disease: A cohort from a primary healthcare electronic database. BMC Cardiovasc. Disord. 2018, 18, 85. [Google Scholar] [CrossRef] [PubMed]
- Ota, H.; Eto, M.; Ogawa, S.; Iijima, K.; Akishita, M.; Ouchi, Y. SIRT1/eNOS Axis as a Potential Target against Vascular Senescence, Dysfunction and Atherosclerosis. J. Atheroscler. Thromb. 2010, 17, 431–435. [Google Scholar] [CrossRef] [PubMed]
- Moreira, H.S.; Lima-Leal, G.A.; Santos-Rocha, J.; Gomes-Pereira, L.; Duarte, G.P.; Xavier, F.E. Phosphodiesterase-3 inhibitor cilostazol reverses endothelial dysfunction with ageing in rat mesenteric resistance arteries. Eur. J. Pharmacol. 2018, 822, 59–68. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.C.; Kemp, B.E.; Pearson, R.B.; Smith, A.J.; Misconi, L.; Van Patten, S.M.; Walsh, D.A. A potent synthetic peptide inhibitor of the cAMP-dependent protein kinase. J. Biol. Chem. 1986, 261, 989–992. [Google Scholar] [CrossRef]
- Taylor, M.S.; Okwuchukwuasanya, C.; Nickl, C.K.; Tegge, W.; Brayden, J.E.; Dostmann, W.R.G. Inhibition of cGMP-Dependent Protein Kinase by the Cell-Permeable Peptide DT-2 Reveals a Novel Mechanism of Vasoregulation. Mol. Pharmacol. 2004, 65, 1111–1119. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cyclic Nucleotide Phosphodiesterases (PDEs) | Selectivity for cAMP Hydrolysis | Selectivity for cGMP Hydrolysis | Dual Selectivity Hydrolysis | Synthetic Phenotype | Contractile Phenotype |
|---|---|---|---|---|---|
| PDE1A |  | | | ||
| PDE1B | | | |||
| PDE1C | | | |||
| PDE3A | | | | ||
| PDE3B | | | |||
| PDE4D | | | | ||
| PDE5A | | | | ||
| PDE7A | | | | ||
| PDE7B | | | |||
| PDE8A | | | | ||
| PDE9 | | | | ||
| PDE10A | | ? | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lorigo, M.; Oliveira, N.; Cairrao, E. PDE-Mediated Cyclic Nucleotide Compartmentation in Vascular Smooth Muscle Cells: From Basic to a Clinical Perspective. J. Cardiovasc. Dev. Dis. 2022, 9, 4. https://doi.org/10.3390/jcdd9010004
Lorigo M, Oliveira N, Cairrao E. PDE-Mediated Cyclic Nucleotide Compartmentation in Vascular Smooth Muscle Cells: From Basic to a Clinical Perspective. Journal of Cardiovascular Development and Disease. 2022; 9(1):4. https://doi.org/10.3390/jcdd9010004
Chicago/Turabian StyleLorigo, Margarida, Nelson Oliveira, and Elisa Cairrao. 2022. "PDE-Mediated Cyclic Nucleotide Compartmentation in Vascular Smooth Muscle Cells: From Basic to a Clinical Perspective" Journal of Cardiovascular Development and Disease 9, no. 1: 4. https://doi.org/10.3390/jcdd9010004
APA StyleLorigo, M., Oliveira, N., & Cairrao, E. (2022). PDE-Mediated Cyclic Nucleotide Compartmentation in Vascular Smooth Muscle Cells: From Basic to a Clinical Perspective. Journal of Cardiovascular Development and Disease, 9(1), 4. https://doi.org/10.3390/jcdd9010004