Roles of PDE1 in Pathological Cardiac Remodeling and Dysfunction


{kind=link}
Abstract
:1. Introduction
2. Pathological Cardiac Remodeling and Heart Failure
3. Cyclic Nucleotide Signaling and PDEs in the Heart
3.1. Cyclic 3′,5′ Adenosine Monophosphate (cAMP) Signaling
3.2. Cyclic 3′,5′ Guanosine Monophosphate (cGMP) Signaling
3.3. Different PDE Isozymes in the Heart
4. PDE1 and Pathological Cardiac Remodeling and Dysfunction
4.1. PDE1
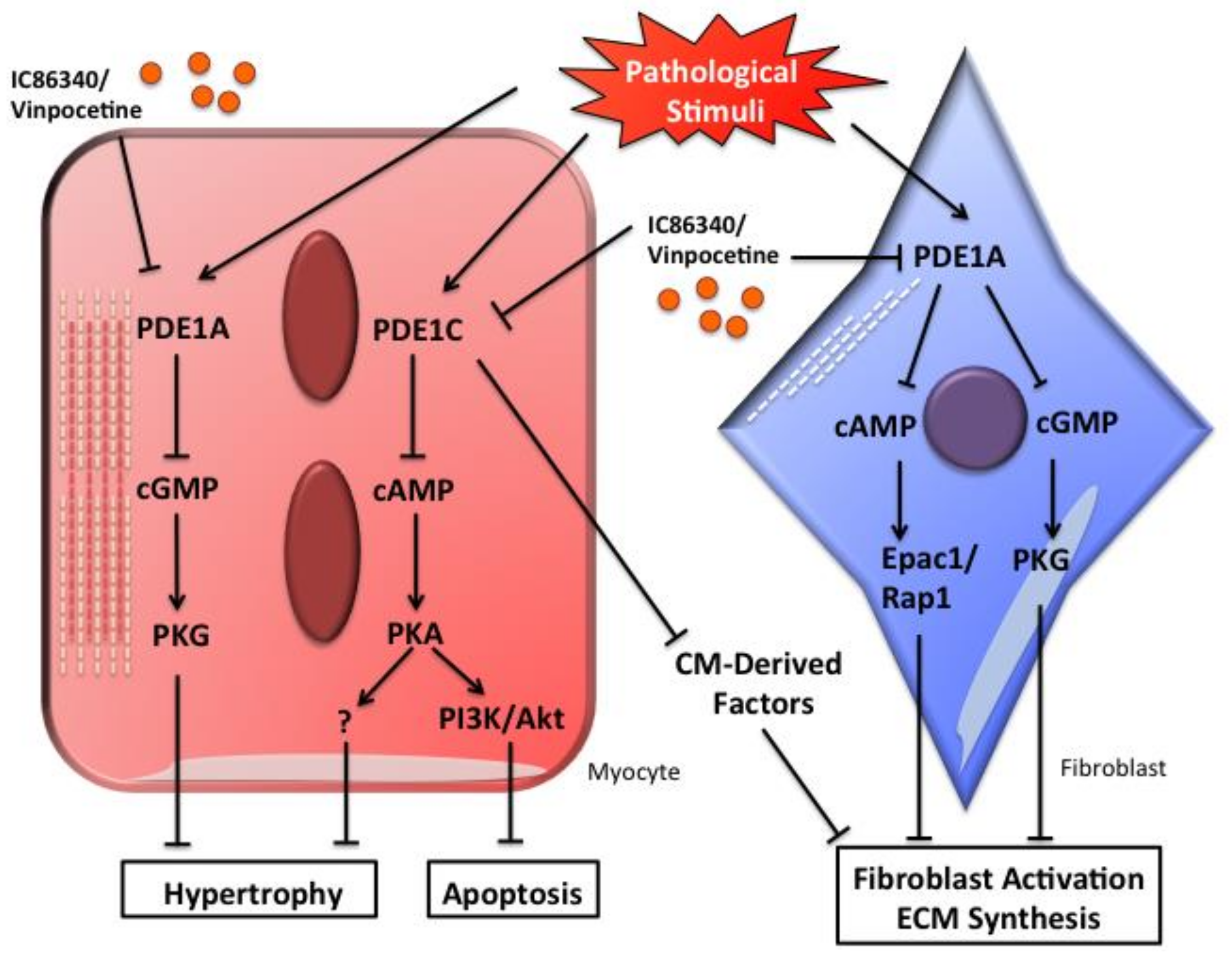
4.2. Role of PDE1A in Cardiac Myocyte Hypertrophy and Fibroblast Activation
4.3. Role of PDE1C in Pathological Cardiac Remodeling and Dysfunction
4.4. PDE1 and Other PDEs: Similarities, Differences, and Potential Interactions
5. Therapeutic Potential of PDE1 Inhibition in Pathological Cardiac Remodeling
6. Conclusions and Perspective
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Frey, N.; Olson, E.N. Cardiac hypertrophy: The good, the bad, and the ugly. Annu. Rev. Physiol. 2003, 65, 45–79. [Google Scholar] [CrossRef] [PubMed]
- Beavo, J.A.; Brunton, L.L. Cyclic nucleotide research—Still expanding after half a century. Nat. Rev. Mol. Cell Biol. 2002, 3, 710–718. [Google Scholar] [CrossRef] [PubMed]
- Maurice, D.H.; Ke, H.; Ahmad, F.; Wang, Y.; Chung, J.; Manganiello, V.C. Advances in targeting cyclic nucleotide phosphodiesterases. Nat. Rev. Drug Discov. 2014, 13, 290–314. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, I.; Fuseler, J.W.; Price, R.L.; Borg, T.K.; Baudino, T.A. Determination of cell types and numbers during cardiac development in the neonatal and adult rat and mouse. Am. J. Physiol. Heart Circ. Physiol. 2007, 293, H1883–H1891. [Google Scholar] [CrossRef] [PubMed]
- Hill, J.A.; Olson, E.N. Cardiac plasticity. N. Engl. J. Med. 2008, 358, 1370–1380. [Google Scholar] [CrossRef] [PubMed]
- McMullen, J.R.; Jennings, G.L. Differences between pathological and physiological cardiac hypertrophy: Novel therapeutic strategies to treat heart failure. Clin. Exp. Pharmacol. Physiol. 2007, 34, 255–262. [Google Scholar] [CrossRef] [PubMed]
- Porter, K.E.; Turner, N.A. Cardiac fibroblasts: At the heart of myocardial remodeling. Pharmacol. Ther. 2009, 123, 255–278. [Google Scholar] [CrossRef] [PubMed]
- Deb, A.; Ubil, E. Cardiac fibroblast in development and wound healing. J. Mol. Cell. Cardiol. 2014, 70, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Chiong, M.; Wang, Z.V.; Pedrozo, Z.; Cao, D.J.; Troncoso, R.; Ibacache, M.; Criollo, A.; Nemchenko, A.; Hill, J.A.; Lavandero, S. Cardiomyocyte death: Mechanisms and translational implications. Cell Death Dis. 2011, 2, e244. [Google Scholar] [CrossRef] [PubMed]
- Nadal-Ginard, B.; Kajstura, J.; Leri, A.; Anversa, P. Myocyte death, growth, and regeneration in cardiac hypertrophy and failure. Circ. Res. 2003, 92, 139–150. [Google Scholar] [CrossRef] [PubMed]
- Balakumar, P.; Jagadeesh, G. Multifarious molecular signaling cascades of cardiac hypertrophy: Can the muddy waters be cleared? Pharmacol. Res. 2010, 62, 365–383. [Google Scholar] [CrossRef] [PubMed]
- Dewenter, M.; von der Lieth, A.; Katus, H.A.; Backs, J. Calcium Signaling and Transcriptional Regulation in Cardiomyocytes. Circ. Res. 2017, 121, 1000–1020. [Google Scholar] [CrossRef] [PubMed]
- De Windt, L.J.; Lim, H.W.; Bueno, O.F.; Liang, Q.; Delling, U.; Braz, J.C.; Glascock, B.J.; Kimball, T.F.; del Monte, F.; Hajjar, R.J.; et al. Targeted inhibition of calcineurin attenuates cardiac hypertrophy in vivo. Proc. Natl. Acad. Sci. USA 2001, 98, 3322–3327. [Google Scholar] [CrossRef] [PubMed]
- Anderson, M.E.; Brown, J.H.; Bers, D.M. CaMKII in myocardial hypertrophy and heart failure. J. Mol. Cell. Cardiol. 2011, 51, 468–473. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Kohlhaas, M.; Backs, J.; Mishra, S.; Phillips, W.; Dybkova, N.; Chang, S.; Ling, H.; Bers, D.M.; Maier, L.S.; et al. CaMKIIdelta isoforms differentially affect calcium handling but similarly regulate HDAC/MEF2 transcriptional responses. J. Biol. Chem. 2007, 282, 35078–35087. [Google Scholar] [CrossRef] [PubMed]
- Berthet, J.; Rall, T.W.; Sutherland, E.W. The relationship of epinephrine and glucagon to liver phosphorylase. IV. Effect of epinephrine and glucagon on the reactivation of phosphorylase in liver homogenates. J. Biol. Chem. 1957, 224, 463–475. [Google Scholar] [PubMed]
- Rall, T.W.; Sutherland, E.W. Formation of a cyclic adenine ribonucleotide by tissue particles. J. Biol. Chem. 1958, 232, 1065–1076. [Google Scholar] [PubMed]
- Kopperud, R.; Krakstad, C.; Selheim, F.; Doskeland, S.O. cAMP effector mechanisms. Novel twists for an ‘old’ signaling system. FEBS Lett. 2003, 546, 121–126. [Google Scholar] [CrossRef]
- Gancedo, J.M. Biological roles of cAMP: Variations on a theme in the different kingdoms of life. Biol. Rev. Camb. Philos. Soc. 2013, 88, 645–668. [Google Scholar] [CrossRef] [PubMed]
- Hanoune, J.; Defer, N. Regulation and role of adenylyl cyclase isoforms. Annu. Rev. Pharmacol. Toxicol. 2001, 41, 145–174. [Google Scholar] [CrossRef] [PubMed]
- Steegborn, C. Structure, mechanism, and regulation of soluble adenylyl cyclases—Similarities and differences to transmembrane adenylyl cyclases. Biochim. Biophys. Acta 2014, 1842, 2535–2547. [Google Scholar] [CrossRef] [PubMed]
- Tresguerres, M.; Levin, L.R.; Buck, J. Intracellular cAMP signaling by soluble adenylyl cyclase. Kidney Int. 2011, 79, 1277–1288. [Google Scholar] [CrossRef] [PubMed]
- Willoughby, D.; Cooper, D.M. Live-cell imaging of cAMP dynamics. Nat. Methods 2008, 5, 29–36. [Google Scholar] [CrossRef] [PubMed]
- Leroy, J.; Abi-Gerges, A.; Nikolaev, V.O.; Richter, W.; Lechene, P.; Mazet, J.L.; Conti, M.; Fischmeister, R.; Vandecasteele, G. Spatiotemporal dynamics of beta-adrenergic cAMP signals and L-type Ca2+ channel regulation in adult rat ventricular myocytes: Role of phosphodiesterases. Circ. Res. 2008, 102, 1091–1100. [Google Scholar] [CrossRef] [PubMed]
- Scriven, D.R.; Dan, P.; Moore, E.D. Distribution of proteins implicated in excitation-contraction coupling in rat ventricular myocytes. Biophys. J. 2000, 79, 2682–2691. [Google Scholar] [CrossRef]
- Trafford, A.W.; Diaz, M.E.; Negretti, N.; Eisner, D.A. Enhanced Ca2+ current and decreased Ca2+ efflux restore sarcoplasmic reticulum Ca2+ content after depletion. Circ. Res. 1997, 81, 477–484. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Piacentino, V., 3rd; Furukawa, S.; Goldman, B.; Margulies, K.B.; Houser, S.R. L-type Ca2+ channel density and regulation are altered in failing human ventricular myocytes and recover after support with mechanical assist devices. Circ. Res. 2002, 91, 517–524. [Google Scholar] [CrossRef] [PubMed]
- Marx, S.O.; Reiken, S.; Hisamatsu, Y.; Gaburjakova, M.; Gaburjakova, J.; Yang, Y.M.; Rosemblit, N.; Marks, A.R. Phosphorylation-dependent regulation of ryanodine receptors: A novel role for leucine/isoleucine zippers. J. Cell Biol. 2001, 153, 699–708. [Google Scholar] [CrossRef] [PubMed]
- Brittsan, A.G.; Kranias, E.G. Phospholamban and cardiac contractile function. J. Mol. Cell. Cardiol. 2000, 32, 2131–2139. [Google Scholar] [CrossRef] [PubMed]
- Guellich, A.; Mehel, H.; Fischmeister, R. Cyclic AMP synthesis and hydrolysis in the normal and failing heart. Pflugers Arch. 2014, 466, 1163–1175. [Google Scholar] [CrossRef] [PubMed]
- Hamad, E.A.; Zhu, W.; Chan, T.O.; Myers, V.; Gao, E.; Li, X.; Zhang, J.; Song, J.; Zhang, X.Q.; Cheung, J.Y.; et al. Cardioprotection of controlled and cardiac-specific over-expression of A(2A)-adenosine receptor in the pressure overload. PLoS ONE 2012, 7, e39919. [Google Scholar] [CrossRef] [PubMed]
- Lukowski, R.; Krieg, T.; Rybalkin, S.D.; Beavo, J.; Hofmann, F. Turning on cGMP-dependent pathways to treat cardiac dysfunctions: Boom, bust, and beyond. Trends Pharmacol. Sci. 2014, 35, 404–413. [Google Scholar] [CrossRef] [PubMed]
- Potter, L.R. Guanylyl cyclase structure, function and regulation. Cell Signal. 2011, 23, 1921–1926. [Google Scholar] [CrossRef] [PubMed]
- Tsai, E.J.; Kass, D.A. Cyclic GMP signaling in cardiovascular pathophysiology and therapeutics. Pharmacol. Ther. 2009, 122, 216–238. [Google Scholar] [CrossRef] [PubMed]
- Castro, L.R.; Verde, I.; Cooper, D.M.; Fischmeister, R. Cyclic guanosine monophosphate compartmentation in rat cardiac myocytes. Circulation 2006, 113, 2221–2228. [Google Scholar] [CrossRef] [PubMed]
- Miller, C.L.; Cai, Y.; Oikawa, M.; Thomas, T.; Dostmann, W.R.; Zaccolo, M.; Fujiwara, K.; Yan, C. Cyclic nucleotide phosphodiesterase 1A: A key regulator of cardiac fibroblast activation and extracellular matrix remodeling in the heart. Basic Res. Cardiol. 2011, 106, 1023–1039. [Google Scholar] [CrossRef] [PubMed]
- Frantz, S.; Klaiber, M.; Baba, H.A.; Oberwinkler, H.; Volker, K.; Gabetaner, B.; Bayer, B.; Abebetaer, M.; Schuh, K.; Feil, R.; et al. Stress-dependent dilated cardiomyopathy in mice with cardiomyocyte-restricted inactivation of cyclic GMP-dependent protein kinase I. Eur Heart J. 2013, 34, 1233–1244. [Google Scholar] [CrossRef] [PubMed]
- Takimoto, E.; Champion, H.C.; Li, M.; Belardi, D.; Ren, S.; Rodriguez, E.R.; Bedja, D.; Gabrielson, K.L.; Wang, Y.; Kass, D.A. Chronic inhibition of cyclic GMP phosphodiesterase 5A prevents and reverses cardiac hypertrophy. Nat. Med. 2005, 11, 214–222. [Google Scholar] [CrossRef] [PubMed]
- Wegener, J.W.; Nawrath, H.; Wolfsgruber, W.; Kuhbandner, S.; Werner, C.; Hofmann, F.; Feil, R. cGMP-dependent protein kinase I mediates the negative inotropic effect of cGMP in the murine myocardium. Circ. Res. 2002, 90, 18–20. [Google Scholar] [CrossRef] [PubMed]
- Patrucco, E.; Domes, K.; Sbroggio, M.; Blaich, A.; Schlossmann, J.; Desch, M.; Rybalkin, S.D.; Beavo, J.A.; Lukowski, R.; Hofmann, F. Roles of cGMP-dependent protein kinase I (cGKI) and PDE5 in the regulation of Ang II-induced cardiac hypertrophy and fibrosis. Proc. Natl. Acad. Sci. USA 2014, 111, 12925–12929. [Google Scholar] [CrossRef] [PubMed]
- Lu, Z.; Xu, X.; Hu, X.; Lee, S.; Traverse, J.H.; Zhu, G.; Fassett, J.; Tao, Y.; Zhang, P.; dos Remedios, C.; et al. Oxidative stress regulates left ventricular PDE5 expression in the failing heart. Circulation 2010, 121, 1474–1483. [Google Scholar] [CrossRef] [PubMed]
- Nagayama, T.; Hsu, S.; Zhang, M.; Koitabashi, N.; Bedja, D.; Gabrielson, K.L.; Takimoto, E.; Kass, D.A. Sildenafil stops progressive chamber, cellular, and molecular remodeling and improves calcium handling and function in hearts with pre-existing advanced hypertrophy caused by pressure overload. J. Am. Coll. Cardiol. 2009, 53, 207–215. [Google Scholar] [CrossRef] [PubMed]
- Takimoto, E.; Belardi, D.; Tocchetti, C.G.; Vahebi, S.; Cormaci, G.; Ketner, E.A.; Moens, A.L.; Champion, H.C.; Kass, D.A. Compartmentalization of cardiac β-adrenergic inotropy modulation by phosphodiesterase type 5. Circulation 2007, 115, 2159–2167. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.I.; Zhu, G.; Sasaki, T.; Cho, G.S.; Hamdani, N.; Holewinski, R.; Jo, S.H.; Danner, T.; Zhang, M.; Rainer, P.P.; et al. Phosphodiesterase 9A controls nitric-oxide-independent cGMP and hypertrophic heart disease. Nature 2015, 519, 472–476. [Google Scholar] [CrossRef] [PubMed]
- Miller, C.L.; Oikawa, M.; Cai, Y.; Wojtovich, A.P.; Nagel, D.J.; Xu, X.; Xu, H.; Florio, V.; Rybalkin, S.D.; Beavo, J.A.; et al. Role of Ca2+/calmodulin-stimulated cyclic nucleotide phosphodiesterase 1 in mediating cardiomyocyte hypertrophy. Circ. Res. 2009, 105, 956–964. [Google Scholar] [CrossRef] [PubMed]
- Bobin, P.; Belacel-Ouari, M.; Bedioune, I.; Zhang, L.; Leroy, J.; Leblais, V.; Fischmeister, R.; Vandecasteele, G. Cyclic nucleotide phosphodiesterases in heart and vessels: A therapeutic perspective. Arch. Cardiovasc. Dis. 2016, 109, 431–443. [Google Scholar] [CrossRef] [PubMed]
- Omori, K.; Kotera, J. Overview of PDEs and their regulation. Circ. Res. 2007, 100, 309–327. [Google Scholar] [CrossRef] [PubMed]
- Ding, B.; Abe, J.I.; Wei, H.; Huang, Q.; Walsh, R.A.; Molina, C.A.; Zhao, A.; Sadoshima, J.; Blaxall, B.C.; Berk, B.C.; et al. Functional role of phosphodiesterase 3 in cardiomyocyte apoptosis: Implication in heart failure. Circulation 2005, 111, 2469–2476. [Google Scholar] [CrossRef] [PubMed]
- Knight, W.E.; Chen, S.; Zhang, Y.; Oikawa, M.; Wu, M.; Zhou, Q.; Miller, C.L.; Cai, Y.; Mickelsen, D.M.; Moravec, C.; et al. PDE1C deficiency antagonizes pathological cardiac remodeling and dysfunction. Proc. Natl. Acad. Sci. USA 2016, 113, E7116–E7125. [Google Scholar] [CrossRef] [PubMed]
- Sonnenburg, W.K.; Rybalkin, S.D.; Bornfeldt, K.E.; Kwak, K.S.; Rybalkina, I.G.; Beavo, J.A. Identification, quantitation, and cellular localization of PDE1 calmodulin-stimulated cyclic nucleotide phosphodiesterases. Methods 1998, 14, 3–19. [Google Scholar] [CrossRef] [PubMed]
- Stephenson, D.T.; Coskran, T.M.; Wilhelms, M.B.; Adamowicz, W.O.; O’Donnell, M.M.; Muravnick, K.B.; Menniti, F.S.; Kleiman, R.J.; Morton, D. Immunohistochemical localization of phosphodiesterase 2A in multiple mammalian species. J. Histochem. Cytochem. 2009, 57, 933–949. [Google Scholar] [CrossRef] [PubMed]
- Richter, W.; Xie, M.; Scheitrum, C.; Krall, J.; Movsesian, M.A.; Conti, M. Conserved expression and functions of PDE4 in rodent and human heart. Basic Res. Cardiol. 2011, 106, 249–262. [Google Scholar] [CrossRef] [PubMed]
- Takimoto, E.; Champion, H.C.; Belardi, D.; Moslehi, J.; Mongillo, M.; Mergia, E.; Montrose, D.C.; Isoda, T.; Aufiero, K.; Zaccolo, M.; et al. cGMP catabolism by phosphodiesterase 5A regulates cardiac adrenergic stimulation by NOS3-dependent mechanism. Circ. Res. 2005, 96, 100–109. [Google Scholar] [CrossRef] [PubMed]
- Patrucco, E.; Albergine, M.S.; Santana, L.F.; Beavo, J.A. Phosphodiesterase 8A (PDE8A) regulates excitation-contraction coupling in ventricular myocytes. J. Mol. Cell. Cardiol. 2010, 49, 330–333. [Google Scholar] [CrossRef] [PubMed]
- Cheung, W.Y. Cyclic 3′,5′-nucleotide phosphodiesterase. Demonstration of an activator. Biochem. Biophys. Res. Commun. 1970, 38, 533–538. [Google Scholar] [CrossRef]
- Bender, A.T.; Beavo, J.A. Cyclic nucleotide phosphodiesterases: Molecular regulation to clinical use. Pharmacol. Rev. 2006, 58, 488–520. [Google Scholar] [CrossRef] [PubMed]
- Yan, C.; Kim, D.; Aizawa, T.; Berk, B.C. Functional interplay between angiotensin II and nitric oxide: Cyclic GMP as a key mediator. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 26–36. [Google Scholar] [CrossRef] [PubMed]
- Sonnenburg, W.K.; Seger, D.; Kwak, K.S.; Huang, J.; Charbonneau, H.; Beavo, J.A. Identification of inhibitory and calmodulin-binding domains of the PDE1A1 and PDE1A2 calmodulin-stimulated cyclic nucleotide phosphodiesterases. J. Biol. Chem. 1995, 270, 30989–31000. [Google Scholar] [CrossRef] [PubMed]
- Chan, S.; Yan, C. PDE1 isozymes, key regulators of pathological vascular remodeling. Curr. Opin. Pharmacol. 2011, 11, 720–724. [Google Scholar] [CrossRef] [PubMed]
- Yan, C.; Zhao, A.Z.; Bentley, J.K.; Beavo, J.A. The calmodulin-dependent phosphodiesterase gene PDE1C encodes several functionally different splice variants in a tissue-specific manner. J. Biol. Chem. 1996, 271, 25699–25706. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Wolda, S.L.; Frazier, A.L.; Florio, V.A.; Martins, T.J.; Snyder, P.B.; Harris, E.A.; McCaw, K.N.; Farrell, C.A.; Steiner, B.; et al. Identification and characterisation of a human calmodulin-stimulated phosphodiesterase PDE1B1. Cell. Signal. 1997, 9, 519–529. [Google Scholar] [CrossRef]
- Vandeput, F.; Wolda, S.L.; Krall, J.; Hambleton, R.; Uher, L.; McCaw, K.N.; Radwanski, P.B.; Florio, V.; Movsesian, M.A. Cyclic Nucleotide Phosphodiesterase PDE1C1 in Human Cardiac Myocytes. J. Biol. Chem. 2007, 282, 32749–32757. [Google Scholar] [CrossRef] [PubMed]
- Lakics, V.; Karran, E.H.; Boess, F.G. Quantitative comparison of phosphodiesterase mRNA distribution in human brain and peripheral tissues. Neuropharmacology 2010, 59, 367–374. [Google Scholar] [CrossRef] [PubMed]
- Yokoyama, U.; Patel, H.H.; Lai, N.C.; Aroonsakool, N.; Roth, D.M.; Insel, P.A. The cyclic AMP effector Epac integrates pro- and anti-fibrotic signals. Proc. Natl. Acad. Sci. USA 2008, 105, 6386–6391. [Google Scholar] [CrossRef] [PubMed]
- Olmedo, I.; Munoz, C.; Guzman, N.; Catalan, M.; Vivar, R.; Ayala, P.; Humeres, C.; Aranguiz, P.; Garcia, L.; Velarde, V.; et al. EPAC expression and function in cardiac fibroblasts and myofibroblasts. Toxicol. Appl. Pharmacol. 2013, 272, 414–422. [Google Scholar] [CrossRef] [PubMed]
- Insel, P.A.; Murray, F.; Yokoyama, U.; Romano, S.; Yun, H.; Brown, L.; Snead, A.; Lu, D.; Aroonsakool, N. cAMP and Epac in the regulation of tissue fibrosis. Br. J. Pharmacol. 2012, 166, 447–456. [Google Scholar] [CrossRef] [PubMed]
- Buxton, I.L.; Duan, D. Cyclic GMP/protein kinase G phosphorylation of Smad3 blocks transforming growth factor-beta-induced nuclear Smad translocation: A key antifibrogenic mechanism of atrial natriuretic peptide. Circ. Res. 2008, 102, 151–153. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Wang, D.; Lucas, J.; Oparil, S.; Xing, D.; Cao, X.; Novak, L.; Renfrow, M.B.; Chen, Y.F. Atrial natriuretic peptide inhibits transforming growth factor beta-induced Smad signaling and myofibroblast transformation in mouse cardiac fibroblasts. Circ. Res. 2008, 102, 185–192. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Yamada, S.; LaRiviere, W.B.; Ye, H.; Bakeberg, J.L.; Irazabal, M.V.; Chebib, F.T.; van Deursen, J.; Harris, P.C.; Sussman, C.R.; et al. Generation and phenotypic characterization of Pde1a mutant mice. PLoS ONE 2017, 12, e0181087. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Morley, M.; Brandimarto, J.; Hannenhalli, S.; Hu, Y.; Ashley, E.A.; Tang, W.H.; Moravec, C.S.; Margulies, K.B.; Cappola, T.P.; et al. RNA-Seq identifies novel myocardial gene expression signatures of heart failure. Genomics 2015, 105, 83–89. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Rybalkin, S.D.; Pi, X.; Wang, Y.; Zhang, C.; Munzel, T.; Beavo, J.A.; Berk, B.C.; Yan, C. Upregulation of phosphodiesterase 1A1 expression is associated with the development of nitrate tolerance. Circulation 2001, 104, 2338–2343. [Google Scholar] [CrossRef] [PubMed]
- Ockaili, R.; Salloum, F.; Hawkins, J.; Kukreja, R.C. Sildenafil (Viagra) induces powerful cardioprotective effect via opening of mitochondrial K(ATP) channels in rabbits. Am. J. Physiol. Heart Circ. Physiol. 2002, 283, H1263–H1269. [Google Scholar] [CrossRef] [PubMed]
- Das, A.; Xi, L.; Kukreja, R.C. Phosphodiesterase-5 inhibitor sildenafil preconditions adult cardiac myocytes against necrosis and apoptosis. Essential role of nitric oxide signaling. J. Biol. Chem. 2005, 280, 12944–12955. [Google Scholar] [CrossRef] [PubMed]
- Fisher, P.W.; Salloum, F.; Das, A.; Hyder, H.; Kukreja, R.C. Phosphodiesterase-5 inhibition with sildenafil attenuates cardiomyocyte apoptosis and left ventricular dysfunction in a chronic model of doxorubicin cardiotoxicity. Circulation 2005, 111, 1601–1610. [Google Scholar] [CrossRef] [PubMed]
- Giannetta, E.; Isidori, A.M.; Galea, N.; Carbone, I.; Mandosi, E.; Vizza, C.D.; Naro, F.; Morano, S.; Fedele, F.; Lenzi, A. Chronic Inhibition of cGMP phosphodiesterase 5A improves diabetic cardiomyopathy: A randomized, controlled clinical trial using magnetic resonance imaging with myocardial tagging. Circulation 2012, 125, 2323–2333. [Google Scholar] [CrossRef] [PubMed]
- Das, A.; Durrant, D.; Salloum, F.N.; Xi, L.; Kukreja, R.C. PDE5 inhibitors as therapeutics for heart disease, diabetes and cancer. Pharmacol. Ther. 2015, 147, 12–21. [Google Scholar] [CrossRef] [PubMed]
- Ding, B.; Abe, J.; Wei, H.; Xu, H.; Che, W.; Aizawa, T.; Liu, W.; Molina, C.A.; Sadoshima, J.; Blaxall, B.C.; et al. A positive feedback loop of phosphodiesterase 3 (PDE3) and inducible cAMP early repressor (ICER) leads to cardiomyocyte apoptosis. Proc. Natl. Acad. Sci. USA 2005, 102, 14771–14776. [Google Scholar] [CrossRef] [PubMed]
- Yan, C.; Miller, C.L.; Abe, J. Regulation of phosphodiesterase 3 and inducible cAMP early repressor in the heart. Circ. Res. 2007, 100, 489–501. [Google Scholar] [CrossRef] [PubMed]
- Oikawa, M.; Wu, M.; Lim, S.; Knight, W.E.; Miller, C.L.; Cai, Y.; Lu, Y.; Blaxall, B.C.; Takeishi, Y.; Abe, J.; et al. Cyclic nucleotide phosphodiesterase 3A1 protects the heart against ischemia-reperfusion injury. J. Mol. Cell. Cardiol. 2013, 64, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Sprenger, J.U.; Bork, N.I.; Herting, J.; Fischer, T.H.; Nikolaev, V.O. Interactions of Calcium Fluctuations during Cardiomyocyte Contraction with Real-Time cAMP Dynamics Detected by FRET. PLoS ONE 2016, 11, e0167974. [Google Scholar] [CrossRef] [PubMed]
- Kraynik, S.M.; Miyaoka, R.S.; Beavo, J.A. PDE3 and PDE4 isozyme-selective inhibitors are both required for synergistic activation of brown adipose tissue. Mol. Pharmacol. 2013, 83, 1155–1165. [Google Scholar] [CrossRef] [PubMed]
- Beltejar, M.G.; Lau, H.T.; Golkowski, M.G.; Ong, S.E.; Beavo, J.A. Analyses of PDE-regulated phosphoproteomes reveal unique and specific cAMP-signaling modules in T cells. Proc. Natl. Acad. Sci. USA 2017, 114, E6240–E6249. [Google Scholar] [CrossRef] [PubMed]
- Laursen, M.; Beck, L.; Kehler, J.; Christoffersen, C.T.; Bundgaard, C.; Mogensen, S.; Mow, T.J.; Pinilla, E.; Knudsen, J.S.; Hedegaard, E.R.; et al. Novel selective PDE type 1 inhibitors cause vasodilatation and lower blood pressure in rats. Br. J. Pharmacol. 2017, 174, 2563–2575. [Google Scholar] [CrossRef] [PubMed]
- Nagel, D.J.; Aizawa, T.; Jeon, K.-I.; Liu, W.; Mohan, A.; Wei, H.; Miano, J.M.; Florio, V.A.; Gao, P.; Korshunov, V.A.; et al. Role of nuclear Ca2+/calmodulin-stimulated phosphodiesterase 1A in vascular smooth muscle cell growth and survival. Circ. Res. 2006, 98, 777–784. [Google Scholar] [CrossRef] [PubMed]
- Rybalkin, S.D.; Bornfeldt, K.E.; Sonnenburg, W.K.; Rybalkina, I.G.; Kwak, K.S.; Hanson, K.; Krebs, E.G.; Beavo, J.A. Calmodulin-stimulated cyclic nucleotide phosphodiesterase (PDE1C) is induced in human arterial smooth muscle cells of the synthetic, proliferative phenotype. J. Clin. Investig. 1997, 100, 2611–2621. [Google Scholar] [CrossRef] [PubMed]
- Cai, Y.; Nagel, D.J.; Zhou, Q.; Cygnar, K.D.; Zhao, H.; Li, F.; Pi, X.; Knight, P.A.; Yan, C. Role of cAMP-phosphodiesterase 1C signaling in regulating growth factor receptor stability, vascular smooth muscle cell growth, migration, and neointimal hyperplasia. Circ. Res. 2015, 116, 1120–1132. [Google Scholar] [CrossRef] [PubMed]
- Bautista Nino, P.K.; Durik, M.; Danser, A.H.; de Vries, R.; Musterd-Bhaggoe, U.M.; Meima, M.E.; Kavousi, M.; Ghanbari, M.; Hoeijmakers, J.H.; O’Donnell, C.J.; et al. Phosphodiesterase 1 regulation is a key mechanism in vascular aging. Clin. Sci. 2015, 129, 1061–1075. [Google Scholar] [CrossRef] [PubMed]
- Yan, C. Cyclic nucleotide phosphodiesterase 1 and vascular aging. Clin. Sci. 2015, 129, 1077–1081. [Google Scholar] [CrossRef] [PubMed]
- Saadane, N.; Alpert, L.; Chalifour, L.E. Expression of immediate early genes, GATA-4, and Nkx-2.5 in adrenergic-induced cardiac hypertrophy and during regression in adult mice. Br. J. Pharmacol. 1999, 127, 1165–1176. [Google Scholar] [CrossRef] [PubMed]
- Jincai, W.; Tingfang, D.; Yongheng, Z.; Zhongmin, L.; Kaihua, Z.; Xiaohong, L. Effects of vinpocetine and ozagrel on behavioral recovery of rats after global brain ischemia. J. Clin. Neurosci. 2014, 21, 661–663. [Google Scholar] [CrossRef] [PubMed]
- Bonoczk, P.; Panczel, G.; Nagy, Z. Vinpocetine increases cerebral blood flow and oxygenation in stroke patients: A near infrared spectroscopy and transcranial Doppler study. Eur. J. Ultrasound 2002, 15, 85–91. [Google Scholar] [CrossRef]
- Vas, A.; Gulyas, B.; Szabo, Z.; Bonoczk, P.; Csiba, L.; Kiss, B.; Karpati, E.; Panczel, G.; Nagy, Z. Clinical and non-clinical investigations using positron emission tomography, near infrared spectroscopy and transcranial Doppler methods on the neuroprotective drug vinpocetine: A summary of evidences. J. Neurol. Sci. 2002, 203–204, 259–262. [Google Scholar] [CrossRef]
- Zhang, Y.S.; Li, J.D.; Yan, C. An update on vinpocetine: New discoveries and clinical implications. Eur. J. Pharmacol. 2018, 819, 30–34. [Google Scholar] [CrossRef] [PubMed]
- Cai, Y.; Knight, W.E.; Guo, S.; Li, J.D.; Knight, P.A.; Yan, C. Vinpocetine suppresses pathological vascular remodeling by inhibiting vascular smooth muscle cell proliferation and migration. J. Pharmacol. Exp. Ther. 2012, 343, 479–488. [Google Scholar] [CrossRef] [PubMed]
- Cai, Y.; Li, J.-D.; Yan, C. Vinpocetine Attenuates Lipid Accumulation and Atherosclerosis Formation. Biochem. Biophys. Res. Commun. 2013, in press. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, J.; Peng, W.; Li, H.; Lu, Y.; Wang, K.; Fan, F.; Li, S.; Xu, Y. Inhibitory effects of vinpocetine on the progression of atherosclerosis are mediated by Akt/NF-κB dependent mechanisms in apoE−/− mice. PLoS ONE 2013, 8, e82509. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.P.; Zhang, Y.S.; Xu, X.; Zhou, Q.; Li, J.D.; Yan, C. Vinpocetine Attenuates Pathological Cardiac Remodeling by Inhibiting Cardiac Hypertrophy and Fibrosis. Cardiovasc. Drugs Ther. 2017, 31, 157–166. [Google Scholar] [CrossRef] [PubMed]
- Jeon, K.I.; Xu, X.; Aizawa, T.; Lim, J.H.; Jono, H.; Kwon, D.S.; Abe, J.; Berk, B.C.; Li, J.D.; Yan, C. Vinpocetine inhibits NF-kappaB-dependent inflammation via an IKK-dependent but PDE-independent mechanism. Proc. Natl. Acad. Sci. USA 2010, 107, 9795–9800. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Miyazawa, K.W.; Pinho-Ribeiro, F.A.; Zarpelon, A.C.; Staurengo-Ferrari, L.; Silva, R.L.; Alves-Filho, J.C.; Cunha, T.M.; Cunha, F.Q.; Casagrande, R.; Verri, W.A., Jr. Vinpocetine reduces lipopolysaccharide-induced inflammatory pain and neutrophil recruitment in mice by targeting oxidative stress, cytokines and NF-κB. Chem. Biol. Interact. 2015, 237, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Evgenov, O.V.; Busch, C.J.; Evgenov, N.V.; Liu, R.; Petersen, B.; Falkowski, G.E.; Petho, B.; Vas, A.M.; Bloch, K.D.; Zapol, W.M.; et al. Inhibition of phosphodiesterase 1 augments the pulmonary vasodilator response to inhaled nitric oxide in awake lambs with acute pulmonary hypertension. Am. J. Physiol. Lung Cell. Mol. Physiol. 2006, 290, L723–L729. [Google Scholar] [CrossRef] [PubMed]
- Lukyanenko, Y.O.; Younes, A.; Lyashkov, A.E.; Tarasov, K.V.; Riordon, D.R.; Lee, J.; Sirenko, S.G.; Kobrinsky, E.; Ziman, B.; Tarasova, Y.S.; et al. Ca2+/calmodulin-activated phosphodiesterase 1A is highly expressed in rabbit cardiac sinoatrial nodal cells and regulates pacemaker function. J. Mol. Cell. Cardiol. 2016, 98, 73–82. [Google Scholar] [CrossRef] [PubMed]
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, S.; Knight, W.E.; Yan, C. Roles of PDE1 in Pathological Cardiac Remodeling and Dysfunction. J. Cardiovasc. Dev. Dis. 2018, 5, 22. https://doi.org/10.3390/jcdd5020022
Chen S, Knight WE, Yan C. Roles of PDE1 in Pathological Cardiac Remodeling and Dysfunction. Journal of Cardiovascular Development and Disease. 2018; 5(2):22. https://doi.org/10.3390/jcdd5020022
Chicago/Turabian StyleChen, Si, Walter E. Knight, and Chen Yan. 2018. "Roles of PDE1 in Pathological Cardiac Remodeling and Dysfunction" Journal of Cardiovascular Development and Disease 5, no. 2: 22. https://doi.org/10.3390/jcdd5020022
APA StyleChen, S., Knight, W. E., & Yan, C. (2018). Roles of PDE1 in Pathological Cardiac Remodeling and Dysfunction. Journal of Cardiovascular Development and Disease, 5(2), 22. https://doi.org/10.3390/jcdd5020022