Polymorphisms/Mutations in A-Kinase Anchoring Proteins (AKAPs): Role in the Cardiovascular System


Abstract
1. Introduction
- (i)
- All AKAPs, though structurally different, have 14–18 α-helix amphipathic amino acid sequence that binds to regulatory subunits of PKA.
- (ii)
- They have a targeting domain that tethers AKAPs to specific subcellular organelles, like mitochondria, the nucleus, and plasma membrane, among others.
- (iii)
- Lastly, all AKAPs contain multiple binding domains by which they bind to other kinases than PKA, phosphatases, phosphodiesterases, and so on.
2. Role of AKAPs in Cardiovascular Physiology
3. Polymorphisms/Mutations in AKAPs and CVDs
3.1. DAKAP2 (AKAP10)
3.2. Yotiao (AKAP9)
3.3. AKAP-Lbc (AKAP13)
3.4. Other AKAPs
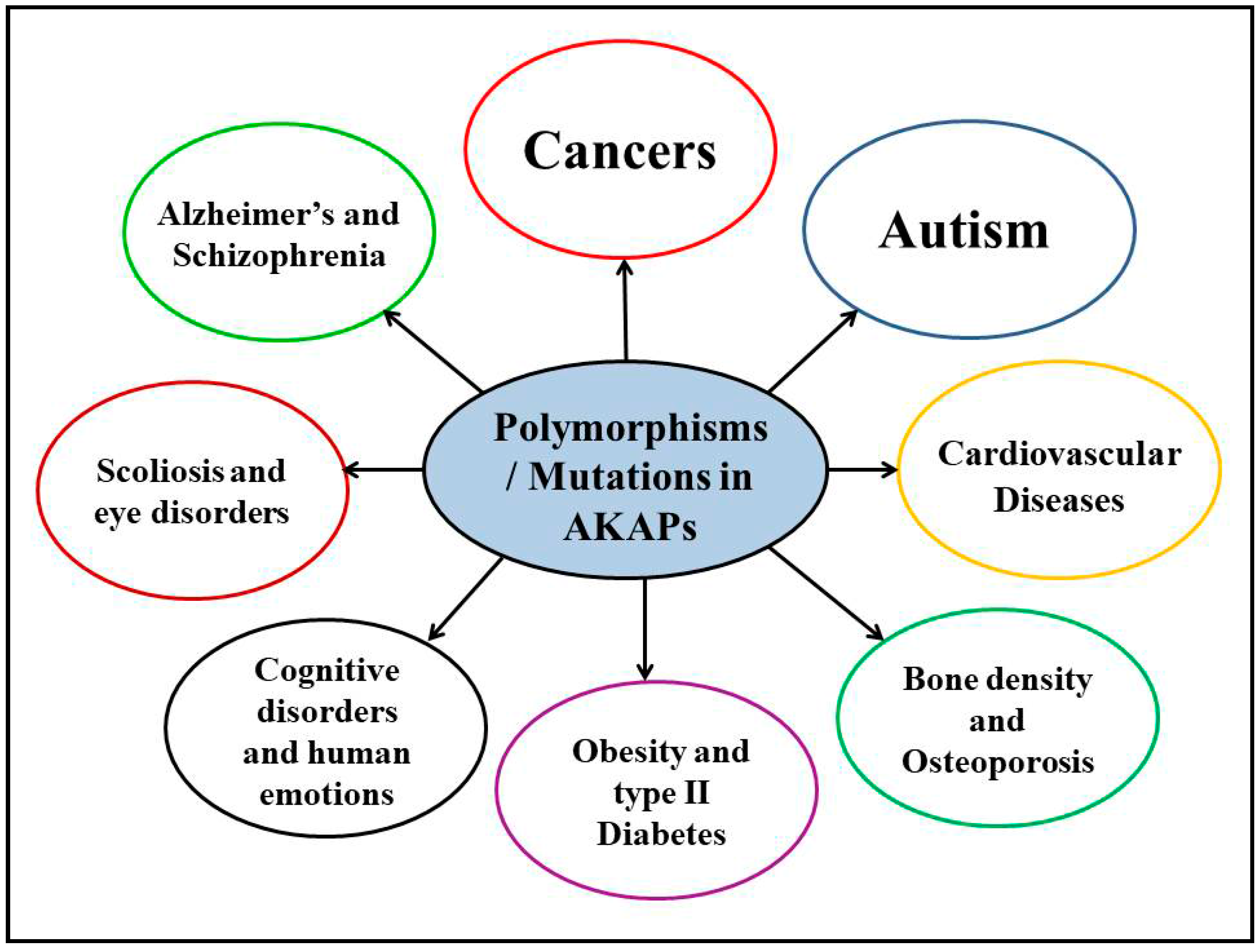
4. Polymorphisms/Mutations in AKAPs and Other Human Diseases
4.1. Neurological Disorders
4.2. Cancers
4.3. Other Human Disorders
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Diviani, D.; Reggi, E.; Arambasic, M.; Caso, S.; Maric, D. Emerging roles of A-kinase anchoring proteins in cardiovascular pathophysiology. Biochim. Biophys. Acta 2016, 1863, 1926–1936. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, I.; Minamino, T. Physiological and pathological cardiac hypertrophy. J. Mol. Cell Cardiol. 2016, 97, 245–262. [Google Scholar] [CrossRef] [PubMed]
- Rababa’h, A.; Singh, S.; Suryavanshi, S.V.; Altarabsheh, S.E.; Deo, S.V.; McConnell, B.K. Compartmentalization role of A-kinase anchoring proteins (AKAPs) in mediating protein kinase A (PKA) signaling and cardiomyocyte hypertrophy. Int. J. Mol. Sci. 2014, 16, 218–229. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Singh, S.; Suryavanshi, S.V.; Ding, W.; Shen, X.; Wijaya, C.S.; Gao, W.D.; McConnell, B.K. Force development and intracellular Ca2+ in intact cardiac muscles from gravin mutant mice. Eur. J. Pharmacol. 2017, 807, 117–126. [Google Scholar] [CrossRef] [PubMed]
- Carnegie, G.K.; Means, C.K.; Scott, J.D. A-kinase anchoring proteins: From protein complexes to physiology and disease. IUBMB Life 2009, 61, 394–406. [Google Scholar] [CrossRef] [PubMed]
- Rababa’h, A.; Craft, J.W., Jr.; Wijaya, C.S.; Atrooz, F.; Fan, Q.; Singh, S.; Guillory, A.N.; Katsonis, P.; Lichtarge, O.; McConnell, B.K. Protein kinase A and phosphodiesterase-4D3 binding to coding polymorphisms of cardiac muscle anchoring protein (mAKAP). J. Mol. Biol. 2013, 425, 3277–3288. [Google Scholar]
- Kapiloff, M.S.; Chandrasekhar, K.D. A-kinase anchoring proteins: Temporal and spatial regulation of intracellular signal transduction in the cardiovascular system. J. Cardiovasc. Pharmacol. 2011, 58, 337–338. [Google Scholar] [CrossRef] [PubMed]
- Wong, W.; Scott, J.D. AKAP signalling complexes: Focal points in space and time. Nat. Rev. Mol. Cell Biol. 2004, 5, 959–970. [Google Scholar] [CrossRef] [PubMed]
- Kritzer, M.D.; Li, J.; Dodge-Kafka, K.; Kapiloff, M.S. AKAPs: The architectural underpinnings of local camp signaling. J. Mol. Cell Cardiol. 2012, 52, 351–358. [Google Scholar] [CrossRef] [PubMed]
- Guillory, A.N.; Yin, X.; Wijaya, C.S.; Diaz Diaz, A.C.; Rababa’h, A.; Singh, S.; Atrooz, F.; Sadayappan, S.; McConnell, B.K. Enhanced cardiac function in gravin mutant mice involves alterations in the beta-adrenergic receptor signaling cascade. PLoS ONE 2013, 8, e74784. [Google Scholar] [CrossRef] [PubMed]
- McConnell, B.; Suryavanshi, S.; Fa’ak, F.; Diaz, A.D.; Singh, S. Disruption of gravin’s scaffolding protects against isoproterenol induced heart failure. FASEB J. 2016, 30, 718. [Google Scholar]
- Kritzer, M.D.; Li, J.; Passariello, C.L.; Gayanilo, M.; Thakur, H.; Dayan, J.; Dodge-Kafka, K.; Kapiloff, M.S. The scaffold protein muscle A-kinase anchoring protein beta orchestrates cardiac myocyte hypertrophic signaling required for the development of heart failure. Circ. Heart Fail. 2014, 7, 663–672. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Li, J.; Drum, B.M.; Chen, Y.; Yin, H.; Guo, X.; Luckey, S.W.; Gilbert, M.L.; McKnight, G.S.; Scott, J.D.; et al. Loss of AKAP150 promotes pathological remodelling and heart failure propensity by disrupting calcium cycling and contractile reserve. Cardiovasc. Res. 2017, 113, 147–159. [Google Scholar] [CrossRef] [PubMed]
- Dema, A.; Perets, E.; Schulz, M.S.; Deak, V.A.; Klussmann, E. Pharmacological targeting of AKAP-directed compartmentalized camp signalling. Cell Signal. 2015, 27, 2474–2487. [Google Scholar] [CrossRef] [PubMed]
- Michel, J.J.; Scott, J.D. AKAP mediated signal transduction. Annu. Rev. Pharmacol. Toxicol. 2002, 42, 235–257. [Google Scholar] [CrossRef] [PubMed]
- Troger, J.; Moutty, M.C.; Skroblin, P.; Klussmann, E. A-kinase anchoring proteins as potential drug targets. Br. J. Pharmacol. 2012, 166, 420–433. [Google Scholar] [CrossRef] [PubMed]
- Cavin, S.; Maric, D.; Diviani, D. A-kinase anchoring protein-Lbc promotes pro-fibrotic signaling in cardiac fibroblasts. Biochim. Biophys. Acta 2014, 1843, 335–345. [Google Scholar] [CrossRef] [PubMed]
- Del Vescovo, C.D.; Cotecchia, S.; Diviani, D. A-kinase-anchoring protein-Lbc anchors ikappab kinase beta to support interleukin-6-mediated cardiomyocyte hypertrophy. Mol. Cell Biol. 2013, 33, 14–27. [Google Scholar] [CrossRef] [PubMed]
- Mayers, C.M.; Wadell, J.; McLean, K.; Venere, M.; Malik, M.; Shibata, T.; Driggers, P.H.; Kino, T.; Guo, X.C.; Koide, H.; et al. The rho guanine nucleotide exchange factor AKAP13 (brx) is essential for cardiac development in mice. J. Biol. Chem. 2010, 285, 12344–12354. [Google Scholar] [CrossRef] [PubMed]
- Schiattarella, G.G.; Cattaneo, F.; Pironti, G.; Magliulo, F.; Carotenuto, G.; Pirozzi, M.; Polishchuk, R.; Borzacchiello, D.; Paolillo, R.; Oliveti, M.; et al. AKAP1 deficiency promotes mitochondrial aberrations and exacerbates cardiac injury following permanent coronary ligation via enhanced mitophagy and apoptosis. PLoS ONE 2016, 11, e0154076. [Google Scholar] [CrossRef] [PubMed]
- Passariello, C.L.; Li, J.; Dodge-Kafka, K.; Kapiloff, M.S. mAKAP—A master scaffold for cardiac remodeling. J. Cardiovasc. Pharmacol. 2015, 65, 218–225. [Google Scholar] [CrossRef] [PubMed]
- Redden, J.M.; Dodge-Kafka, K.L. AKAP phosphatase complexes in the heart. J. Cardiovasc. Pharmacol. 2011, 58, 354–362. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Redden, J.M.; Kapiloff, M.S.; Dodge-Kafka, K.L. The large isoforms of A-kinase anchoring protein 18 mediate the phosphorylation of inhibitor-1 by protein kinase A and the inhibition of protein phosphatase 1 activity. Mol. Pharmacol. 2011, 79, 533–540. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Chen, L.; Kass, R.S.; Dessauer, C.W. The A-kinase anchoring protein Yotiao facilitates complex formation between adenylyl cyclase type 9 and the IKs potassium channel in heart. J. Biol. Chem. 2012, 287, 29815–29824. [Google Scholar] [CrossRef] [PubMed]
- Tingley, W.G.; Pawlikowska, L.; Zaroff, J.G.; Kim, T.; Nguyen, T.; Young, S.G.; Vranizan, K.; Kwok, P.Y.; Whooley, M.A.; Conklin, B.R. Gene-trapped mouse embryonic stem cell-derived cardiac myocytes and human genetics implicate AKAP10 in heart rhythm regulation. Proc. Natl. Acad. Sci. USA 2007, 104, 8461–8466. [Google Scholar] [CrossRef] [PubMed]
- Chung, J.; Wittig, J.G.; Ghamari, A.; Maeda, M.; Dailey, T.A.; Bergonia, H.; Kafina, M.D.; Coughlin, E.E.; Minogue, C.E.; Hebert, A.S.; et al. Erythropoietin signaling regulates heme biosynthesis. Elife 2017, 6. [Google Scholar] [CrossRef] [PubMed]
- Perrino, C.; Schroder, J.N.; Lima, B.; Villamizar, N.; Nienaber, J.J.; Milano, C.A.; Naga Prasad, S.V. Dynamic regulation of phosphoinositide 3-kinase-γ activity and β-adrenergic receptor trafficking in end-stage human heart failure. Circulation 2007, 116, 2571–2579. [Google Scholar] [CrossRef] [PubMed]
- Perino, A.; Ghigo, A.; Ferrero, E.; Morello, F.; Santulli, G.; Baillie, G.S.; Damilano, F.; Dunlop, A.J.; Pawson, C.; Walser, R.; et al. Integrating cardiac PIP3 and cAMP signaling through a PKA anchoring function of p110γ. Mol. Cell 2011, 42, 84–95. [Google Scholar] [CrossRef] [PubMed]
- Kammerer, S.; Burns-Hamuro, L.L.; Ma, Y.; Hamon, S.C.; Canaves, J.M.; Shi, M.M.; Nelson, M.R.; Sing, C.F.; Cantor, C.R.; Taylor, S.S.; et al. Amino acid variant in the kinase binding domain of dual-specific a kinase-anchoring protein 2: A disease susceptibility polymorphism. Proc. Natl. Acad. Sci. USA 2003, 100, 4066–4071. [Google Scholar] [CrossRef] [PubMed]
- Neumann, S.A.; Tingley, W.G.; Conklin, B.R.; Shrader, C.J.; Peet, E.; Muldoon, M.F.; Jennings, J.R.; Ferrell, R.E.; Manuck, S.B. AKAP10 (i646v) functional polymorphism predicts heart rate and heart rate variability in apparently healthy, middle-aged European-Americans. Psychophysiology 2009, 46, 466–472. [Google Scholar] [CrossRef] [PubMed]
- Nishihama, K.; Yamada, Y.; Matsuo, H.; Segawa, T.; Watanabe, S.; Kato, K.; Yajima, K.; Hibino, T.; Yokoi, K.; Ichihara, S.; et al. Association of gene polymorphisms with myocardial infarction in individuals with or without conventional coronary risk factors. Int. J. Mol. Med. 2007, 19, 129–141. [Google Scholar] [CrossRef] [PubMed]
- Loniewska, B.; Kaczmarczyk, M.; Clark, J.S.; Goracy, I.; Horodnicka-Jozwa, A.; Ciechanowicz, A. Association of functional genetic variants of A-kinase anchoring protein 10 with QT interval length in full-term polish newborns. Arch. Med. Sci. 2015, 11, 149–154. [Google Scholar] [CrossRef] [PubMed]
- Loniewska, B.; Kaczmarczyk, M.; Clark, J.S.; Binczak-Kuleta, A.; Adler, G.; Kordek, A.; Horodnicka-Jozwa, A.; Dawid, G.; Rudnicki, J.; Ciechanowicz, A. Association of 1936a > g in AKAP10 (A-kinase anchoring protein 10) and blood pressure in polish full-term newborns. Blood Press 2013, 22, 51–56. [Google Scholar] [CrossRef] [PubMed]
- Loniewska, B.; Kaczmarczyk, M.; Clark, J.S.; Kordek, A.; Ciechanowicz, A. Polymorphism 1936a > g in the AKAP10 gene (encoding A-kinase-anchoring protein 10) is associated with higher cholesterol cord blood concentration in polish full-term newsborns. J. Perinat. Med. 2013, 41, 205–210. [Google Scholar] [CrossRef] [PubMed]
- Fu, F.; Deng, Q.; Lei, T.Y.; Li, R.; Jing, X.Y.; Yang, X.; Liao, C. Clinical application of SNP array analysis in fetuses with ventricular septal defects and normal karyotypes. Arch. Gynecol. Obstet. 2017, 296, 929–940. [Google Scholar] [CrossRef] [PubMed]
- Wilke, R.A.; Lin, D.W.; Roden, D.M.; Watkins, P.B.; Flockhart, D.; Zineh, I.; Giacomini, K.M.; Krauss, R.M. Identifying genetic risk factors for serious adverse drug reactions: Current progress and challenges. Nat. Rev. Drug Discov. 2007, 6, 904–916. [Google Scholar] [CrossRef] [PubMed]
- Ramirez, A.H.; Shaffer, C.M.; Delaney, J.T.; Sexton, D.P.; Levy, S.E.; Rieder, M.J.; Nickerson, D.A.; George, A.L., Jr.; Roden, D.M. Novel rare variants in congenital cardiac arrhythmia genes are frequent in drug-induced torsades de pointes. Pharmacogenomics J. 2013, 13, 325–329. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, P.J.; Crotti, L.; Insolia, R. Long-QT syndrome: From genetics to management. Circ. Arrhythm. Electrophysiol. 2012, 5, 868–877. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Marquardt, M.L.; Tester, D.J.; Sampson, K.J.; Ackerman, M.J.; Kass, R.S. Mutation of an A-kinase-anchoring protein causes long-QT syndrome. Proc. Natl. Acad. Sci. USA 2007, 104, 20990–20995. [Google Scholar] [CrossRef] [PubMed]
- Moss, A.J.; Kass, R.S. Long QT syndrome: From channels to cardiac arrhythmias. J. Clin. Investig. 2005, 115, 2018–2024. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Sampson, K.J.; Kass, R.S. Cardiac delayed rectifier potassium channels in health and disease. Card. Electrophysiol. Clin. 2016, 8, 307–322. [Google Scholar] [CrossRef] [PubMed]
- De Villiers, C.P.; van der Merwe, L.; Crotti, L.; Goosen, A.; George, A.L., Jr.; Schwartz, P.J.; Brink, P.A.; Moolman-Smook, J.C.; Corfield, V.A. AKAP9 is a genetic modifier of congenital long-QT syndrome type 1. Circ. Cardiovasc. Genet. 2014, 7, 599–606. [Google Scholar] [CrossRef] [PubMed]
- Dong, C.; Tang, L.; Liu, Z.; Bu, S.; Liu, Q.; Wang, Q.; Mai, Y.; Wang, D.W.; Duan, S. Landscape of the relationship between type 2 diabetes and coronary heart disease through an integrated gene network analysis. Gene 2014, 539, 30–36. [Google Scholar] [CrossRef] [PubMed]
- Hong, K.W.; Lim, J.E.; Oh, B. A regulatory SNP in AKAP13 is associated with blood pressure in Koreans. J. Hum. Genet. 2011, 56, 205–210. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, T.; Kato, K.; Yokoi, K.; Oguri, M.; Watanabe, S.; Metoki, N.; Yoshida, H.; Satoh, K.; Aoyagi, Y.; Nozawa, Y.; et al. Association of gene polymorphisms with chronic kidney disease in Japanese individuals. Int. J. Mol. Med. 2009, 24, 539–547. [Google Scholar] [PubMed]
- Horikoshi, M.; Mgi, R.; van de Bunt, M.; Surakka, I.; Sarin, A.P.; Mahajan, A.; Marullo, L.; Thorleifsson, G.; Hgg, S.; Hottenga, J.J.; et al. Discovery and fine-mapping of glycaemic and obesity-related trait loci using high-density imputation. PLoS Genet. 2015, 11, e1005230. [Google Scholar] [CrossRef] [PubMed]
- Suryavanshi, S.; Jadhav, S.; Anderson, K.; Katsonis, P.; Lichtarge, O.; McConnell, B.K. Abstract 24010: Muscle-specific A-kinase anchoring protein polymorphisms pre-dispose humans to cardiovascular diseases by affecting cyclic AMP/PKA signaling. Circulation 2017, 136, A24010. [Google Scholar]
- Li, W.; Li, Y.; Zhang, L.; Guo, H.; Tian, D.; Li, Y.; Peng, Y.; Zheng, Y.; Dai, Y.; Xia, K.; et al. AKAP2 identified as a novel gene mutated in a Chinese family with adolescent idiopathic scoliosis. J. Med. Genet. 2016, 53, 488–493. [Google Scholar] [CrossRef] [PubMed]
- Panza, E.; Gimelli, G.; Passalacqua, M.; Cohen, A.; Gimelli, S.; Giglio, S.; Ghezzi, C.; Sparatore, B.; Heye, B.; Zuffardi, O.; et al. The breakpoint identified in a balanced de novo translocation t(7;9)(p14.1;q31.3) disrupts the A-kinase (PRKA) anchor protein 2 gene (AKAP2) on chromosome 9 in a patient with kallmann syndrome and bone anomalies. Int. J. Mol. Med. 2007, 19, 429–435. [Google Scholar] [CrossRef] [PubMed]
- Suarez-Rama, J.J.; Arrojo, M.; Sobrino, B.; Amigo, J.; Brenlla, J.; Agra, S.; Paz, E.; Brion, M.; Carracedo, A.; Paramo, M.; et al. Resequencing and association analysis of coding regions at twenty candidate genes suggest a role for rare risk variation at AKAP9 and protective variation at nrxn1 in schizophrenia susceptibility. J. Psychiatr. Res. 2015, 66–67, 38–44. [Google Scholar] [CrossRef] [PubMed]
- Logue, M.W.; Schu, M.; Vardarajan, B.N.; Farrell, J.; Bennett, D.A.; Buxbaum, J.D.; Byrd, G.S.; Ertekin-Taner, N.; Evans, D.; Foroud, T.; et al. Two rare AKAP9 variants are associated with Alzheimer’s disease in African Americans. Alzheimer’s Dement. 2014, 10, 609–618. [Google Scholar] [CrossRef] [PubMed]
- Matsunami, N.; Hensel, C.H.; Baird, L.; Stevens, J.; Otterud, B.; Leppert, T.; Varvil, T.; Hadley, D.; Glessner, J.T.; Pellegrino, R.; et al. Identification of rare DNA sequence variants in high-risk autism families and their prevalence in a large case/control population. Mol. Autism 2014, 5, 5. [Google Scholar] [CrossRef] [PubMed]
- Poelmans, G.; Franke, B.; Pauls, D.L.; Glennon, J.C.; Buitelaar, J.K. AKAPs integrate genetic findings for autism spectrum disorders. Transl. Psychiatry 2013, 3, e270. [Google Scholar] [CrossRef] [PubMed]
- Wilson, G.M.; Flibotte, S.; Chopra, V.; Melnyk, B.L.; Honer, W.G.; Holt, R.A. DNA copy-number analysis in bipolar disorder and schizophrenia reveals aberrations in genes involved in glutamate signaling. Hum. Mol. Genet. 2006, 15, 743–749. [Google Scholar] [CrossRef] [PubMed]
- Richter, S.; Gorny, X.; Machts, J.; Behnisch, G.; Wustenberg, T.; Herbort, M.C.; Munte, T.F.; Seidenbecher, C.I.; Schott, B.H. Effects of AKAP5 pro100leu genotype on working memory for emotional stimuli. PLoS ONE 2013, 8, e55613. [Google Scholar] [CrossRef] [PubMed]
- Richter, S.; Gorny, X.; Marco-Pallares, J.; Kramer, U.M.; Machts, J.; Barman, A.; Bernstein, H.G.; Schule, R.; Schols, L.; Rodriguez-Fornells, A.; et al. A potential role for a genetic variation of AKAP5 in human aggression and anger control. Front. Hum. Neurosci. 2011, 5, 175. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Zhang, H.; Bloss, C.S.; Duvvuri, V.; Kaye, W.; Schork, N.J.; Berrettini, W.; Hakonarson, H.; Price Foundation Collaborative Group. A genome-wide association study on common SNPs and rare CNVs in anorexia nervosa. Mol. Psychiatry 2011, 16, 949–959. [Google Scholar] [CrossRef] [PubMed]
- Seshadri, S.; Fitzpatrick, A.L.; Ikram, M.A.; DeStefano, A.L.; Gudnason, V.; Boada, M.; Bis, J.C.; Smith, A.V.; Carassquillo, M.M.; Lambert, J.C.; et al. Genome-wide analysis of genetic loci associated with alzheimer disease. JAMA 2010, 303, 1832–1840. [Google Scholar] [CrossRef] [PubMed]
- Davies, G.; Armstrong, N.; Bis, J.C.; Bressler, J.; Chouraki, V.; Giddaluru, S.; Hofer, E.; Ibrahim-Verbaas, C.A.; Kirin, M.; Lahti, J.; et al. Genetic contributions to variation in general cognitive function: A meta-analysis of genome-wide association studies in the charge consortium (N = 53,949). Mol. Psychiatry 2015, 20, 183–192. [Google Scholar] [CrossRef] [PubMed]
- Andrews, S.J.; Das, D.; Anstey, K.J.; Easteal, S. Association of AKAP6 and mir2113 with cognitive performance in a population-based sample of older adults. Genes Brain Behav. 2017, 16, 472–478. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Huang, J.Y.; Chen, Y.N.; Yuan, F.; Zhang, H.; Yan, F.H.; Wang, M.J.; Wang, G.; Su, M.; Lu, G.; et al. Whole genome and transcriptome sequencing of matched primary and peritoneal metastatic gastric carcinoma. Sci. Rep. 2015, 5, 13750. [Google Scholar] [CrossRef] [PubMed]
- Jo, Y.S.; Kim, M.S.; Yoo, N.J.; Lee, S.H. Frameshift mutations of AKAP9 gene in gastric and colorectal cancers with high microsatellite instability. Pathol. Oncol. Res. 2016, 22, 587–592. [Google Scholar] [CrossRef] [PubMed]
- Frank, B.; Wiestler, M.; Kropp, S.; Hemminki, K.; Spurdle, A.B.; Sutter, C.; Wappenschmidt, B.; Chen, X.; Beesley, J.; Hopper, J.L.; et al. Association of a common AKAP9 variant with breast cancer risk: A collaborative analysis. J. Natl. Cancer Inst. 2008, 100, 437–442. [Google Scholar] [CrossRef] [PubMed]
- Milne, R.L.; Burwinkel, B.; Michailidou, K.; Arias-Perez, J.I.; Zamora, M.P.; Menendez-Rodriguez, P.; Hardisson, D.; Mendiola, M.; Gonzalez-Neira, A.; Pita, G.; et al. Common non-synonymous SNPs associated with breast cancer susceptibility: Findings from the breast cancer association consortium. Hum. Mol. Genet. 2014, 23, 6096–6111. [Google Scholar] [CrossRef] [PubMed]
- Rudd, M.F.; Webb, E.L.; Matakidou, A.; Sellick, G.S.; Williams, R.D.; Bridle, H.; Eisen, T.; Houlston, R.S.; Consortium, G. Variants in the GH-IGF axis confer susceptibility to lung cancer. Genome Res. 2006, 16, 693–701. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Zhang, D.; Wang, R.; Rui, Y.; Zhou, J.; Wang, R.; Zhou, B.; Huang, X.; Yang, L.; Li, Y.; et al. A-kinase anchoring proteins 10 expression in relation to 2073a/g polymorphism and tumor progression in patients with colorectal cancer. Pathol. Oncol. Res. 2013, 19, 521–527. [Google Scholar] [CrossRef] [PubMed]
- Wirtenberger, M.; Schmutzhard, J.; Hemminki, K.; Meindl, A.; Sutter, C.; Schmutzler, R.K.; Wappenschmidt, B.; Kiechle, M.; Arnold, N.; Weber, B.H.; et al. The functional genetic variant ile646val located in the kinase binding domain of the A-kinase anchoring protein 10 is associated with familial breast cancer. Carcinogenesis 2007, 28, 423–426. [Google Scholar] [CrossRef] [PubMed]
- Wirtenberger, M.; Tchatchou, S.; Hemminki, K.; Klaes, R.; Schmutzler, R.K.; Bermejo, J.L.; Chen, B.; Wappenschmidt, B.; Meindl, A.; Bartram, C.R.; et al. Association of genetic variants in the rho guanine nucleotide exchange factor AKAP13 with familial breast cancer. Carcinogenesis 2006, 27, 593–598. [Google Scholar] [CrossRef] [PubMed]
- Kresse, S.H.; Rydbeck, H.; Skarn, M.; Namlos, H.M.; Barragan-Polania, A.H.; Cleton-Jansen, A.M.; Serra, M.; Liestol, K.; Hogendoorn, P.C.; Hovig, E.; et al. Integrative analysis reveals relationships of genetic and epigenetic alterations in osteosarcoma. PLoS ONE 2012, 7, e48262. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Ye, C.; Guo, X.; Wen, W.; Long, J.; Gao, Y.T.; Shu, X.O.; Zheng, W.; Cai, Q. Evaluation of potential regulatory function of breast cancer risk locus at 6q25.1. Carcinogenesis 2016, 37, 163–168. [Google Scholar] [CrossRef] [PubMed]
- Visser, L.; Westerveld, G.H.; Xie, F.; van Daalen, S.K.; van der Veen, F.; Lombardi, M.P.; Repping, S. A comprehensive gene mutation screen in men with asthenozoospermia. Fertil. Steril. 2011, 95, 1020–1024.e9. [Google Scholar] [CrossRef] [PubMed]
- Baccetti, B.; Collodel, G.; Estenoz, M.; Manca, D.; Moretti, E.; Piomboni, P. Gene deletions in an infertile man with sperm fibrous sheath dysplasia. Hum. Reprod. 2005, 20, 2790–2794. [Google Scholar] [CrossRef] [PubMed]
- Lin, Q.; Zhou, N.; Zhang, N.; Zhu, B.; Hu, S.; Zhou, Z.; Qi, Y. Genetic variations and polymorphisms in the ezrin gene are associated with age-related cataract. Mol. Vis. 2013, 19, 1572–1579. [Google Scholar] [PubMed]
- Zhang, L.; Choi, H.J.; Estrada, K.; Leo, P.J.; Li, J.; Pei, Y.F.; Zhang, Y.; Lin, Y.; Shen, H.; Liu, Y.Z.; et al. Multistage genome-wide association meta-analyses identified two new loci for bone mineral density. Hum. Mol. Genet. 2014, 23, 1923–1933. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| AKAPs | SNPs/Mutations and Heart Disease | Reference |
|---|---|---|
| AKAP6 | SNP Val839Ala; cardiac arrhythmia | [37] |
| SNP rs12885467; higher BMI | [46] | |
| AKAP7 | SNP Gln112Arg; cardiac arrhythmia | [37] |
| AKAP9 | SNP Gln3531Glu; cardiac arrhythmia | [37] |
| Mutation Ser1570Leu; long-QT syndrome | [39] | |
| Four SNPs; long-QT Syndrome Type 1 | [42] | |
| AKAP10 | SNP Ile646Val; decrease in PR interval | [29] |
| Mutations; cardiac arrhythmia | [25] | |
| SNP Ile646Val; myocardial infarction | [31] | |
| SNP Ile646Val; blood pressure | [33] | |
| SNP Ile646Val; hypercholesterolemia | [34] | |
| SNP Ile646Val; heart rate variability | [30] | |
| SNP Ile646Val; long QTc interval length | [32] | |
| Copy number variations; ventricular septal defects | [35] | |
| AKAP12 | SNP with multiple alleles; chronic kidney disease | [45] |
| AKAP13 | Genetic locus; coronary artery disease | [43] |
| SNP; high blood pressure | [44] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Suryavanshi, S.V.; Jadhav, S.M.; McConnell, B.K. Polymorphisms/Mutations in A-Kinase Anchoring Proteins (AKAPs): Role in the Cardiovascular System. J. Cardiovasc. Dev. Dis. 2018, 5, 7. https://doi.org/10.3390/jcdd5010007
Suryavanshi SV, Jadhav SM, McConnell BK. Polymorphisms/Mutations in A-Kinase Anchoring Proteins (AKAPs): Role in the Cardiovascular System. Journal of Cardiovascular Development and Disease. 2018; 5(1):7. https://doi.org/10.3390/jcdd5010007
Chicago/Turabian StyleSuryavanshi, Santosh V., Shweta M. Jadhav, and Bradley K. McConnell. 2018. "Polymorphisms/Mutations in A-Kinase Anchoring Proteins (AKAPs): Role in the Cardiovascular System" Journal of Cardiovascular Development and Disease 5, no. 1: 7. https://doi.org/10.3390/jcdd5010007
APA StyleSuryavanshi, S. V., Jadhav, S. M., & McConnell, B. K. (2018). Polymorphisms/Mutations in A-Kinase Anchoring Proteins (AKAPs): Role in the Cardiovascular System. Journal of Cardiovascular Development and Disease, 5(1), 7. https://doi.org/10.3390/jcdd5010007