The Popeye Domain Containing Genes and Their Function as cAMP Effector Proteins in Striated Muscle


Abstract
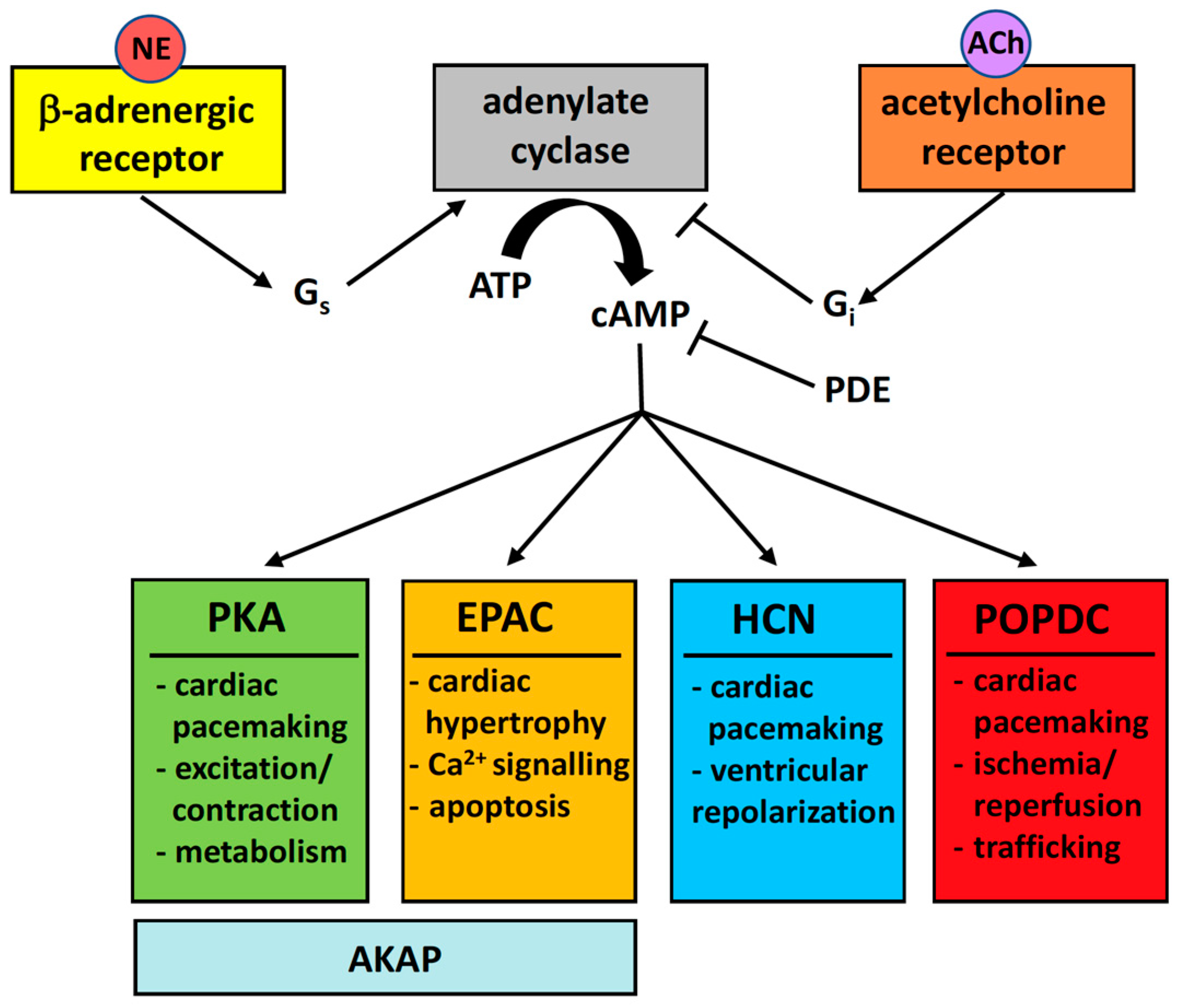
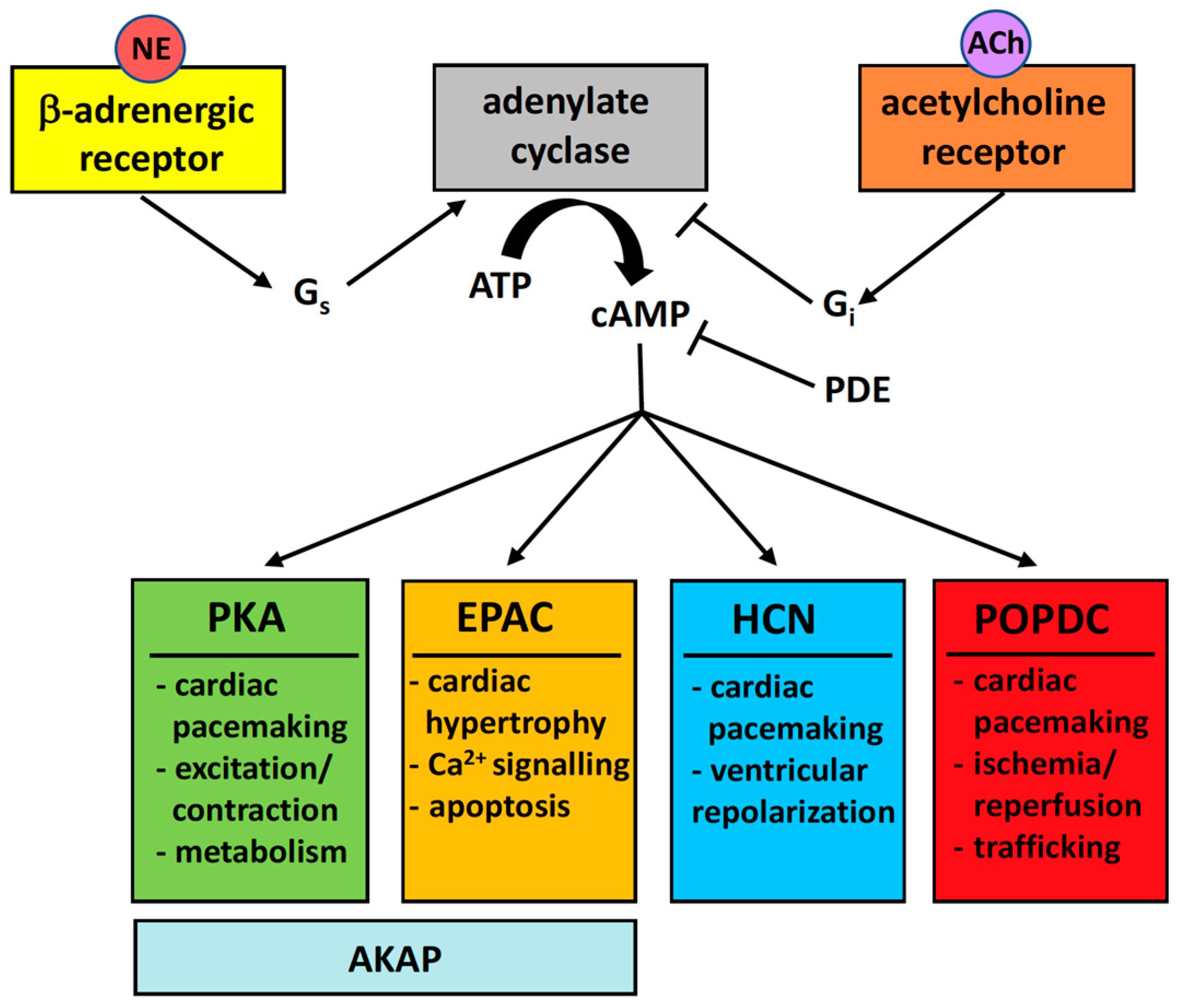
:1. Introduction
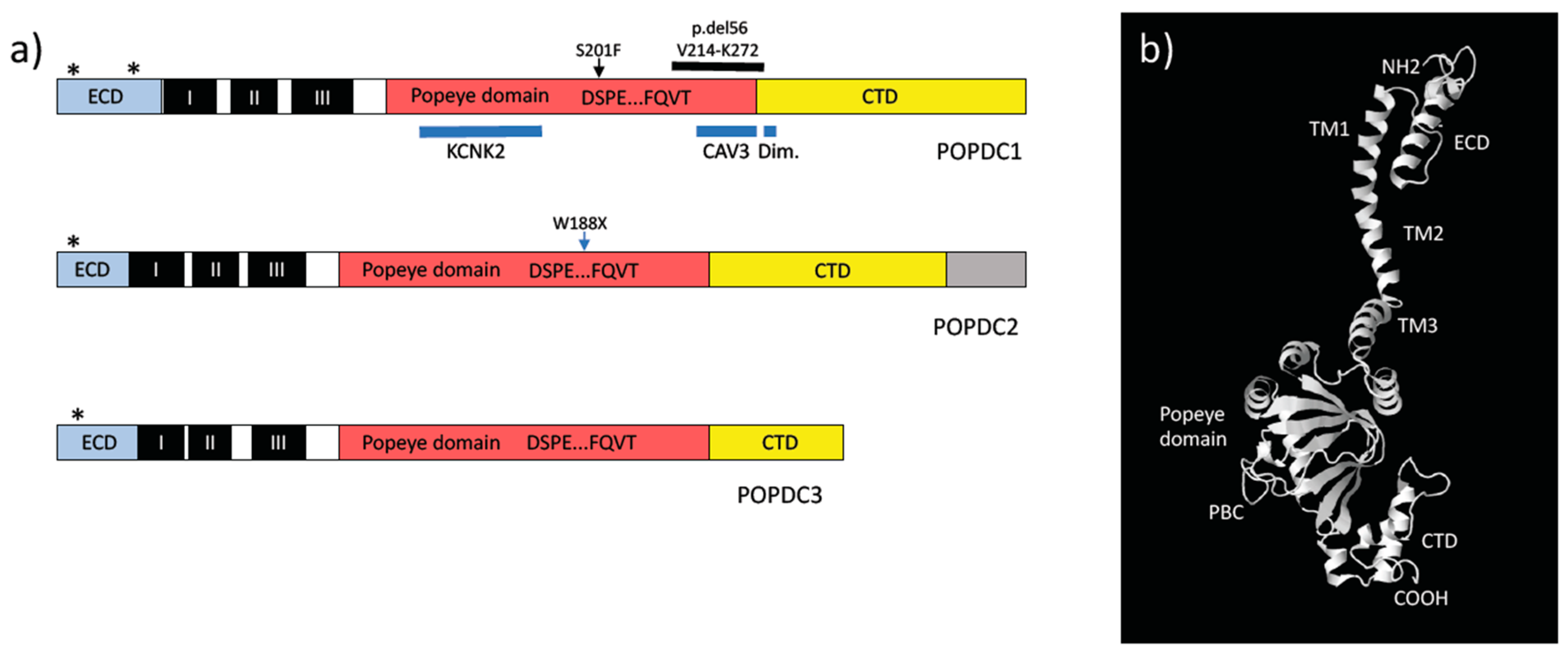
2. The Popeye Gene Family
3. POPDC Proteins
4. The Popeye Domain Is a Novel cAMP-Binding Domain
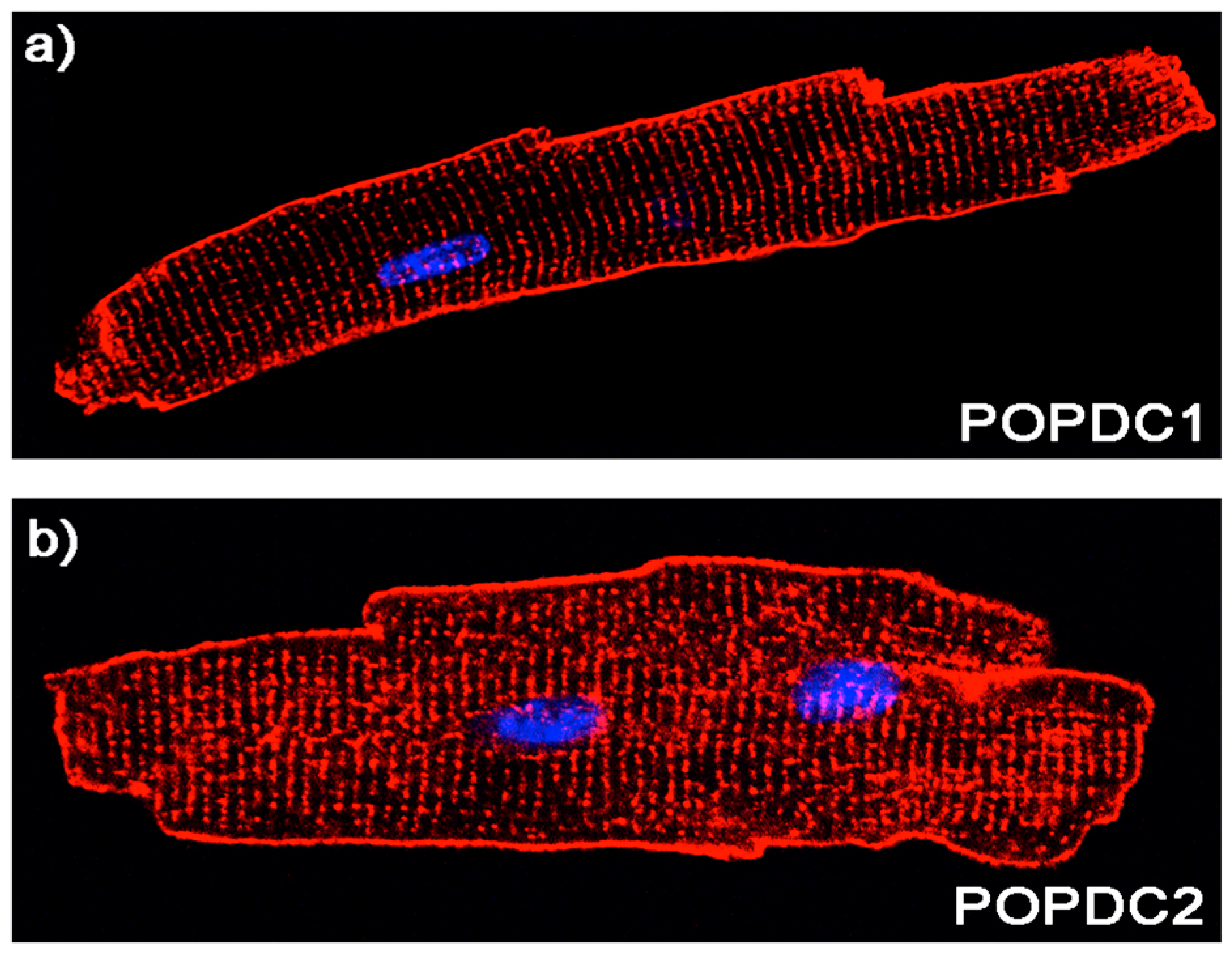
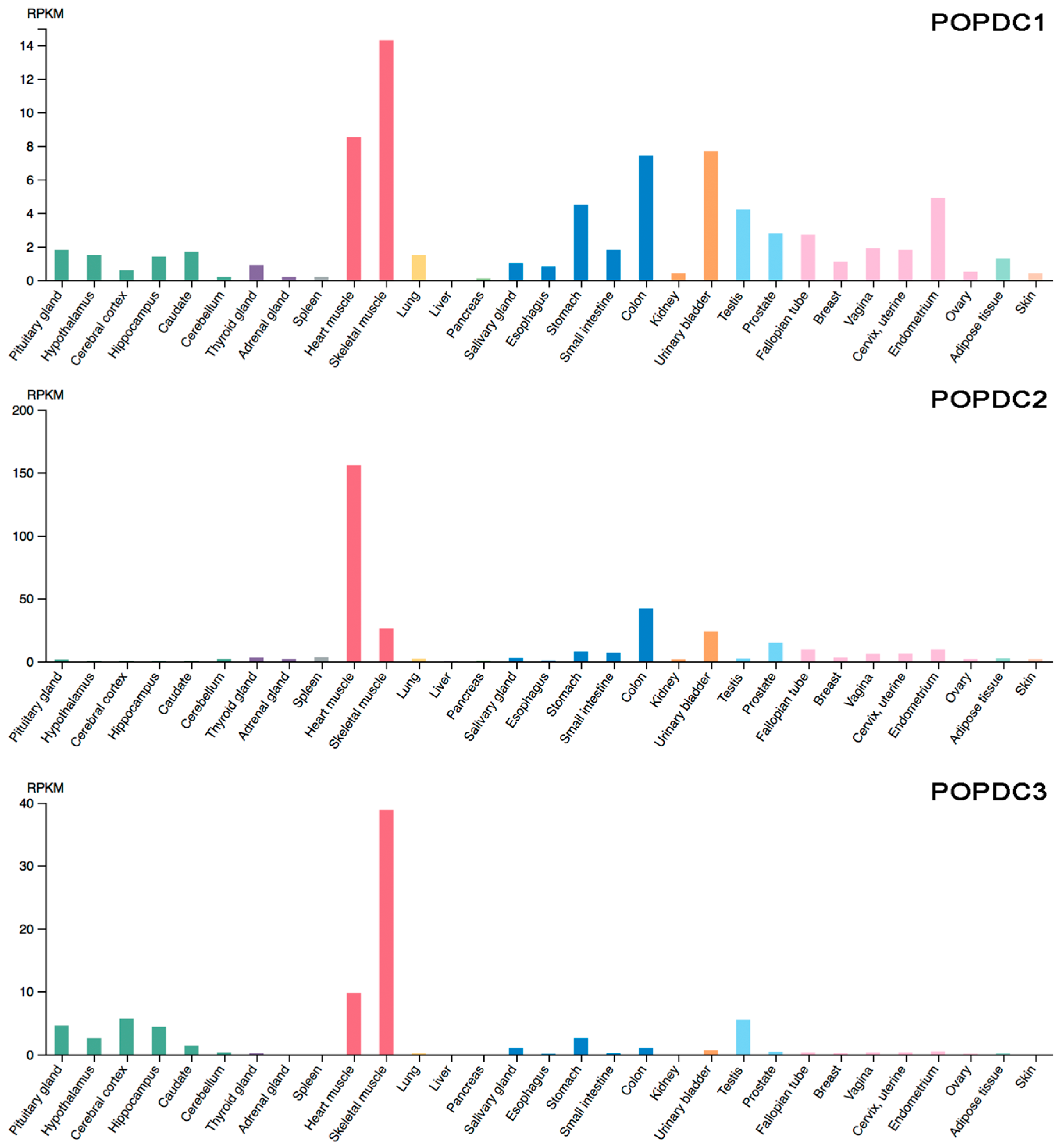
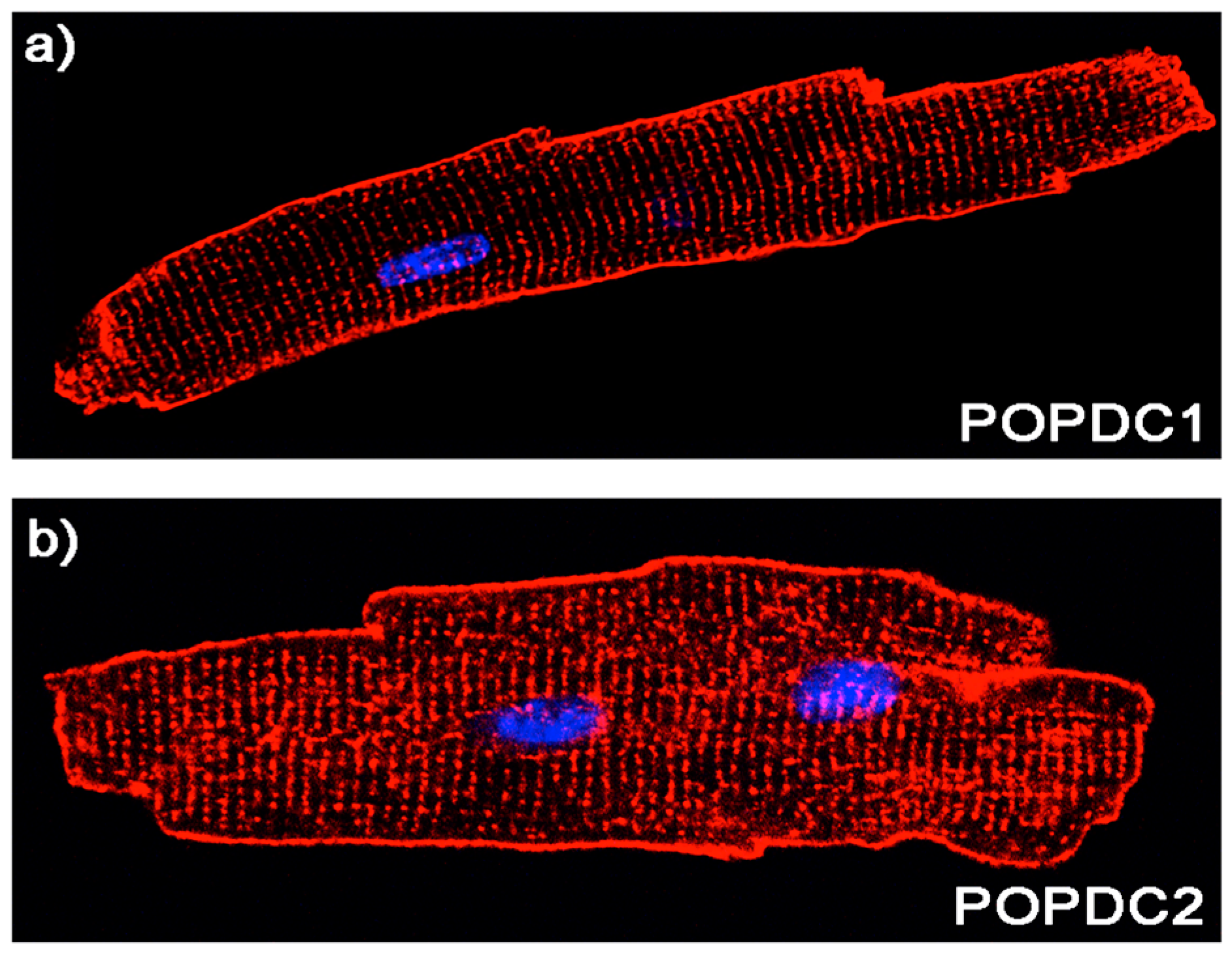
5. Expression Pattern of POPDC Genes
6. Animal Models to Define the Function POPDC Genes
7. Evidence for a Role of POPDC Genes in Human Heart and Skeletal Muscle
8. Conclusions
Acknowledgments
Conflicts of Interest
References
- Lundby, A.; Andersen, M.N.; Steffensen, A.B.; Horn, H.; Kelstrup, C.D.; Francavilla, C.; Jensen, L.J.; Schmitt, N.; Thomsen, M.B.; Olsen, J.V. In vivo phosphoproteomics analysis reveals the cardiac targets of beta-adrenergic receptor signaling. Sci. Signal. 2013, 6. [Google Scholar] [CrossRef] [PubMed]
- Baldwin, T.A.; Dessauer, C.W. Function of adenylyl cyclase in heart: The AKAP connection. J. Cardiovasc. Dev. Dis. 2018, 5. [Google Scholar] [CrossRef] [PubMed]
- Ercu, M.; Klussmann, E. Roles of A-kinase anchoring proteins and phosphodiesterases in the cardiovascular system. J. Cardiovasc. Dev. Dis. 2018, 5. [Google Scholar] [CrossRef] [PubMed]
- Smith, F.D.; Esseltine, J.L.; Nygren, P.J.; Veesler, D.; Byrne, D.P.; Vonderach, M.; Strashnov, I.; Eyers, C.E.; Eyers, P.A.; Langeberg, L.K.; et al. Local protein kinase A action proceeds through intact holoenzymes. Science 2017, 356, 1288–1293. [Google Scholar] [CrossRef] [PubMed]
- Lezoualc’h, F.; Fazal, L.; Laudette, M.; Conte, C. Cyclic AMP sensor Epac proteins and their role in cardiovascular function and disease. Circ. Res. 2016, 118, 881–897. [Google Scholar] [CrossRef] [PubMed]
- Morel, E.; Marcantoni, A.; Gastineau, M.; Birkedal, R.; Rochais, F.; Garnier, A.; Lompre, A.M.; Vandecasteele, G.; Lezoualc’h, F. cAMP-binding protein Epac induces cardiomyocyte hypertrophy. Circ. Res. 2005, 97, 1296–1304. [Google Scholar] [CrossRef] [PubMed]
- Pereira, L.; Rehmann, H.; Lao, D.H.; Erickson, J.R.; Bossuyt, J.; Chen, J.; Bers, D.M. Novel Epac fluorescent ligand reveals distinct Epac1 vs. Epac2 distribution and function in cardiomyocytes. Proc. Natl. Acad. Sci. USA 2015, 112, 3991–3996. [Google Scholar] [CrossRef] [PubMed]
- Parnell, E.; Smith, B.O.; Yarwood, S.J. The cAMP sensors, Epac1 and Epac2, display distinct subcellular distributions despite sharing a common nuclear pore localisation signal. Cell. Signal. 2015, 27, 989–996. [Google Scholar] [CrossRef] [PubMed]
- Fazal, L.; Laudette, M.; Paula-Gomes, S.; Pons, S.; Conte, C.; Tortosa, F.; Sicard, P.; Sainte-Marie, Y.; Bisserier, M.; Lairez, O.; et al. Multifunctional mitochondrial Epac1 controls myocardial cell death. Circ. Res. 2017, 120, 645–657. [Google Scholar] [CrossRef] [PubMed]
- Brette, F.; Blandin, E.; Simard, C.; Guinamard, R.; Salle, L. EPAC activator critically regulates action potential duration by decreasing potassium current in rat adult ventricle. J. Mol. Cell. Cardiol. 2013, 57, 96–105. [Google Scholar] [CrossRef] [PubMed]
- Metrich, M.; Lucas, A.; Gastineau, M.; Samuel, J.L.; Heymes, C.; Morel, E.; Lezoualc’h, F. Epac mediates β-adrenergic receptor-induced cardiomyocyte hypertrophy. Circ. Res. 2008, 102, 959–965. [Google Scholar] [CrossRef] [PubMed]
- Herrmann, S.; Layh, B.; Ludwig, A. Novel insights into the distribution of cardiac HCN channels: An expression study in the mouse heart. J. Mol. Cell. Cardiol. 2011, 51, 997–1006. [Google Scholar] [CrossRef] [PubMed]
- DiFrancesco, D.; Tortora, P. Direct activation of cardiac pacemaker channels by intracellular cyclic AMP. Nature 1991, 351, 145–147. [Google Scholar] [CrossRef] [PubMed]
- Vinogradova, T.M.; Lyashkov, A.E.; Zhu, W.; Ruknudin, A.M.; Sirenko, S.; Yang, D.; Deo, S.; Barlow, M.; Johnson, S.; Caffrey, J.L.; et al. High basal protein kinase A-dependent phosphorylation drives rhythmic internal Ca2+ store oscillations and spontaneous beating of cardiac pacemaker cells. Circ. Res. 2006, 98, 505–514. [Google Scholar] [CrossRef] [PubMed]
- Lakatta, E.G.; Maltsev, V.A.; Vinogradova, T.M. A coupled system of intracellular Ca2+ clocks and surface membrane voltage clocks controls the timekeeping mechanism of the heart’s pacemaker. Circ. Res. 2010, 106, 659–673. [Google Scholar] [CrossRef] [PubMed]
- Herrmann, S.; Stieber, J.; Stockl, G.; Hofmann, F.; Ludwig, A. HCN4 provides a ‘depolarization reserve’ and is not required for heart rate acceleration in mice. EMBO J. 2007, 26, 4423–4432. [Google Scholar] [CrossRef] [PubMed]
- Schweizer, P.A.; Duhme, N.; Thomas, D.; Becker, R.; Zehelein, J.; Draguhn, A.; Bruehl, C.; Katus, H.A.; Koenen, M. cAMP sensitivity of HCN pacemaker channels determines basal heart rate but is not critical for autonomic rate control. Circ. Arrhythm. Electrophysiol. 2010, 3, 542–552. [Google Scholar] [CrossRef] [PubMed]
- Kozasa, Y.; Nakashima, N.; Ito, M.; Ishikawa, T.; Kimoto, H.; Ushijima, K.; Makita, N.; Takano, M. HCN4 pacemaker channels attenuate the parasympathetic response and stabilize the spontaneous firing of sinoatrial node. J. Physiol. 2018, 596, 809–825. [Google Scholar] [CrossRef] [PubMed]
- Mesirca, P.; Alig, J.; Torrente, A.G.; Muller, J.C.; Marger, L.; Rollin, A.; Marquilly, C.; Vincent, A.; Dubel, S.; Bidaud, I.; et al. Cardiac arrhythmia induced by genetic silencing of ‘funny’ (f) channels is rescued by GIRK4 inactivation. Nat. Commun. 2014, 5, 4664. [Google Scholar] [CrossRef] [PubMed]
- Fenske, S.; Krause, S.; Biel, M.; Wahl-Schott, C. The role of HCN channels in ventricular repolarization. Trends Cardiovasc. Med. 2011, 21, 216–220. [Google Scholar] [CrossRef] [PubMed]
- Wahl-Schott, C.; Fenske, S.; Biel, M. HCN channels: New roles in sinoatrial node function. Curr. Opin. Pharmacol. 2014, 15, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, F.; Fabritz, L.; Stieber, J.; Schmitt, J.; Kirchhof, P.; Ludwig, A.; Herrmann, S. Ventricular HCN channels decrease the repolarization reserve in the hypertrophic heart. Cardiovasc. Res. 2012, 95, 317–326. [Google Scholar] [CrossRef] [PubMed]
- De Rooij, J.; Zwartkruis, F.J.; Verheijen, M.H.; Cool, R.H.; Nijman, S.M.; Wittinghofer, A.; Bos, J.L. Epac is a Rap1 guanine-nucleotide-exchange factor directly activated by cyclic AMP. Nature 1998, 396, 474–477. [Google Scholar] [CrossRef] [PubMed]
- Kawasaki, H.; Springett, G.M.; Mochizuki, N.; Toki, S.; Nakaya, M.; Matsuda, M.; Housman, D.E.; Graybiel, A.M. A family of cAMP-binding proteins that directly activate Rap1. Science 1998, 282, 2275–2279. [Google Scholar] [CrossRef] [PubMed]
- Reese, D.E.; Zavaljevski, M.; Streiff, N.L.; Bader, D. Bves: A novel gene expressed during coronary blood vessel development. Dev. Biol. 1999, 209, 159–171. [Google Scholar] [CrossRef] [PubMed]
- Andree, B.; Hillemann, T.; Kessler-Icekson, G.; Schmitt-John, T.; Jockusch, H.; Arnold, H.H.; Brand, T. Isolation and characterization of the novel popeye gene family expressed in skeletal muscle and heart. Dev. Biol. 2000, 223, 371–382. [Google Scholar] [CrossRef] [PubMed]
- Froese, A.; Breher, S.S.; Waldeyer, C.; Schindler, R.F.; Nikolaev, V.O.; Rinne, S.; Wischmeyer, E.; Schlueter, J.; Becher, J.; Simrick, S.; et al. Popeye domain containing proteins are essential for stress-mediated modulation of cardiac pacemaking in mice. J. Clin. Investig. 2012, 122, 1119–1130. [Google Scholar] [CrossRef] [PubMed]
- Schindler, R.F.; Scotton, C.; Zhang, J.; Passarelli, C.; Ortiz-Bonnin, B.; Simrick, S.; Schwerte, T.; Poon, K.L.; Fang, M.; Rinne, S.; et al. POPDC1S201F causes muscular dystrophy and arrhythmia by affecting protein trafficking. J. Clin. Investig. 2016, 126, 239–253. [Google Scholar] [CrossRef] [PubMed]
- Tree of the Month: Zebrafish Popeye-Domain-Containing Proteins and Heartbeat Regulation. Available online: http://phylomedb.org/?q=node/659 (accessed on 30 January 2018).
- Brand, T. The popeye domain-containing gene family. Cell Biochem. Biophys. 2005, 43, 95–104. [Google Scholar] [CrossRef]
- Vasavada, T.K.; DiAngelo, J.R.; Duncan, M.K. Developmental expression of Pop1/Bves. J. Histochem. Cytochem. 2004, 52, 371–377. [Google Scholar] [CrossRef] [PubMed]
- Knight, R.F.; Bader, D.M.; Backstrom, J.R. Membrane topology of Bves/Pop1A, a cell adhesion molecule that displays dynamic changes in cellular distribution during development. J. Biol. Chem. 2003, 278, 32872–32879. [Google Scholar] [CrossRef] [PubMed]
- Alcalay, Y.; Hochhauser, E.; Kliminski, V.; Dick, J.; Zahalka, M.A.; Parnes, D.; Schlesinger, H.; Abassi, Z.; Shainberg, A.; Schindler, R.F.; et al. Popeye domain containing 1 (Popdc1/Bves) is a caveolae-associated protein involved in ischemia tolerance. PLoS ONE 2013, 8, e71100. [Google Scholar] [CrossRef] [PubMed]
- Kawaguchi, M.; Hager, H.A.; Wada, A.; Koyama, T.; Chang, M.S.; Bader, D.M. Identification of a novel intracellular interaction domain essential for Bves function. PLoS ONE 2008, 3, e2261. [Google Scholar] [CrossRef] [PubMed]
- Kelley, L.A.; Mezulis, S.; Yates, C.M.; Wass, M.N.; Sternberg, M.J. The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 2015, 10, 845–858. [Google Scholar] [CrossRef] [PubMed]
- First Glance in Jmol. Available online: http://bioinformatics.org/firstglance/fgij/ (accessed on 13 February 2018).
- Russ, P.K.; Pino, C.J.; Williams, C.S.; Bader, D.M.; Haselton, F.R.; Chang, M.S. Bves modulates tight junction associated signaling. PLoS ONE 2011, 6, e14563. [Google Scholar] [CrossRef] [PubMed]
- Kannan, N.; Wu, J.; Anand, G.S.; Yooseph, S.; Neuwald, A.F.; Venter, J.C.; Taylor, S.S. Evolution of allostery in the cyclic nucleotide binding module. Genome Biol. 2007, 8, R264. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.M.; Ten Eyck, L.F.; Goodsell, D.S.; Haste, N.M.; Kornev, A.; Taylor, S.S. The cAMP binding domain: An ancient signaling module. Proc. Natl. Acad. Sci. USA 2005, 102, 45–50. [Google Scholar] [CrossRef] [PubMed]
- Lorenz, R.; Moon, E.W.; Kim, J.J.; Schmidt, S.H.; Sankaran, B.; Pavlidis, I.V.; Kim, C.; Herberg, F.W. Mutations of PKA cyclic nucleotide-binding domains reveal novel aspects of cyclic nucleotide selectivity. Biochem. J. 2017, 474, 2389–2403. [Google Scholar] [CrossRef] [PubMed]
- Froese, A.; Brand, T. Expression pattern of Popdc2 during mouse embryogenesis and in the adult. Dev. Dyn. 2008, 237, 780–787. [Google Scholar] [CrossRef] [PubMed]
- Breher, S.S.; Mavridou, E.; Brenneis, C.; Froese, A.; Arnold, H.H.; Brand, T. Popeye domain containing gene 2 (Popdc2) is a myocyte-specific differentiation marker during chick heart development. Dev. Dyn. 2004, 229, 695–702. [Google Scholar] [CrossRef] [PubMed]
- Torlopp, A.; Breher, S.S.; Schluter, J.; Brand, T. Comparative analysis of mRNA and protein expression of Popdc1 (Bves) during early development in the chick embryo. Dev. Dyn. 2006, 235, 691–700. [Google Scholar] [CrossRef] [PubMed]
- Wada, A.; Reese, D.; Bader, D. Bves: Prototype of a new class of cell adhesion molecules expressed during coronary artery development. Development 2001, 128, 2085–2093. [Google Scholar] [PubMed]
- Smith, T.K.; Bader, D.M. Characterization of Bves expression during mouse development using newly generated immunoreagents. Dev. Dyn. 2006, 235, 1701–1708. [Google Scholar] [CrossRef] [PubMed]
- The Human Protein Atlas. Available online: https://www.proteinatlas.org/ENSG00000112276-BVES/tissue (accessed on 19 February 2018).
- The Human Protein Atlas. Available online: https://www.proteinatlas.org/ENSG00000121577-POPDC2/tissue (accessed on 19 February 2018).
- The Human Protein Atlas. Available online: https://www.proteinatlas.org/ENSG00000132429-POPDC3/tissue (accessed on 19 February 2018).
- Korfali, N.; Wilkie, G.S.; Swanson, S.K.; Srsen, V.; de Las Heras, J.; Batrakou, D.G.; Malik, P.; Zuleger, N.; Kerr, A.R.; Florens, L.; et al. The nuclear envelope proteome differs notably between tissues. Nucleus 2012, 3, 552–564. [Google Scholar] [CrossRef] [PubMed]
- Schindler, R.; Simrick, S.; Brand, T. Nuclear localization of members of popeye domain containing (Popdc) protein family. Cardiovasc. Res. 2012, 93, S98. [Google Scholar]
- Soni, S.; Raaijmakers, A.J.; Raaijmakers, L.M.; Damen, J.M.; van Stuijvenberg, L.; Vos, M.A.; Heck, A.J.; van Veen, T.A.; Scholten, A. A proteomics approach to identify new putative cardiac intercalated disk proteins. PLoS ONE 2016, 11, e0152231. [Google Scholar] [CrossRef] [PubMed]
- Andrée, B.; Fleige, A.; Arnold, H.H.; Brand, T. Mouse pop1 is required for muscle regeneration in adult skeletal muscle. Mol. Cell. Biol. 2002, 22, 1504–1512. [Google Scholar] [CrossRef] [PubMed]
- Kirchmaier, B.C.; Poon, K.L.; Schwerte, T.; Huisken, J.; Winkler, C.; Jungblut, B.; Stainier, D.Y.; Brand, T. The popeye domain containing 2 (Popdc2) gene in zebrafish is required for heart and skeletal muscle development. Dev. Biol. 2012, 363, 438–450. [Google Scholar] [CrossRef] [PubMed]
- Nelson, I.; Beuvin, M.; Ben-Yaou, R.; Masson, C.; Boland, A.; Schindler, R.; Brand, T.; Eymard, B.; Bonne, G. Novel recessive splice site mutation in Popdc1 (Bves) is associated with first-degree atrioventricular block and muscular dystrophy. Neuromuscul. Disord. 2017, 27, S139–S140. [Google Scholar] [CrossRef]
- Rinné, S.; Ortiz-Bonnin, B.; Stallmeyer, B.; Schindler, R.F.R.; Kiper, A.K.; Dittmann, S.; Friedrich, C.; Zumhagen, S.; Simrick, S.L.; Gonzalez, W.; et al. Conduction disorder caused by a mutation in POPDC2, a novel modulator of the cardias sodium channel SCN5A. Acta Physiol. 2016, 216, 42. [Google Scholar]
- Wu, J.; Schuessler, R.B.; Rodefeld, M.D.; Saffitz, J.E.; Boineau, J.P. Morphological and membrane characteristics of spider and spindle cells isolated from rabbit sinus node. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2001, 280, H1232–H1240. [Google Scholar] [CrossRef] [PubMed]
- Honore, E. The neuronal background K2P channels: Focus on TREK1. Nat. Rev. Neurosci. 2007, 8, 251–261. [Google Scholar] [CrossRef] [PubMed]
- Unudurthi, S.D.; Wu, X.; Qian, L.; Amari, F.; Onal, B.; Li, N.; Makara, M.A.; Smith, S.A.; Snyder, J.; Fedorov, V.V.; et al. Two-pore K+ channel TREK-1 regulates sinoatrial node membrane excitability. J. Am. Heart Assoc. 2016, 5, e002865. [Google Scholar] [CrossRef] [PubMed]
- Mangoni, M.E.; Nargeot, J. Genesis and regulation of the heart automaticity. Physiol. Rev. 2008, 88, 919–982. [Google Scholar] [CrossRef] [PubMed]
- Lubelwana Hafver, T.; Wanichawan, P.; Manfra, O.; de Souza, G.A.; Lunde, M.; Martinsen, M.; Louch, W.E.; Mathias Sejersted, O.; Carlson, C.R. Mapping the in vitro interactome of cardiac sodium (Na+)-calcium (Ca2+) exchanger 1 (NCX1). Proteomics 2017, 17. [Google Scholar] [CrossRef] [PubMed]
- Torrente, A.G.; Zhang, R.; Zaini, A.; Giani, J.F.; Kang, J.; Lamp, S.T.; Philipson, K.D.; Goldhaber, J.I. Burst pacemaker activity of the sinoatrial node in sodium-calcium exchanger knockout mice. Proc. Natl. Acad. Sci. USA 2015, 112, 9769–9774. [Google Scholar] [CrossRef] [PubMed]
- Kliminski, V.; Uziel, O.; Kessler-Icekson, G. Popdc1/Bves functions in the preservation of cardiomyocyte viability while affecting Rac1 activity and Bnip3 expression. J. Cell. Biochem. 2017, 118, 1505–1517. [Google Scholar] [CrossRef] [PubMed]
- Andree, B.; Fleige, A.; Hillemann, T.; Arnold, H.H.; Kessler-Icekson, G.; Brand, T. Molecular and functional analysis of popeye genes: A novel family of transmembrane proteins preferentially expressed in heart and skeletal muscle. Exp. Clin. Cardiol. 2002, 7, 99–103. [Google Scholar] [PubMed]
- Harvey, R.D.; Calaghan, S.C. Caveolae create local signalling domains through their distinct protein content, lipid profile and morphology. J. Mol. Cell. Cardiol. 2012, 52, 366–375. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.C.; Liu, C.Y.; Chen, Y.H.; Chen, R.F.; Huang, C.J.; Wang, I.J. Blood vessel epicardial substance (Bves) regulates epidermal tight junction integrity through atypical protein kinase C. J. Biol. Chem. 2012, 287, 39887–39897. [Google Scholar] [CrossRef] [PubMed]
- Osler, M.E.; Chang, M.S.; Bader, D.M. Bves modulates epithelial integrity through an interaction at the tight junction. J. Cell Sci. 2005, 118, 4667–4678. [Google Scholar] [CrossRef] [PubMed]
- Charvet, B.; Malbouyres, M.; Pagnon-Minot, A.; Ruggiero, F.; Le Guellec, D. Development of the zebrafish myoseptum with emphasis on the myotendinous junction. Cell Tissue Res. 2011, 346, 439–449. [Google Scholar] [CrossRef] [PubMed]
- Charvet, B.; Guiraud, A.; Malbouyres, M.; Zwolanek, D.; Guillon, E.; Bretaud, S.; Monnot, C.; Schulze, J.; Bader, H.L.; Allard, B.; et al. Knockdown of col22a1 gene in zebrafish induces a muscular dystrophy by disruption of the myotendinous junction. Development 2013, 140, 4602–4613. [Google Scholar] [CrossRef] [PubMed]
- Goody, M.F.; Sher, R.B.; Henry, C.A. Hanging on for the ride: Adhesion to the extracellular matrix mediates cellular responses in skeletal muscle morphogenesis and disease. Dev. Biol. 2015, 401, 75–91. [Google Scholar] [CrossRef] [PubMed]
- Brand, T.; Schindler, R. New kids on the block: The popeye domain containing (Popdc) protein family acting as a novel class of cAMP effector proteins in striated muscle. Cell. Signal. 2017, 40, 156–165. [Google Scholar] [CrossRef] [PubMed]
- Magri, F.; Del Bo, R.; D’Angelo, M.G.; Sciacco, M.; Gandossini, S.; Govoni, A.; Napoli, L.; Ciscato, P.; Fortunato, F.; Brighina, E.; et al. Frequency and characterisation of anoctamin 5 mutations in a cohort of italian limb-girdle muscular dystrophy patients. Neuromuscul. Disord. 2012, 22, 934–943. [Google Scholar] [CrossRef] [PubMed]
- Rehmann, H.; Wittinghofer, A.; Bos, J.L. Capturing cyclic nucleotides in action: Snapshots from crystallographic studies. Nat. Rev. Mol. Cell Biol. 2007, 8, 63–73. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | Mutation | Heart | Skeletal Muscle | References |
|---|---|---|---|---|
| mouse | Popdc1-/- | stress-induced sinus bradycardia | regeneration defect | [27,50] |
| ischemia-reperfusion damage | not analyzed | [33] | ||
| Popdc2-/- | Stress-induced sinus bradycardia | not analyzed | [27] | |
| zebrafish | popdc1 morphants | AV-block, pericardial effusion | muscular dystrophy | [28] |
| popdc2 morphants | AV-block, pericardial effusion | muscular dystrophy | [53] | |
| popdc1S191F | AV-block, pericardial effusion | muscular dystrophy | [28] | |
| human | POPDC1S201F | 2nd degree AV-block | limb-girdle MD | [28] |
| POPDC1del56 V217-K272 | 1st degree AV-block, | limb-girdle MD | [54] | |
| POPDC2W188X | 3rd degree AV-block | no known phenotype | [55] |
© 2018 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brand, T. The Popeye Domain Containing Genes and Their Function as cAMP Effector Proteins in Striated Muscle. J. Cardiovasc. Dev. Dis. 2018, 5, 18. https://doi.org/10.3390/jcdd5010018
Brand T. The Popeye Domain Containing Genes and Their Function as cAMP Effector Proteins in Striated Muscle. Journal of Cardiovascular Development and Disease. 2018; 5(1):18. https://doi.org/10.3390/jcdd5010018
Chicago/Turabian StyleBrand, Thomas. 2018. "The Popeye Domain Containing Genes and Their Function as cAMP Effector Proteins in Striated Muscle" Journal of Cardiovascular Development and Disease 5, no. 1: 18. https://doi.org/10.3390/jcdd5010018
APA StyleBrand, T. (2018). The Popeye Domain Containing Genes and Their Function as cAMP Effector Proteins in Striated Muscle. Journal of Cardiovascular Development and Disease, 5(1), 18. https://doi.org/10.3390/jcdd5010018