Using cAMP Sensors to Study Cardiac Nanodomains


{kind=link}
{kind=link}
Abstract
:1. Introduction
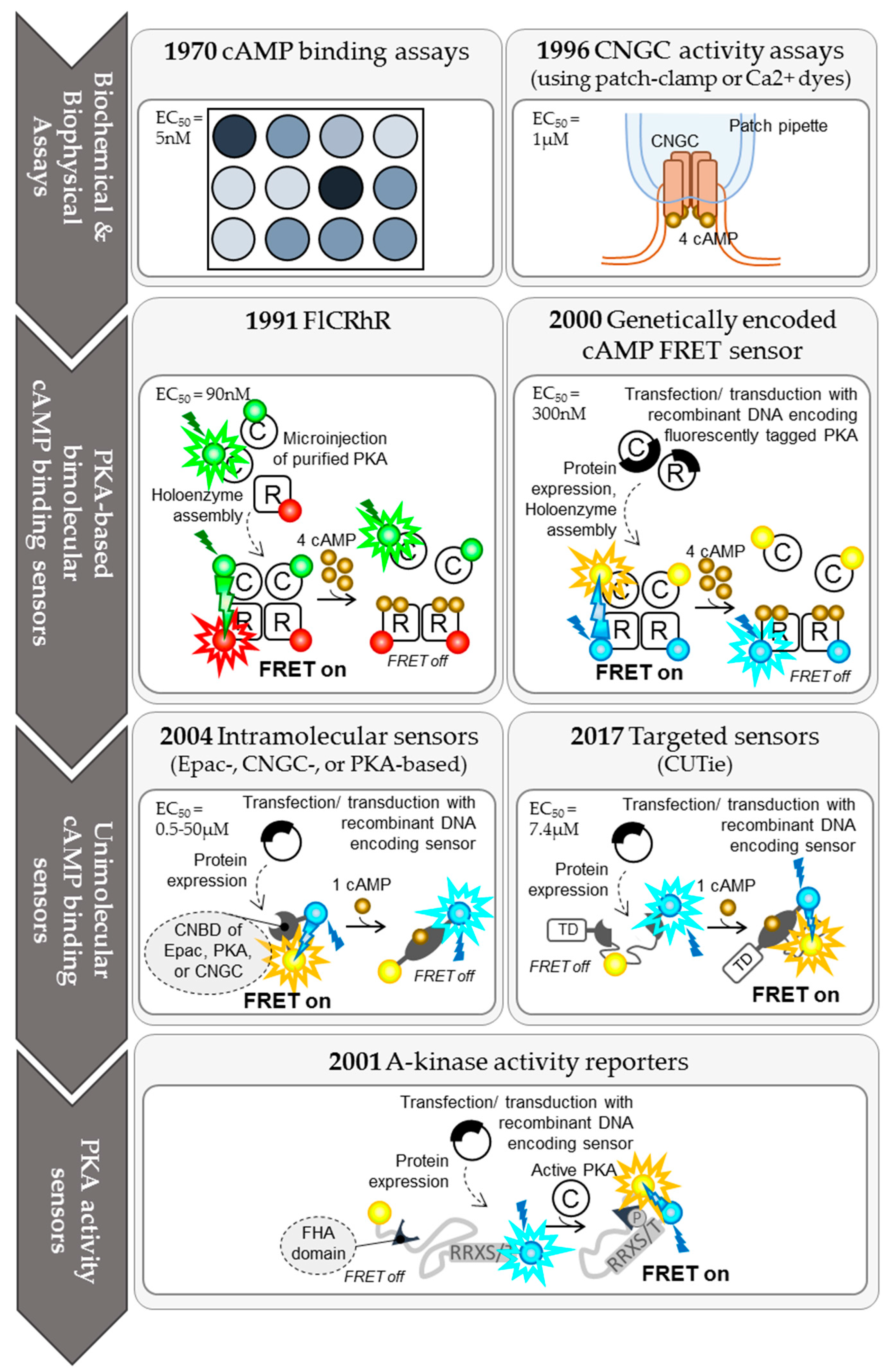
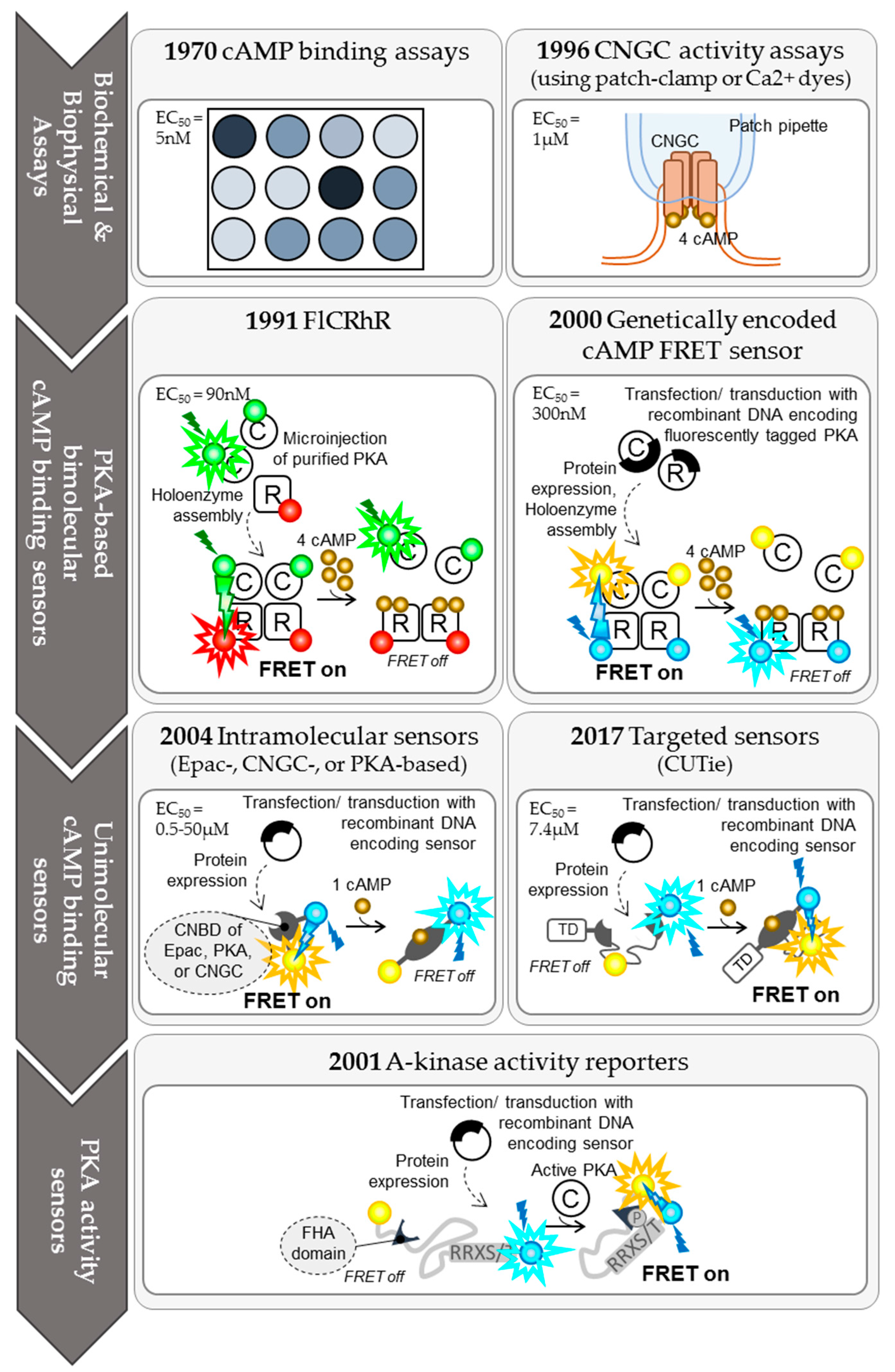
2. The Evolution of cAMP Imaging Techniques and Their Contribution to Our Understanding of the Spatio-Temporal Compartmentation of cAMP Signalling
2.1. Biochemical Protein Binding Assays for cAMP
2.2. Cyclic Nucleotide Gated Channel (CNGC) Activity Measurements
2.3. PKA-Based bi-Molecular FRET Sensors
2.3.1. FlCRhR Probes
2.3.2. Genetically Encoded, Tetrameric cAMP FRET Probes
2.4. Intramolecular FRET Sensors
2.4.1. Epac-Based Single-Chain FRET Sensors
2.4.2. CNGC-Based Single-Chain FRET Sensors
2.4.3. PKA-Based Single-Chain FRET Sensors
2.5. Single-Wavelength Fluorescent Sensors for cAMP
2.6. PKA Activity Sensors
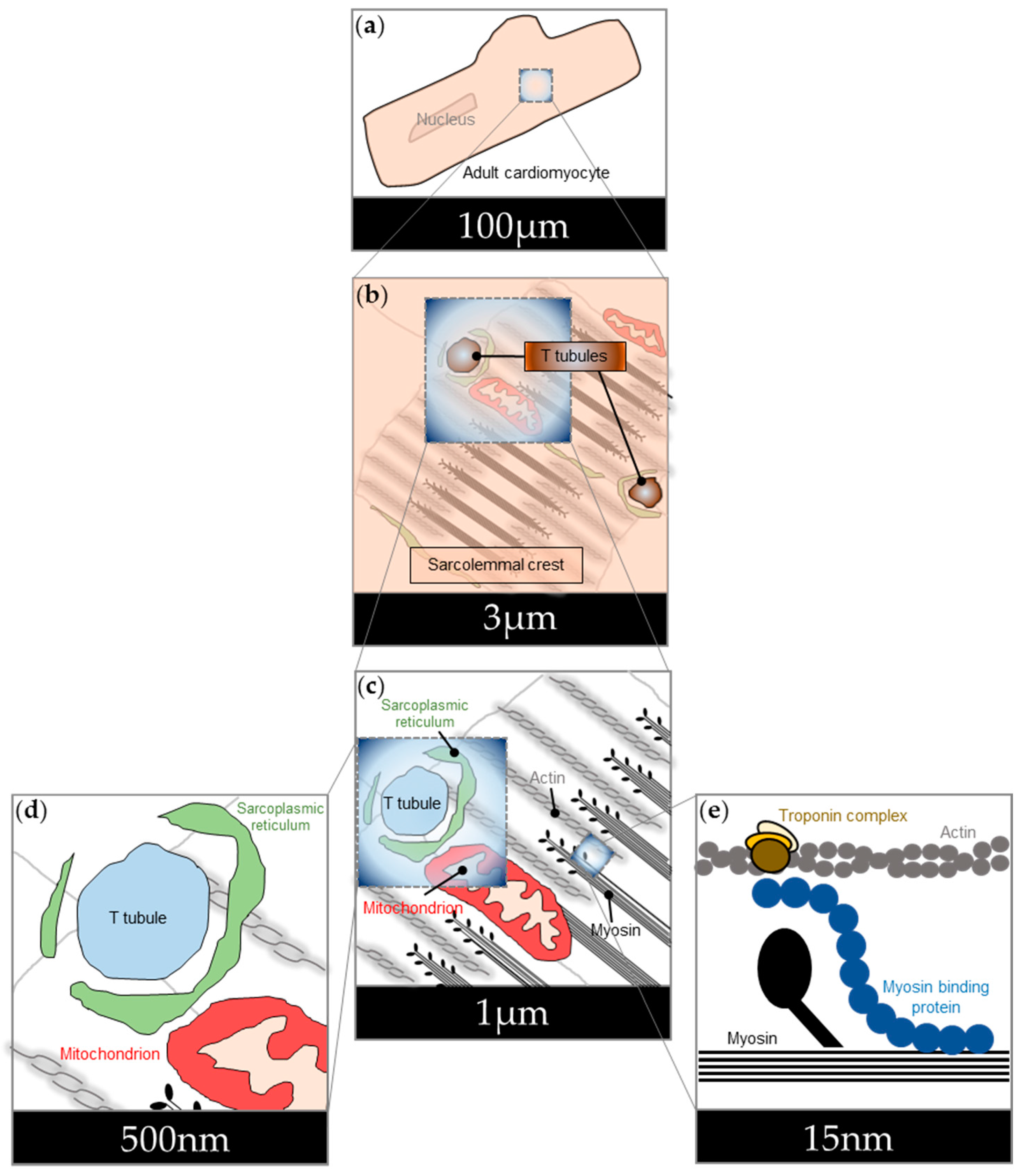
3. Cardiac Nanodomains Studied Using FRET-Based cAMP Sensors
3.1. cAMP Signalling in the Nucleus
3.2. cAMP Signalling at the Sarcolemma
3.3. cAMP Signalling at the Mitochondria
3.4. cAMP Signalling at the Sarcoplasmic Reticulum
3.5. cAMP Signalling at the Sarcomere
3.6. cAMP Signalling at A Kinase Anchoring Proteins (AKAPs)
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Salazar, N.C.; Chen, J.; Rockman, H.A. Cardiac GPCRs: GPCR signaling in healthy and failing hearts. Biochim. Biophys. Acta Biomembr. 2007, 1768, 1006–1018. [Google Scholar] [CrossRef] [PubMed]
- Kaupp, U.B.; Seifert, R. Cyclic nucleotide-gated ion channels. Physiol. Rev. 2002, 82, 769–824. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, M.; Dekker, F.J.; Maarsingh, H. Exchange Protein Directly Activated by cAMP (epac): A Multidomain cAMP Mediator in the Regulation of Diverse Biological Functions. Pharmacol. Rev. 2013, 65, 670–709. [Google Scholar] [CrossRef] [PubMed]
- Schindler, R.F.R.; Brand, T. The Popeye domain containing protein family—A novel class of cAMP effectors with important functions in multiple tissues. Prog. Biophys. Mol. Biol. 2016, 120, 28–36. [Google Scholar] [CrossRef] [PubMed]
- Taylor, S.S.; Kim, C.; Cheng, C.Y.; Brown, S.H.J.; Wu, J.; Kannan, N. Signaling through cAMP and cAMP-dependent protein kinase: Diverse strategies for drug design. Biochim. Biophys. Acta Proteins Proteom. 2008, 1784, 16–26. [Google Scholar] [CrossRef] [PubMed]
- Buxton, I.L.O.; Brunton, L.L. Compartments of cyclic AMP and protein kinase in mammalian cardiomyocytes. J. Biol. Chem. 1983, 258, 10233–10239. [Google Scholar] [PubMed]
- Wang, H.; Robinson, H.; Ke, H. The Molecular Basis for Different Recognition of Substrates by Phosphodiesterase Families 4 and 10. J. Mol. Biol. 2007, 371, 302–307. [Google Scholar] [CrossRef] [PubMed]
- Sunahara, R.K.; Tesmer, J.J.; Gilman, A.G.; Sprang, S.R. Crystal structure of the adenylyl cyclase activator Gsα. Science 1997, 278, 1943–1947. [Google Scholar] [CrossRef] [PubMed]
- Richards, M.; Lomas, O.; Jalink, K.; Ford, K.L.; Vaughan-Jones, R.D.; Lefkimmiatis, K.; Swietach, P. Intracellular tortuosity underlies slow cAMP diffusion in adult ventricular myocytes. Cardiovasc. Res. 2016, 110, 395–407. [Google Scholar] [CrossRef] [PubMed]
- Gesellchen, F.; Stangherlin, A.; Surdo, N.; Terrin, A.; Zoccarato, A.; Zaccolo, M. Measuring spatiotemporal dynamics of cyclic AMP signaling in real-time using FRET-based biosensors. Methods Mol. Biol. 2011, 746, 297–316. [Google Scholar] [CrossRef] [PubMed]
- Sprenger, J.U.; Nikolaev, V.O. Biophysical techniques for detection of cAMP and cGMP in living cells. Int. J. Mol. Sci. 2013, 14, 8025–8046. [Google Scholar] [CrossRef] [PubMed]
- Surdo, N.C.; Berrera, M.; Koschinski, A.; Brescia, M.; Machado, M.R.; Carr, C.; Wright, P.; Gorelik, J.; Morotti, S.; Grandi, E.; et al. FRET biosensor uncovers cAMP nano-domains at β-adrenergic targets that dictate precise tuning of cardiac contractility. Nat. Commun. 2017, 8, 15031. [Google Scholar] [CrossRef] [PubMed]
- Horton, J.K.; Martin, R.C.; Kalinka, S.; Cushing, A.; Kitcher, J.P.; O’Sullivan, M.J.; Baxendale, P.M. Enzyme immunoassays for the estimation of adenosine 3′,5′ cyclic monophosphate and guanosine 3′,5′ cyclic monophosphate in biological fluids. J. Immunol. Methods 1992, 155, 31–40. [Google Scholar] [CrossRef]
- Gilman, A.G. A protein binding assay for adenosine 3’:5’-cyclic monophosphate. Proc. Natl. Acad. Sci. USA 1970, 67, 305–312. [Google Scholar] [CrossRef] [PubMed]
- Pradelles, P.; Grassi, J.; Chabardes, D.; Guiso, N. Enzyme Immunoassays of Adenosine Cyclic 3’,5’-Monophosphate and Guanosine Cyclic 3’,5’-Monophosphate Using Acetylcholinesterase. Anal. Chem. 1989, 61, 447–453. [Google Scholar] [CrossRef] [PubMed]
- Steiner, A.L.; Kipnis, D.M.; Utiger, R.; Parker, C. Radioimmunoassay for the measurement of adenosine 3’,5’-cyclic phosphate. Proc. Natl. Acad. Sci. USA 1969, 64, 367–373. [Google Scholar] [CrossRef] [PubMed]
- Boularan, C.; Gales, C. Cardiac cAMP: Production, hydrolysis, modulation and detection. Front. Pharmacol. 2015, 6, 203. [Google Scholar] [CrossRef] [PubMed]
- Rochais, F.; Vandecasteele, G.; Lefebvre, F.; Lugnier, C.; Lum, H.; Mazet, J.L.; Cooper, D.M.F.; Fischmeister, R. Negative feedback exerted by cAMP-dependent protein kinase and cAMP phosphodiesterase on subsarcolemmal cAMP signals in intact cardiac myocytes: An in vivo study using adenovirus-mediated expression of CNG channels. J. Biol. Chem. 2004, 279, 52095–52105. [Google Scholar] [CrossRef] [PubMed]
- Fagan, K.A.; Schaack, J.; Zweifach, A.; Cooper, D.M.F. Adenovirus encoded cyclic nucleotide-gated channels: A new methodology for monitoring cAMP in living cells. FEBS Lett. 2001, 500, 85–90. [Google Scholar] [CrossRef]
- Rich, T.C.; Fagan, K.A.; Tse, T.E.; Schaack, J.; Cooper, D.M.; Karpen, J.W. A uniform extracellular stimulus triggers distinct cAMP signals in different compartments of a simple cell. Proc. Natl. Acad. Sci. USA 2001, 98, 13049–13054. [Google Scholar] [CrossRef] [PubMed]
- Jurevicius, J.; Fischmeister, R. cAMP compartmentation is responsible for a local activation of cardiac Ca2+ channels by β-adrenergic agonists. Proc. Natl. Acad. Sci. USA 1996, 93, 295–299. [Google Scholar] [CrossRef] [PubMed]
- Rich, T.C.; Tse, T.E.; Rohan, J.G.; Schaack, J.; Karpen, J.W. In vivo assessment of local phosphodiesterase activity using tailored cyclic nucleotide-gated channels as cAMP sensors. J. Gen. Physiol. 2001, 118, 63–78. [Google Scholar] [CrossRef] [PubMed]
- Adams, S.R.; Harootunian, A.T.; Buechler, Y.J.; Taylor, S.S.; Tsien, R.Y. Fluorescence ratio imaging of cyclic AMP in single cells. Nature 1991, 349, 694–697. [Google Scholar] [CrossRef] [PubMed]
- Goaillard, J.M.; Vincent, P.; Fischmeister, R. Simultaneous measurements of intracellular cAMP and l-type Ca2+ current in single frog ventricular myocytes. J. Physiol. 2001, 530, 79–91. [Google Scholar] [CrossRef] [PubMed]
- Zaccolo, M.; De Giorgi, F.; Cho, C.Y.; Feng, L.; Knapp, T.; Negulescu, P.A.; Taylor, S.S.; Tsien, R.Y.; Pozzan, T. A genetically encoded, fluorescent indicator for cyclic AMP in living cells. Nat. Cell Biol. 2000, 2, 25–29. [Google Scholar] [CrossRef] [PubMed]
- Zaccolo, M.; Pozzan, T. Discrete Microdomains with High Concentration of cAMP in Stimulated Rat Neonatal Cardiac Myocytes. Science 2002, 295, 1711–1715. [Google Scholar] [CrossRef] [PubMed]
- Kokkonen, K.; Kass, D.A. Nanodomain Regulation of Cardiac Cyclic Nucleotide Signaling by Phosphodiesterases. Annu. Rev. Pharmacol. Toxicol. 2017, 57, 455–479. [Google Scholar] [CrossRef] [PubMed]
- Mongillo, M.; McSorley, T.; Evellin, S.; Sood, A.; Lissandron, V.; Terrin, A.; Huston, E.; Hannawacker, A.; Lohse, M.J.; Pozzan, T.; et al. Fluorescence Resonance Energy Transfer Based Analysis of cAMP Dynamics in Live Neonatal Rat Cardiac Myocytes Reveals Distinct Functions of Compartmentalized Phosphodiesterases. Circ. Res. 2004, 95, 67–75. [Google Scholar] [CrossRef] [PubMed]
- Lehnart, S.E.; Wehrens, X.H.T.; Reiken, S.; Warrier, S.; Belevych, A.E.; Harvey, R.D.; Richter, W.; Jin, S.L.C.; Conti, M.; Marks, A.R. Phosphodiesterase 4D deficiency in the ryanodine-receptor complex promotes heart failure and arrhythmias. Cell 2005, 123, 25–35. [Google Scholar] [CrossRef] [PubMed]
- Warrier, S.; Belevych, A.E.; Ruse, M.; Eckert, R.L.; Zaccolo, M.; Pozzan, T.; Harvey, R.D. Beta-adrenergic- and muscarinic receptor-induced changes in cAMP activity in adult cardiac myocytes detected with FRET-based biosensor. Am. J. Physiol. Cell Physiol. 2005, 289, C455–C461. [Google Scholar] [CrossRef] [PubMed]
- Terrin, A.; Di Benedetto, G.; Pertegato, V.; Cheung, Y.F.; Baillie, G.; Lynch, M.J.; Elvassore, N.; Prinz, A.; Herberg, F.W.; Houslay, M.D.; et al. PGE1 stimulation of HEK293 cells generates multiple contiguous domains with different [cAMP]: Role of compartmentalized phosphodiesterases. J. Cell Biol. 2006, 175, 441–451. [Google Scholar] [CrossRef] [PubMed]
- Nikolaev, V.O.; Bünemann, M.; Hein, L.; Hannawacker, A.; Lohse, M.J. Novel single chain cAMP sensors for receptor-induced signal propagation. J. Biol. Chem. 2004, 279, 37215–37218. [Google Scholar] [CrossRef] [PubMed]
- Nikolaev, V.O.; Bünemann, M.; Schmitteckert, E.; Lohse, M.J.; Engelhardt, S. Cyclic AMP imaging in adult cardiac myocytes reveals far-reaching β1-adrenergic but locally confined β2-adrenergic receptor-mediated signaling. Circ. Res. 2006, 99, 1084–1091. [Google Scholar] [CrossRef] [PubMed]
- Koschinski, A.; Zaccolo, M. Activation of PKA in cell requires higher concentration of cAMP than in vitro: Implications for compartmentalization of cAMP signalling. Sci. Rep. 2017, 7, 14090. [Google Scholar] [CrossRef] [PubMed]
- Tsalkova, T.; Blumenthal, D.K.; Mei, F.C.; White, M.A.; Cheng, X. Mechanism of Epac activation. Structural and functional analyses of Epac2 hinge mutants with constitutive and reduced activities. J. Biol. Chem. 2009, 284, 23644–23651. [Google Scholar] [CrossRef] [PubMed]
- Consonni, S.V.; Gloerich, M.; Spanjaard, E.; Bos, J.L. cAMP regulates DEP domain-mediated binding of the guanine nucleotide exchange factor Epac1 to phosphatidic acid at the plasma membrane. Proc. Natl. Acad. Sci. USA 2012, 109, 3814–3819. [Google Scholar] [CrossRef] [PubMed]
- DiPilato, L.M.; Cheng, X.; Zhang, J. Fluorescent indicators of cAMP and Epac activation reveal differential dynamics of cAMP signaling within discrete subcellular compartments. Proc. Natl. Acad. Sci. USA 2004, 101, 16513–16518. [Google Scholar] [CrossRef] [PubMed]
- Ponsioen, B.; Zhao, J.; Riedl, J.; Zwartkruis, F.; van der Krogt, G.; Zaccolo, M.; Moolenaar, W.H.; Bos, J.L.; Jalink, K. Detecting cAMP-induced Epac activation by fluorescence resonance energy transfer: Epac as a novel cAMP indicator. EMBO Rep. 2004, 5, 1176–1180. [Google Scholar] [CrossRef] [PubMed]
- Violin, J.D.; DiPilato, L.M.; Yildirim, N.; Elston, T.C.; Zhang, J.; Lefkowitz, R.J. β2-Adrenergic receptor signaling and desensitization elucidated by quantitative modeling of real time cAMP dynamics. J. Biol. Chem. 2008, 283, 2949–2961. [Google Scholar] [CrossRef] [PubMed]
- DiPilato, L.M.; Zhang, J. The role of membrane microdomains in shaping beta2-adrenergic receptor-mediated cAMP dynamics. Mol. Biosyst. 2009, 5, 832–837. [Google Scholar] [CrossRef] [PubMed]
- Klarenbeek, J.; Goedhart, J.; Van Batenburg, A.; Groenewald, D.; Jalink, K. Fourth-generation Epac-based FRET sensors for cAMP feature exceptional brightness, photostability and dynamic range: Characterization of dedicated sensors for FLIM, for ratiometry and with high affinity. PLoS ONE 2015, 10, e0122513. [Google Scholar] [CrossRef] [PubMed]
- Di Benedetto, G.; Zoccarato, A.; Lissandron, V.; Terrin, A.; Li, X.; Houslay, M.D.; Baillie, G.S.; Zaccolo, M. Protein kinase A type I and type II define distinct intracellular signaling compartments. Circ. Res. 2008, 103, 836–844. [Google Scholar] [CrossRef] [PubMed]
- Nikolaev, V.O.; Moshkov, A.; Lyon, A.R.; Miragoli, M.; Novak, P.; Paur, H.; Lohse, M.J.; Korchev, Y.E.; Harding, S.E.; Gorelik, J.; et al. β2-adrenergic receptor redistribution in heart failure changes cAMP compartmentation. Science 2010, 327, 1653–1657. [Google Scholar] [CrossRef] [PubMed]
- Llopis, J.; McCaffery, J.M.; Miyawaki, A.; Farquhar, M.G.; Tsien, R.Y. Measurement of cytosolic, mitochondrial, and Golgi pH in single living cells with green fluorescent proteins. Proc. Natl. Acad. Sci. USA 1998, 95, 6803–6808. [Google Scholar] [CrossRef] [PubMed]
- Di Benedetto, G.; Scalzotto, E.; Mongillo, M.; Pozzan, T. Mitochondrial Ca2+ uptake induces cyclic AMP generation in the matrix and modulates organelle ATP levels. Cell Metab. 2013, 17, 965–975. [Google Scholar] [CrossRef] [PubMed]
- Sprenger, J.U.; Perera, R.K.; Steinbrecher, J.H.; Lehnart, S.E.; Maier, L.S.; Hasenfuss, G.; Nikolaev, V.O. In vivo model with targeted cAMP biosensor reveals changes in receptor-microdomain communication in cardiac disease. Nat. Commun. 2015, 6, 6965. [Google Scholar] [CrossRef] [PubMed]
- Herget, S.; Lohse, M.J.; Nikolaev, V.O. Real-time monitoring of phosphodiesterase inhibition in intact cells. Cell. Signal. 2008, 20, 1423–1431. [Google Scholar] [CrossRef] [PubMed]
- Packer, M.; Carver, J.R.; Rodeheffer, R.J.; Ivanhoe, R.J.; DiBianco, R.; Zeldis, S.M.; Hendrix, G.H.; Bommer, W.J.; Elkayam, U.; Kukin, M.L.; et al. Effect of oral milrinone on mortality in severe chronic heart failure. N. Engl. J. Med. 1991, 325, 1468–1475. [Google Scholar] [CrossRef] [PubMed]
- Harbeck, M.C.; Chepurny, O.; Nikolaev, V.O.; Lohse, M.J.; Holz, G.G.; Roe, M.W. Simultaneous optical measurements of cytosolic Ca2+ and cAMP in single cells. Sci. Signal. 2006, 2006. [Google Scholar] [CrossRef] [PubMed]
- Odaka, H.; Arai, S.; Inoue, T.; Kitaguchi, T. Genetically-encoded yellow fluorescent cAMP indicator with an expanded dynamic range for dual-color imaging. PLoS ONE 2014, 9, e100252. [Google Scholar] [CrossRef] [PubMed]
- Hackley, C.R.; Mazzoni, E.O.; Blau, J. cAMPr: A single-wavelength fluorescent sensor for cyclic AMP. Sci. Signal. 2018, 11, 255–268. [Google Scholar] [CrossRef] [PubMed]
- Beavo, J.A.; Brunton, L.L. Cyclic nucleotide research—Still expanding after half a century. Nat. Rev. Mol. Cell Biol. 2002, 3, 710–718. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Ma, Y.; Taylor, S.S.; Tsien, R.Y. Genetically encoded reporters of protein kinase A activity reveal impact of substrate tethering. Proc. Natl. Acad. Sci. USA 2001, 98, 14997–15002. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Hupfeld, C.J.; Taylor, S.S.; Olefsky, J.M.; Tsien, R.Y. Insulin disrupts β-adrenergic signalling to protein kinase a in adipocytes. Nature 2005, 437, 569–573. [Google Scholar] [CrossRef] [PubMed]
- Allen, M.D.; Zhang, J. Subcellular dynamics of protein kinase A activity visualized by FRET-based reporters. Biochem. Biophys. Res. Commun. 2006, 348, 716–721. [Google Scholar] [CrossRef] [PubMed]
- Depry, C.; Allen, M.D.; Zhang, J. Visualization of PKA activity in plasma membrane microdomains. Mol. Biosyst. 2011, 7, 52–58. [Google Scholar] [CrossRef] [PubMed]
- Koschinski, A.; Zaccolo, M. A novel approach combining real-time imaging and the patch-clamp technique to calibrate FRET-based reporters for cAMP in their cellular microenvironment. In cAMP Signaling: Methods and Protocols; Humana Press: New York, NY, USA, 2015; pp. 25–40. ISBN 9781493925377. [Google Scholar]
- Chang, C.R.; Blackstone, C. Cyclic AMP-dependent protein kinase phosphorylation of Drp1 regulates its GTPase activity and mitochondrial morphology. J. Biol. Chem. 2007, 282, 21583–21587. [Google Scholar] [CrossRef] [PubMed]
- Hartzell, H.C.; Glass, D.B. Phosphorylation of purified cardiac muscle C-protein by purified cAMP-dependent and endogenous Ca2+-calmodulin-dependent protein kinases. J. Biol. Chem. 1984, 259, 15587–15596. [Google Scholar] [PubMed]
- Solaro, R.J.; Moir, A.J.G.; Perry, S.V. Phosphorylation of troponin I and the inotropic effect of adrenaline in the perfused rabbit heart. Nature 1976, 262, 615–617. [Google Scholar] [CrossRef] [PubMed]
- Marx, S.O.; Reiken, S.; Hisamatsu, Y.; Jayaraman, T.; Burkhoff, D.; Rosemblit, N.; Marks, A.R. PKA Phosphorylation Dissociates FKBP12.6 from the Calcium Release Channel (Ryanodine Receptor). Cell 2000, 101, 365–376. [Google Scholar] [CrossRef]
- Burgers, P.P.; van der Heyden, M.A.; Kok, B.; Heck, A.J.; Scholten, A. A systematic evaluation of protein kinase A-A-kinase anchoring protein interaction motifs. Biochemistry 2015, 54, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Welch, E.J.; Jones, B.W.; Scott, J.D. Networking with AKAPs: Context-dependent regulation of anchored enzymes. Mol. Interv. 2010, 10, 86–97. [Google Scholar] [CrossRef] [PubMed]
- Kinderman, F.S.; Kim, C.; von Daake, S.; Ma, Y.; Pham, B.Q.; Spraggon, G.; Xuong, N.H.; Jennings, P.A.; Taylor, S.S. A Dynamic Mechanism for AKAP Binding to RII Isoforms of cAMP-Dependent Protein Kinase. Mol. Cell 2006, 24, 397–408. [Google Scholar] [CrossRef] [PubMed]
- Maurice, D.H.; Ke, H.; Ahmad, F.; Wang, Y.; Chung, J.; Manganiello, V.C. Advances in targeting cyclic nucleotide phosphodiesterases. Nat. Rev. Drug Discov. 2014, 13, 290–314. [Google Scholar] [CrossRef] [PubMed]
- Akazawa, H.; Komuro, I. Roles of cardiac transcription factors in cardiac hypertrophy. Circ. Res. 2003, 92, 1079–1088. [Google Scholar] [CrossRef] [PubMed]
- Zippin, J.H.; Chen, Y.; Nahirney, P.; Kamenetsky, M.; Wuttke, M.S.; Fischman, D.A.; Levin, L.R.; Buck, J. Compartmentalization of bicarbonate-sensitive adenylyl cyclase in distinct signaling microdomains. FASEB J. 2003, 17, 82–84. [Google Scholar] [CrossRef] [PubMed]
- Zippin, J.H.; Farrell, J.; Huron, D.; Kamenetsky, M.; Hess, K.C.; Fischman, D.A.; Levin, L.R.; Buck, J. Bicarbonate-responsive “soluble” adenylyl cyclase defines a nuclear cAMP microdomain. J. Cell Biol. 2004, 164, 527–534. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, S.R.; Miyashiro, K.; Latt, H.; Ostrom, R.S.; Harvey, R.D. Compartmentalized cAMP responses to prostaglandin EP 2 receptor activation in human airway smooth muscle cells. Br. J. Pharmacol. 2017, 174, 2784–2796. [Google Scholar] [CrossRef] [PubMed]
- Sample, V.; DiPilato, L.M.; Yang, J.H.; Ni, Q.; Saucerman, J.J.; Zhang, J. Regulation of nuclear PKA revealed by spatiotemporal manipulation of cyclic AMP. Nat. Chem. Biol. 2012, 8, 375–382. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.H.; Polanowska-Grabowska, R.K.; Smith, J.S.; Shields, C.W.; Saucerman, J.J. PKA catalytic subunit compartmentation regulates contractile and hypertrophic responses to β-adrenergic signaling. J. Mol. Cell. Cardiol. 2014, 66, 83–93. [Google Scholar] [CrossRef] [PubMed]
- Saucerman, J.J.; Zhang, J.; Martin, J.C.; Peng, L.X.; Stenbit, A.E.; Tsien, R.Y.; McCulloch, A.D. Systems analysis of PKA-mediated phosphorylation gradients in live cardiac myocytes. Proc. Natl. Acad. Sci. USA 2006, 103, 12923–12928. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, S.R.; MacDougall, D.A.; Tyser, R.; Pugh, S.D.; Calaghan, S.C.; Harvey, R.D. Effects of cholesterol depletion on compartmentalized cAMP responses in adult cardiac myocytes. J. Mol. Cell. Cardiol. 2011, 50, 500–509. [Google Scholar] [CrossRef] [PubMed]
- Wright, P.T.; Nikolaev, V.O.; O’Hara, T.; Diakonov, I.; Bhargava, A.; Tokar, S.; Schobesberger, S.; Shevchuk, A.I.; Sikkel, M.B.; Wilkinson, R.; et al. Caveolin-3 regulates compartmentation of cardiomyocyte beta2-adrenergic receptor-mediated cAMP signaling. J. Mol. Cell. Cardiol. 2014, 67, 38–48. [Google Scholar] [CrossRef] [PubMed]
- Mika, D.; Richter, W.; Westenbroek, R.E.; Catterall, W.A.; Conti, M. PDE4B mediates local feedback regulation of 1-adrenergic cAMP signaling in a sarcolemmal compartment of cardiac myocytes. J. Cell Sci. 2014, 127, 1033–1042. [Google Scholar] [CrossRef] [PubMed]
- Doenst, T.; Nguyen, T.D.; Abel, E.D. Cardiac metabolism in heart failure: Implications beyond atp production. Circ. Res. 2013, 113, 709–724. [Google Scholar] [CrossRef] [PubMed]
- Neubauer, S. The Failing Heart—An Engine Out of Fuel. N. Engl. J. Med. 2007, 356, 1140–1151. [Google Scholar] [CrossRef] [PubMed]
- Rizzuto, R.; Simpson, A.W.M.; Brini, M.; Pozzan, T. Rapid changes of mitochondrial Ca2+ revealed by specifically targeted recombinant aequorin. Nature 1992, 358, 325–327. [Google Scholar] [CrossRef] [PubMed]
- Rizzuto, R.; Brini, M.; Murgia, M.; Pozzan, T. Microdomains with high Ca2+ close to IP3-sensitive channels that are sensed by neighboring mitochondria. Science 1993, 262, 744–747. [Google Scholar] [CrossRef] [PubMed]
- Rizzuto, R. Close Contacts with the Endoplasmic Reticulum as Determinants of Mitochondrial Ca2+ Responses. Science 1998, 280, 1763–1766. [Google Scholar] [CrossRef] [PubMed]
- Jouaville, L.S.; Pinton, P.; Bastianutto, C.; Rutter, G.A.; Rizzuto, R. Regulation of mitochondrial ATP synthesis by calcium: Evidence for a long-term metabolic priming. Proc. Natl. Acad. Sci. USA 1999, 96, 13807–13812. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Gong, Q.; Stice, J.P.; Knowlton, A.A. Mitochondrial OPA1, apoptosis, and heart failure. Cardiovasc. Res. 2009, 84, 91–99. [Google Scholar] [CrossRef] [PubMed]
- Cribbs, J.T.; Strack, S. Reversible phosphorylation of Drp1 by cyclic AMP-dependent protein kinase and calcineurin regulates mitochondrial fission and cell death. EMBO Rep. 2007, 8, 939–944. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Taylor, S.S. A molecular switch for targeting between endoplasmic reticulum (ER) and mitochondria: Conversion of a mitochondria-targeting element into an ER-targeting signal in DAKAP1. J. Biol. Chem. 2008, 283, 11743–11751. [Google Scholar] [CrossRef] [PubMed]
- Lefkimmiatis, K.; Leronni, D.; Hofer, A.M. The inner and outer compartments of mitochondria are sites of distinct cAMP/PKA signaling dynamics. J. Cell Biol. 2013, 202, 453–462. [Google Scholar] [CrossRef] [PubMed]
- Monterisi, S.; Lobo, M.J.; Livie, C.; Castle, J.C.; Weinberger, M.; Baillie, G.; Surdo, N.C.; Musheshe, N.; Stangherlin, A.; Gottlieb, E.; et al. PDE2A2 regulates mitochondria morphology and apoptotic cell death via local modulation of cAMP/PKA signalling. Elife 2017, 6. [Google Scholar] [CrossRef] [PubMed]
- Moss, R.L.; Razumova, M.; Fitzsimons, D.P. Myosin crossbridge activation of cardiac thin filaments: Implications for myocardial function in health and disease. Circ. Res. 2004, 94, 1290–1300. [Google Scholar] [CrossRef] [PubMed]
- Wehrens, X.H.T.; Lehnart, S.E.; Reiken, S.; Vest, J.A.; Wronska, A.; Marks, A.R. Ryanodine receptor/calcium release channel PKA phosphorylation: A critical mediator of heart failure progression. Proc. Natl. Acad. Sci. USA 2006, 103, 511–518. [Google Scholar] [CrossRef] [PubMed]
- Ceholski, D.K.; Trieber, C.A.; Holmes, C.F.B.; Young, H.S. Lethal, hereditary mutants of phospholamban elude phosphorylation by protein kinase A. J. Biol. Chem. 2012, 287, 26596–26605. [Google Scholar] [CrossRef] [PubMed]
- Piacentino, V.; Weber, C.R.; Chen, X.; Weisser-Thomas, J.; Margulies, K.B.; Bers, D.M.; Houser, S.R. Cellular basis of abnormal calcium transients of failing human ventricular myocytes. Circ. Res. 2003, 92, 651–658. [Google Scholar] [CrossRef] [PubMed]
- Barbagallo, F.; Xu, B.; Reddy, G.R.; West, T.; Wang, Q.; Fu, Q.; Li, M.; Shi, Q.; Ginsburg, K.S.; Ferrier, W.; et al. Genetically Encoded Biosensors Reveal PKA Hyperphosphorylation on the Myofilaments in Rabbit Heart Failure. Circ. Res. 2016, 119, 931–943. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Zhang, J.; Xiang, Y.K. FRET-based direct detection of dynamic protein kinase a activity on the sarcoplasmic reticulum in cardiomyocytes. Biochem. Biophys. Res. Commun. 2011, 404, 581–586. [Google Scholar] [CrossRef] [PubMed]
- Yin, X.; Cuello, F.; Mayr, U.; Hao, Z.; Hornshaw, M.; Ehler, E.; Avkiran, M.; Mayr, M. Proteomics analysis of the cardiac myofilament subproteome reveals dynamic alterations in phosphatase subunit distribution. Mol. Cell. Proteom. 2010, 9, 497–509. [Google Scholar] [CrossRef] [PubMed]
- Kensler, R.W.; Craig, R.; Moss, R.L. Phosphorylation of cardiac myosin binding protein C releases myosin heads from the surface of cardiac thick filaments. Proc. Natl. Acad. Sci. USA 2017, 114, E1355–E1364. [Google Scholar] [CrossRef] [PubMed]
- Ray, K.P.; England, P.J. Phosphorylation of the inhibitory subunit of troponin and its effect on the calcium dependence of cardiac myofibril adenosine triphosphatase. FEBS Lett. 1976, 70, 11–16. [Google Scholar] [CrossRef]
- Scott, J.D.; Santana, L.F. A-Kinase Anchoring Proteins: Getting to the heart of the matter. Circulation 2010, 121, 1264–1271. [Google Scholar] [CrossRef] [PubMed]
- Carr, D.W.; Hausken, Z.E.; Fraser, I.D.C.; Stofko-Hahn, R.E.; Scott, J.D. Association of the type II cAMP-dependent protein kinase with a human thyroid RII-anchoring protein: Cloning and characterization of the RII-binding domain. J. Biol. Chem. 1992, 267, 13376–13382. [Google Scholar] [PubMed]
- Carr, D.W.; Stofko-Hahn, R.E.; Fraser, I.D.C.; Bishop, S.M.; Acott, T.S.; Brennan, R.G.; Scott, J.D. Interaction of the regulatory subunit (RII) of cAMP-dependent protein kinase with RII-anchoring proteins occurs through an amphipathic helix binding motif. J. Biol. Chem. 1991, 266, 14188–14192. [Google Scholar] [PubMed]
- Zoccarato, A.; Surdo, N.C.; Aronsen, J.M.; Fields, L.A.; Mancuso, L.; Dodoni, G.; Stangherlin, A.; Livie, C.; Jiang, H.; Sin, Y.Y.; et al. Cardiac Hypertrophy Is Inhibited by a Local Pool of cAMP Regulated by Phosphodiesterase 2. Circ. Res. 2015, 117, 707–719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheridan, C.M.; Heist, E.K.; Beals, C.R.; Crabtree, G.R.; Gardner, P. Protein kinase A negatively modulates the nuclear accumulation of NF-ATc1 by priming for subsequent phosphorylation by glycogen synthase kinase-3. J. Biol. Chem. 2002, 277, 48664–48676. [Google Scholar] [CrossRef] [PubMed]
- Molkentin, J.D.; Lu, J.R.; Antos, C.L.; Markham, B.; Richardson, J.; Robbins, J.; Grant, S.R.; Olson, E.N. A calcineurin-dependent transcriptional pathway for cardiac hypertrophy. Cell 1998, 93, 215–228. [Google Scholar] [CrossRef]
- Stangherlin, A.; Gesellchen, F.; Zoccarato, A.; Terrin, A.; Fields, L.A.; Berrera, M.; Surdo, N.C.; Craig, M.A.; Smith, G.; Hamilton, G.; et al. CGMP signals modulate camp levels in a compartment-specific manner to regulate catecholamine-dependent signaling in cardiac myocytes. Circ. Res. 2011, 108, 929–939. [Google Scholar] [CrossRef] [PubMed]
- Mongillo, M.; Tocchetti, C.G.; Terrin, A.; Lissandron, V.; Cheung, Y.F.; Dostmann, W.R.; Pozzan, T.; Kass, D.A.; Paolocci, N.; Houslay, M.D.; et al. Compartmentalized phosphodiesterase-2 activity blunts β-adrenergic cardiac inotropy via an NO/cGMP-dependent pathway. Circ. Res. 2006, 98, 226–234. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Nooh, M.M.; Bahouth, S.W. Role of AKAP79/150 protein in β1-adrenergic receptor trafficking and signaling in mammalian cells. J. Biol. Chem. 2013, 288, 33797–33812. [Google Scholar] [CrossRef] [PubMed]
- Neary, M.T.; Ng, K.-E.; Ludtmann, M.H.R.; Hall, A.R.; Piotrowska, I.; Ong, S.-B.; Hausenloy, D.J.; Mohun, T.J.; Abramov, A.Y.; Breckenridge, R.A. Hypoxia signaling controls postnatal changes in cardiac mitochondrial morphology and function. J. Mol. Cell. Cardiol. 2014, 74, 340–352. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, T.; Martone, M.E.; Yu, Z.; Thor, A.; Doi, M.; Holst, M.J.; Ellisman, M.H.; Hoshijima, M. Three-dimensional electron microscopy reveals new details of membrane systems for Ca2+ signaling in the heart. J. Cell Sci. 2009, 122, 1005–1013. [Google Scholar] [CrossRef] [PubMed]
- Luther, P.K.; Winkler, H.; Taylor, K.; Zoghbi, M.E.; Craig, R.; Padron, R.; Squire, J.M.; Liu, J. Direct visualization of myosin-binding protein C bridging myosin and actin filaments in intact muscle. Proc. Natl. Acad. Sci. USA 2011, 108, 11423–11428. [Google Scholar] [CrossRef] [PubMed]
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schleicher, K.; Zaccolo, M. Using cAMP Sensors to Study Cardiac Nanodomains. J. Cardiovasc. Dev. Dis. 2018, 5, 17. https://doi.org/10.3390/jcdd5010017
Schleicher K, Zaccolo M. Using cAMP Sensors to Study Cardiac Nanodomains. Journal of Cardiovascular Development and Disease. 2018; 5(1):17. https://doi.org/10.3390/jcdd5010017
Chicago/Turabian StyleSchleicher, Katharina, and Manuela Zaccolo. 2018. "Using cAMP Sensors to Study Cardiac Nanodomains" Journal of Cardiovascular Development and Disease 5, no. 1: 17. https://doi.org/10.3390/jcdd5010017
APA StyleSchleicher, K., & Zaccolo, M. (2018). Using cAMP Sensors to Study Cardiac Nanodomains. Journal of Cardiovascular Development and Disease, 5(1), 17. https://doi.org/10.3390/jcdd5010017