
MVP-Associated Filamin A Mutations Affect FlnA-PTPN12 (PTP-PEST) Interactions

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Experimental Section
2.1. cDNAs, Mutagenesis, and Reagents
2.2. Cell Culture and Transfection
2.3. Migration Assays: Wound Healing Assays
2.4. Yeast Two-Hybrid Library Screening
2.5. Yeast Two-Hybrid Interaction Assays
2.6. Co-Immunoprecipitation and Immunoblotting
2.7. Pulldown Experiments
3. Results
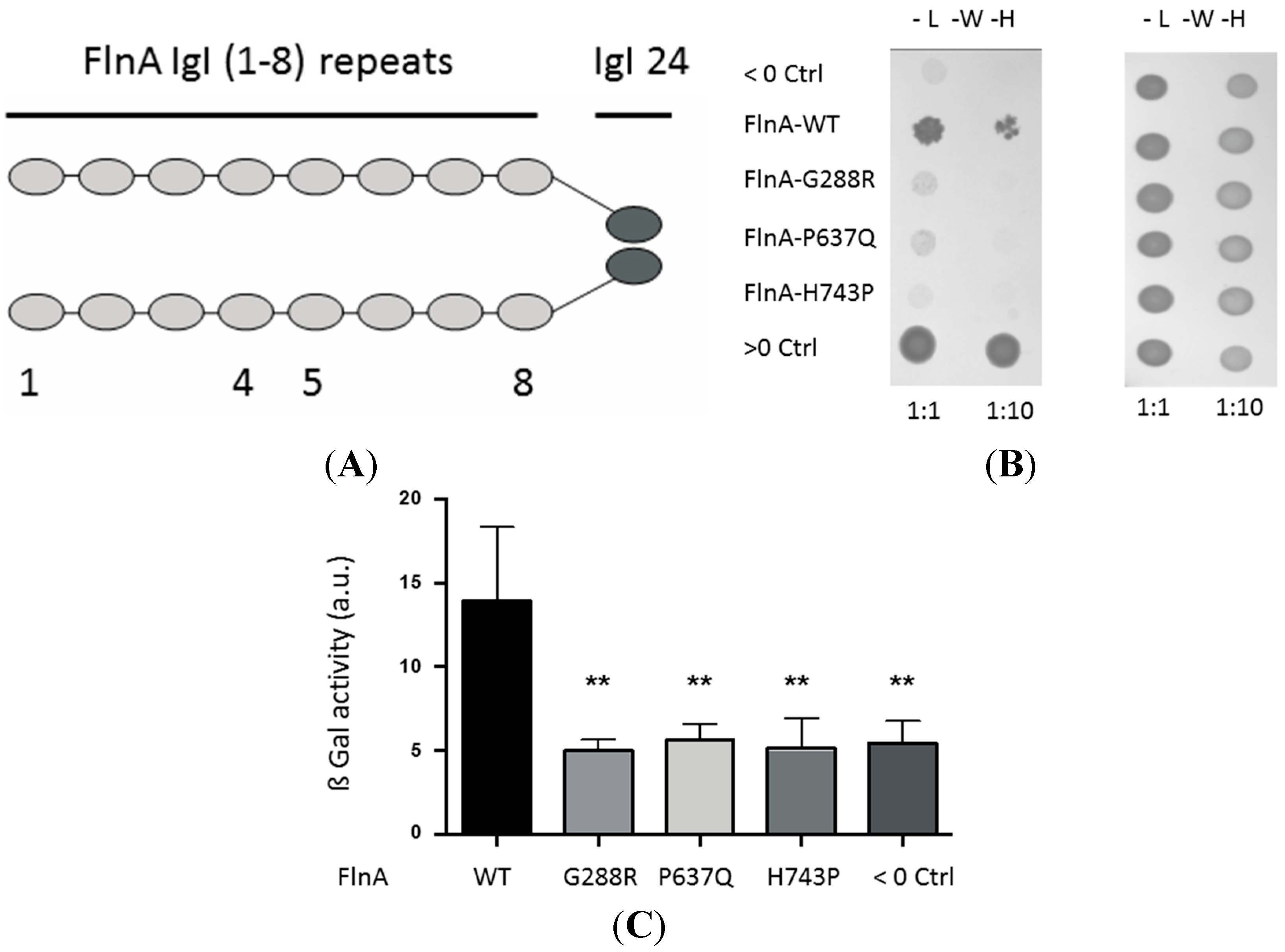
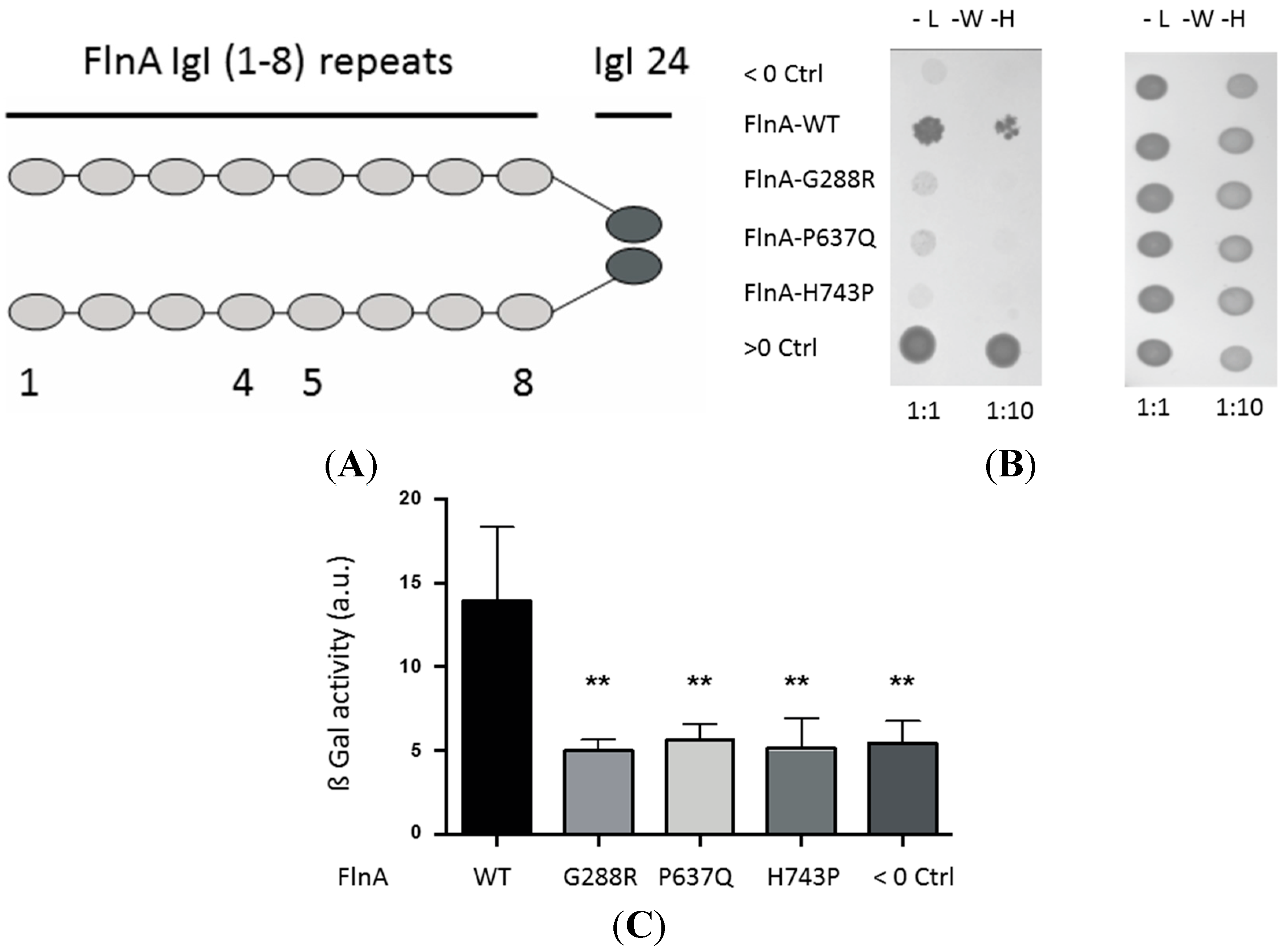
3.1. A Yeast Two-Hybrid Library Screen Identified PTPN12 as A FlnA Binding Partner
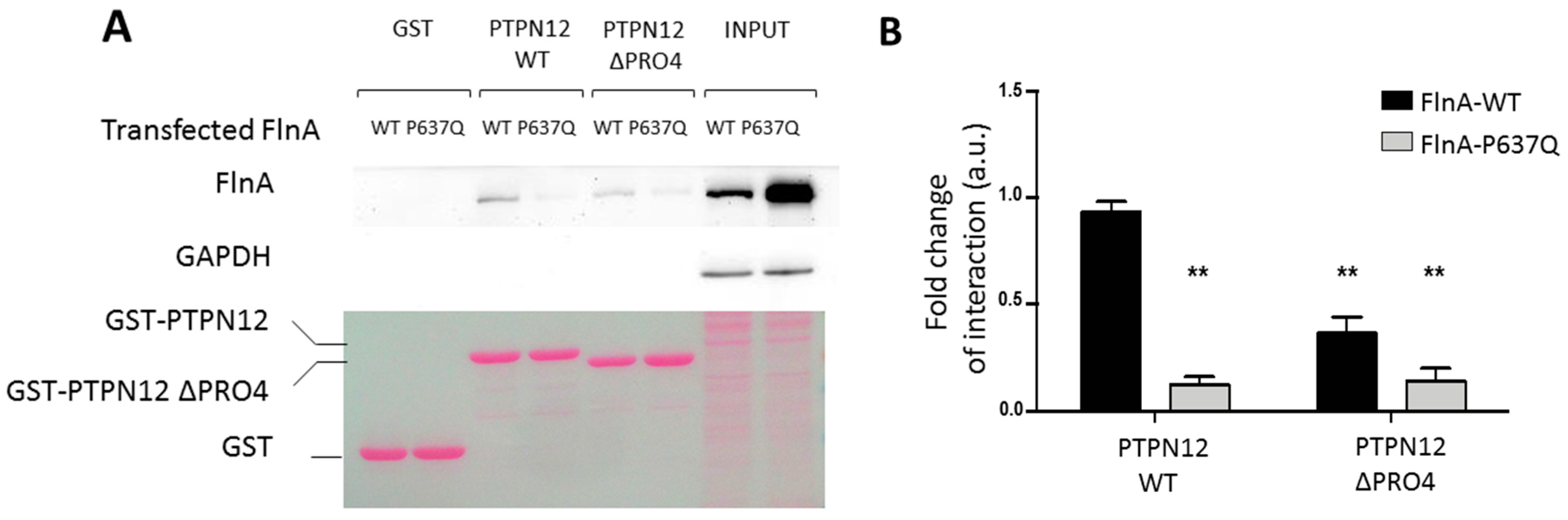
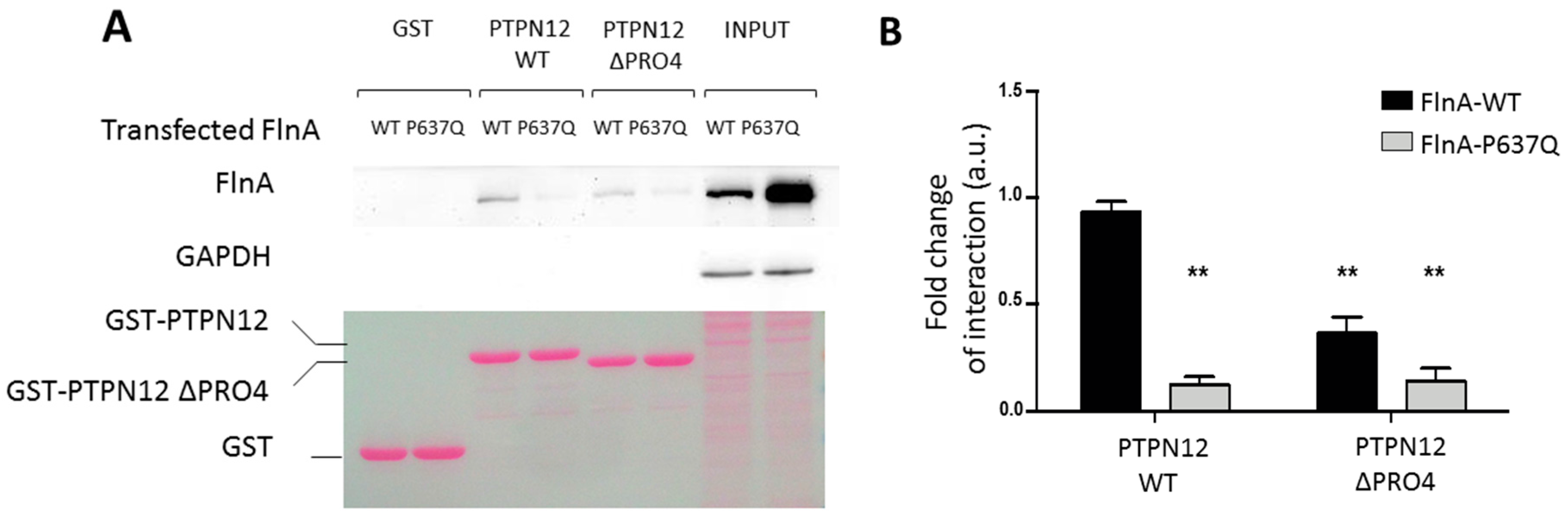
3.2. MVP-Associated FlnA Mutations Impede its Interactions with PTPN12
3.2.1. Yeast Two-Hybrid Assay
3.2.2. Pulldown and Co-Immunoprecipitation Assays
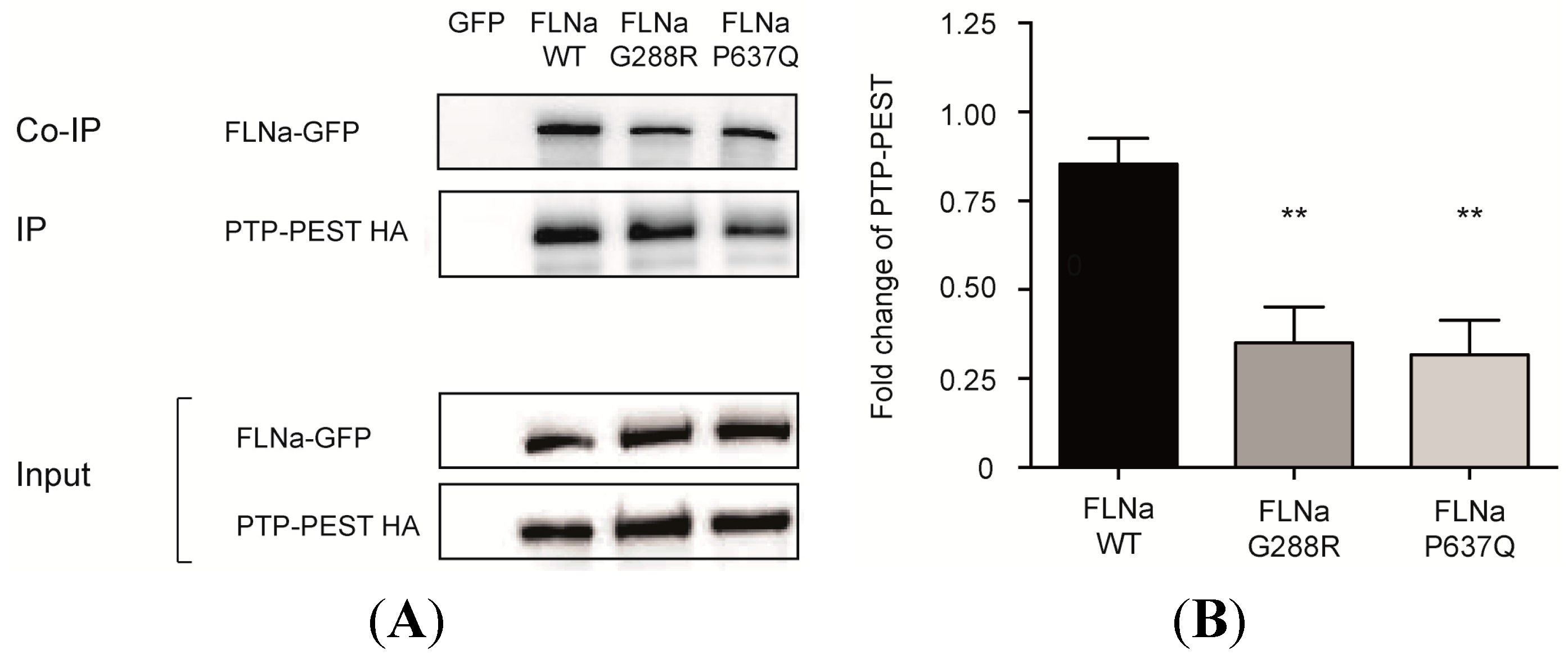
3.3. PTPN12-FlnA Interactions Participate to p190RhoGAP and Src Regulation
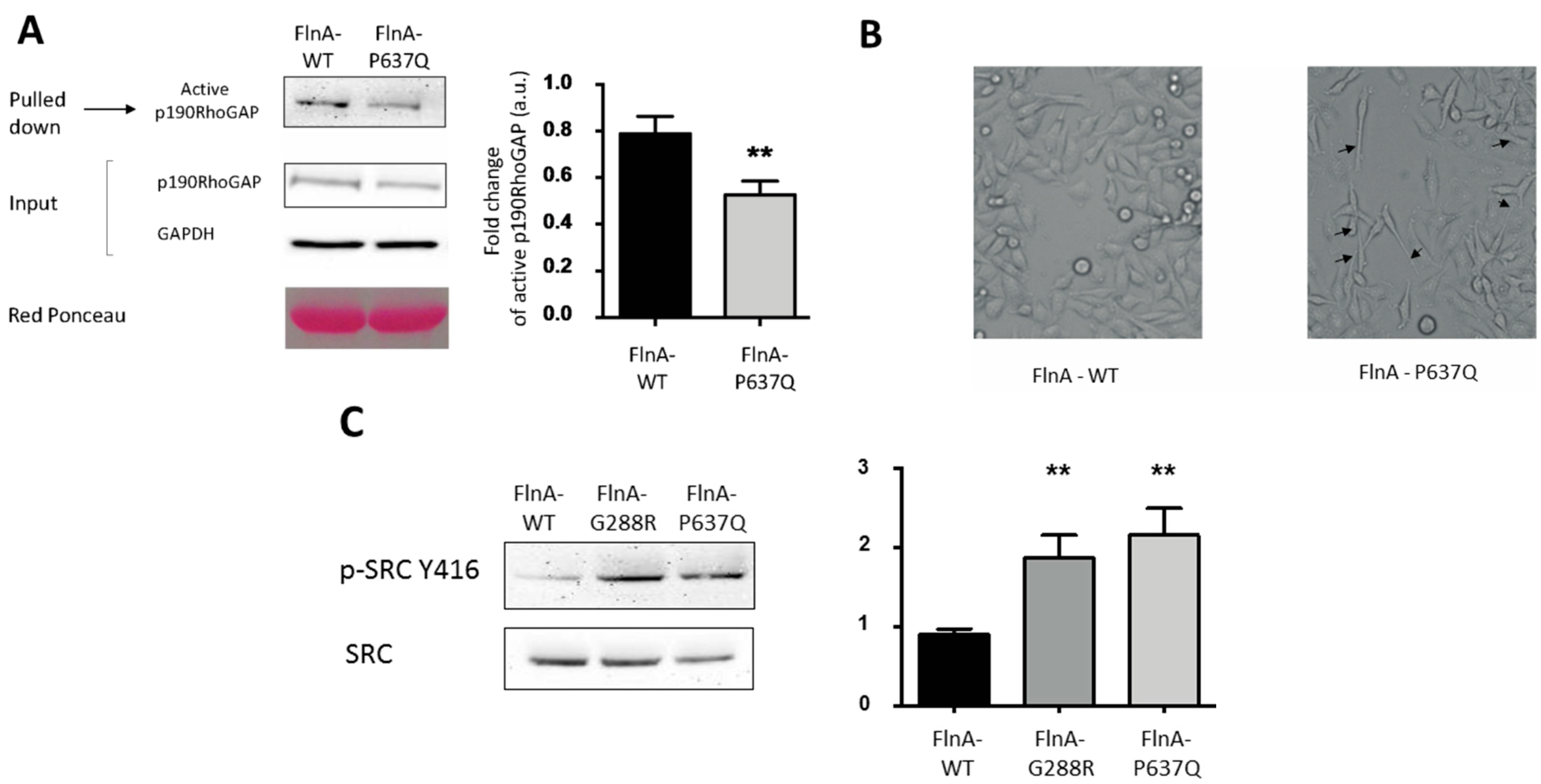
4. Discussion
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Delling, F.N.; Rong, J.; Larson, M.G.; Lehman, B.; Osypiuk, E.; Stantchev, P.; Slaugenhaupt, S.A.; Benjamin, E.J.; Levine, R.A.; Vasan, R.S. Familial clustering of mitral valve prolapse in the community. Circulation 2015, 131, 263–268. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.; Godfrey, M.; Vitale, E.; Hori, H.; Mattei, M.G.; Sarfarazi, M.; Tsipouras, P.; Ramirez, F.; Hollister, D.W. Linkage of marfan syndrome and a phenotypically related disorder to two different fibrillin genes. Nature 1991, 352, 330–334. [Google Scholar] [CrossRef] [PubMed]
- Singh, K.K.; Rommel, K.; Mishra, A.; Karck, M.; Haverich, A.; Schmidtke, J.; Arslan-Kirchner, M. TGFBR1 and TGFBR2 mutations in patients with features of marfan syndrome and loeys-dietz syndrome. Hum. Mutat. 2006, 27, 770–777. [Google Scholar] [CrossRef] [PubMed]
- Malfait, F.; Coucke, P.; Symoens, S.; Loeys, B.; Nuytinck, L.; De Paepe, A. The molecular basis of classic ehlers-danlos syndrome: A comprehensive study of biochemical and molecular findings in 48 unrelated patients. Hum. Mutat. 2005, 25, 28–37. [Google Scholar] [CrossRef] [PubMed]
- Kyndt, F.; Gueffet, J.P.; Probst, V.; Jaafar, P.; Legendre, A.; Le Bouffant, F.; Toquet, C.; Roy, E.; McGregor, L.; Lynch, S.A.; et al. Mutations in the gene encoding filamin a as a cause for familial cardiac valvular dystrophy. Circulation 2007, 115, 40–49. [Google Scholar] [CrossRef] [PubMed]
- Kyndt, F.; Schott, J.J.; Trochu, J.N.; Baranger, F.; Herbert, O.; Scott, V.; Fressinaud, E.; David, A.; Moisan, J.P.; Bouhour, J.B.; et al. Mapping of X-linked myxomatous valvular dystrophy to chromosome xq28. Am. J. Hum. Genet. 1998, 62, 627–632. [Google Scholar] [CrossRef] [PubMed]
- Holm, T.M.; Habashi, J.P.; Doyle, J.J.; Bedja, D.; Chen, Y.; van Erp, C.; Lindsay, M.E.; Kim, D.; Schoenhoff, F.; Cohn, R.D.; et al. Noncanonical TGFβ signaling contributes to aortic aneurysm progression in marfan syndrome mice. Science 2011, 332, 358–361. [Google Scholar] [CrossRef] [PubMed]
- Lacro, R.V.; Dietz, H.C.; Sleeper, L.A.; Yetman, A.T.; Bradley, T.J.; Colan, S.D.; Pearson, G.D.; Selamet Tierney, E.S.; Levine, J.C.; Atz, A.M.; et al. Atenolol versus losartan in children and young adults with marfan's syndrome. N. Engl. J. Med. 2014, 371, 2061–2071. [Google Scholar] [CrossRef] [PubMed]
- Robertson, S.P.; Jenkins, Z.A.; Morgan, T.; Ades, L.; Aftimos, S.; Boute, O.; Fiskerstrand, T.; Garcia-Minaur, S.; Grix, A.; Green, A.; et al. Frontometaphyseal dysplasia: Mutations in flna and phenotypic diversity. Am. J. Med. Genet. A 2006, 140, 1726–1736. [Google Scholar] [CrossRef] [PubMed]
- Sheen, V.L.; Dixon, P.H.; Fox, J.W.; Hong, S.E.; Kinton, L.; Sisodiya, S.M.; Duncan, J.S.; Dubeau, F.; Scheffer, I.E.; Schachter, S.C.; et al. Mutations in the X-linked filamin 1 gene cause periventricular nodular heterotopia in males as well as in females. Hum. Mol. Genet. 2001, 10, 1775–1783. [Google Scholar] [CrossRef] [PubMed]
- Norris, R.A.; Moreno-Rodriguez, R.; Wessels, A.; Merot, J.; Bruneval, P.; Chester, A.H.; Yacoub, M.H.; Hagege, A.; Slaugenhaupt, S.A.; Aikawa, E.; et al. Expression of the familial cardiac valvular dystrophy gene, filamin-a, during heart morphogenesis. Dev. Dyn. 2010, 239, 2118–2127. [Google Scholar] [CrossRef] [PubMed]
- Sauls, K.; de Vlaming, A.; Harris, B.S.; Williams, K.; Wessels, A.; Levine, R.A.; Slaugenhaupt, S.A.; Goodwin, R.L.; Pavone, L.M.; Merot, J.; et al. Developmental basis for filamin-a-associated myxomatous mitral valve disease. Cardiovasc. Res. 2012, 96, 109–119. [Google Scholar] [CrossRef] [PubMed]
- Stossel, T.P.; Condeelis, J.; Cooley, L.; Hartwig, J.H.; Noegel, A.; Schleicher, M.; Shapiro, S.S. Filamins as integrators of cell mechanics and signalling. Nat. Rev. Mol. Cell. Biol. 2001, 2, 138–145. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Walsh, C.A. The many faces of filamin: A versatile molecular scaffold for cell motility and signalling. Nat. Cell. Biol. 2004, 6, 1034–1038. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, F.; Osborn, T.M.; Hartemink, C.A.; Hartwig, J.H.; Stossel, T.P. Structural basis of filamin a functions. J. Cell. Biol. 2007, 179, 1011–1025. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, F.; Stossel, T.P.; Hartwig, J.H. The filamins: Organizers of cell structure and function. Cell. Adh. Migr. 2011, 5, 160–169. [Google Scholar] [CrossRef] [PubMed]
- Zhou, A.X.; Hartwig, J.H.; Akyurek, L.M. Filamins in cell signaling, transcription and organ development. Trends Cell. Biol. 2010, 20, 113–123. [Google Scholar] [CrossRef] [PubMed]
- Gawecka, J.E.; Griffiths, G.S.; Ek-Rylander, B.; Ramos, J.W.; Matter, M.L. R-ras regulates migration through an interaction with filamin a in melanoma cells. PLoS ONE 2010, 5, e11269. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Nakamura, F.; Lee, W.; Hong, C.; Perez-Sala, D.; McCulloch, C.A. Regulation of cell adhesion to collagen via β1 integrins is dependent on interactions of filamin a with vimentin and protein kinase c epsilon. Exp. Cell Res. 2010, 316, 1829–1844. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Lu, Z. Regulation of tumor cell migration by protein tyrosine phosphatase (PTP)-proline-, glutamate-, serine-,and threonine-rich sequence (PEST). Chin. J. Cancer 2013, 32, 75–83. [Google Scholar] [CrossRef] [PubMed]
- Rhee, I.; Zhong, M.C.; Reizis, B.; Cheong, C.; Veillette, A. Control of dendritic cell migration, t cell-dependent immunity, and autoimmunity by protein tyrosine phosphatase ptpn12 expressed in dendritic cells. Mol. Cell. Biol. 2014, 34, 888–899. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.S.; Cheerathodi, M.; Chaki, S.P.; Reyes, S.B.; Zheng, Y.; Lu, Z.; DerMardirossian, C.; Lacy-Hulbert, A.; Rivera, G.M.; McCarty, J.H. Ptp-pest and β8 integrin control spatiotemporal patterns of rhogdi1 activation in migrating cells. Mol. Cell. Biol. 2015, 35, 1401–1413. [Google Scholar] [CrossRef] [PubMed]
- Boudin, H.; Doan, A.; Xia, J.; Shigemoto, R.; Huganir, R.L.; Worley, P.; Craig, A.M. Presynaptic clustering of mglur7a requires the pick1 pdz domain binding site. Neuron 2000, 28, 485–497. [Google Scholar] [CrossRef]
- Duval, D.; Lardeux, A.; Le Tourneau, T.; Norris, R.A.; Markwald, R.R.; Sauzeau, V.; Probst, V.; Le Marec, H.; Levine, R.; Schott, J.J.; et al. Valvular dystrophy associated filamin a mutations reveal a new role of its first repeats in small-gtpase regulation. Biochim. Biophys. Acta 2014, 1843, 234–244. [Google Scholar] [CrossRef] [PubMed]
- Fromont-Racine, M.; Rain, J.C.; Legrain, P. Toward a functional analysis of the yeast genome through exhaustive two-hybrid screens. Nature Genet. 1997, 16, 277–282. [Google Scholar] [CrossRef] [PubMed]
- Formstecher, E.; Aresta, S.; Collura, V.; Hamburger, A.; Meil, A.; Trehin, A.; Reverdy, C.; Betin, V.; Maire, S.; Brun, C.; et al. Protein interaction mapping: A drosophila case study. Genome Res. 2005, 15, 376–384. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, F.; Heikkinen, O.; Pentikainen, O.T.; Osborn, T.M.; Kasza, K.E.; Weitz, D.A.; Kupiainen, O.; Permi, P.; Kilpelainen, I.; Ylanne, J.; et al. Molecular basis of filamin a-filgap interaction and its impairment in congenital disorders associated with filamin a mutations. PLoS ONE 2009, 4, e4928. [Google Scholar] [CrossRef] [PubMed]
- Playford, M.P.; Lyons, P.D.; Sastry, S.K.; Schaller, M.D. Identification of a filamin docking site on ptp-pest. J. Biol. Chem. 2006, 281, 34104–34112. [Google Scholar] [CrossRef] [PubMed]
- Sastry, S.K.; Rajfur, Z.; Liu, B.P.; Cote, J.F.; Tremblay, M.L.; Burridge, K. Ptp-pest couples membrane protrusion and tail retraction via VAV2 and p190RhoGAP. J. Biol. Chem. 2006, 281, 11627–11636. [Google Scholar] [CrossRef] [PubMed]
- Guilluy, C.; Dubash, A.D.; Garcia-Mata, R. Analysis of rhoa and rho gef activity in whole cells and the cell nucleus. Nat. Protoc. 2011, 6, 2050–2060. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Yang, W.; Xia, Y.; Hawke, D.; Liu, D.X.; Lu, Z. Ras-induced and extracellular signal-regulated kinase 1 and 2 phosphorylation-dependent isomerization of protein tyrosine phosphatase (ptp)-pest by pin1 promotes fak dephosphorylation by ptp-pest. Mol. Cell. Biol. 2011, 31, 4258–4269. [Google Scholar] [CrossRef] [PubMed]
- Falet, H.; Pollitt, A.Y.; Begonja, A.J.; Weber, S.E.; Duerschmied, D.; Wagner, D.D.; Watson, S.P.; Hartwig, J.H. A novel interaction between flna and syk regulates platelet itam-mediated receptor signaling and function. J. Exp. Med. 2010, 207, 1967–1979. [Google Scholar] [CrossRef] [PubMed]
- Ithychanda, S.S.; Hsu, D.; Li, H.; Yan, L.; Liu, D.D.; Das, M.; Plow, E.F.; Qin, J. Identification and characterization of multiple similar ligand-binding repeats in filamin: Implication on filamin-mediated receptor clustering and cross-talk. J. Biol. Chem. 2009, 284, 35113–35121. [Google Scholar] [CrossRef] [PubMed]
- Heikkinen, O.K.; Ruskamo, S.; Konarev, P.V.; Svergun, D.I.; Iivanainen, T.; Heikkinen, S.M.; Permi, P.; Koskela, H.; Kilpelainen, I.; Ylanne, J. Atomic structures of two novel immunoglobulin-like domain pairs in the actin cross-linking protein filamin. J. Biol. Chem. 2009, 284, 25450–25458. [Google Scholar] [CrossRef] [PubMed]
- Sethi, R.; Ylanne, J. Small-angle x-ray scattering reveals compact domain-domain interactions in the N-terminal region of filamin c. PLoS ONE 2014, 9, e107457. [Google Scholar] [CrossRef] [PubMed]
- Sethi, R.; Seppala, J.; Tossavainen, H.; Ylilauri, M.; Ruskamo, S.; Pentikainen, O.T.; Pentikainen, U.; Permi, P.; Ylanne, J. A novel structural unit in the N-terminal region of filamins. J. Biol. Chem. 2014, 289, 8588–8598. [Google Scholar] [CrossRef] [PubMed]
- Sirois, J.; Cote, J.F.; Charest, A.; Uetani, N.; Bourdeau, A.; Duncan, S.A.; Daniels, E.; Tremblay, M.L. Essential function of ptp-pest during mouse embryonic vascularization, mesenchyme formation, neurogenesis and early liver development. Mech. Dev. 2006, 123, 869–880. [Google Scholar] [CrossRef] [PubMed]
- Souza, C.M.; Davidson, D.; Rhee, I.; Gratton, J.P.; Davis, E.C.; Veillette, A. The phosphatase ptp-pest/ptpn12 regulates endothelial cell migration and adhesion, but not permeability, and controls vascular development and embryonic viability. J. Biol. Chem. 2012, 287, 43180–43190. [Google Scholar] [CrossRef] [PubMed]
- Angers-Loustau, A.; Cote, J.F.; Charest, A.; Dowbenko, D.; Spencer, S.; Lasky, L.A.; Tremblay, M.L. Protein tyrosine phosphatase-pest regulates focal adhesion disassembly, migration, and cytokinesis in fibroblasts. J. Cell. Biol. 1999, 144, 1019–1031. [Google Scholar] [CrossRef] [PubMed]
- Ohta, Y.; Hartwig, J.H.; Stossel, T.P. Filgap, a rho- and rock-regulated gap for rac binds filamin a to control actin remodelling. Nat. Cell. Biol. 2006, 8, 803–814. [Google Scholar] [CrossRef] [PubMed]
- Ehrlicher, A.J.; Nakamura, F.; Hartwig, J.H.; Weitz, D.A.; Stossel, T.P. Mechanical strain in actin networks regulates filgap and integrin binding to filamin A. Nature 2011, 478, 260–263. [Google Scholar] [CrossRef] [PubMed]
- Razinia, Z.; Makela, T.; Ylanne, J.; Calderwood, D.A. Filamins in mechanosensing and signaling. Annu. Rev. Biophys. 2012, 41, 227–246. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Duval, D.; Labbé, P.; Bureau, L.; Tourneau, T.L.; Norris, R.A.; Markwald, R.R.; Levine, R.; Schott, J.-J.; Mérot, J. MVP-Associated Filamin A Mutations Affect FlnA-PTPN12 (PTP-PEST) Interactions. J. Cardiovasc. Dev. Dis. 2015, 2, 233-247. https://doi.org/10.3390/jcdd2030233
Duval D, Labbé P, Bureau L, Tourneau TL, Norris RA, Markwald RR, Levine R, Schott J-J, Mérot J. MVP-Associated Filamin A Mutations Affect FlnA-PTPN12 (PTP-PEST) Interactions. Journal of Cardiovascular Development and Disease. 2015; 2(3):233-247. https://doi.org/10.3390/jcdd2030233
Chicago/Turabian StyleDuval, Damien, Pauline Labbé, Léa Bureau, Thierry Le Tourneau, Russell A. Norris, Roger R. Markwald, Robert Levine, Jean-Jacques Schott, and Jean Mérot. 2015. "MVP-Associated Filamin A Mutations Affect FlnA-PTPN12 (PTP-PEST) Interactions" Journal of Cardiovascular Development and Disease 2, no. 3: 233-247. https://doi.org/10.3390/jcdd2030233
APA StyleDuval, D., Labbé, P., Bureau, L., Tourneau, T. L., Norris, R. A., Markwald, R. R., Levine, R., Schott, J.-J., & Mérot, J. (2015). MVP-Associated Filamin A Mutations Affect FlnA-PTPN12 (PTP-PEST) Interactions. Journal of Cardiovascular Development and Disease, 2(3), 233-247. https://doi.org/10.3390/jcdd2030233