Mast Cells in Cardiac Remodeling: Focus on the Right Ventricle
 ,
,  and
and 
Abstract
1. Introduction
2. Right Ventricular Remodeling
3. Mast Cell Biology
4. Mast Cell-Deficient Lines to Study Cardiac Physiology and Remodeling
5. Mast Cells in Healthy Hearts
6. Mast Cells in Right Ventricular Physiology and Remodeling
7. Mechanisms of Mast Cell-Mediated Effects on the Heart
7.1. Effects of Mast Cells on Cardiomyocytes
7.2. Mast Cells and Extracellular Matrix Modulation
7.3. Mast Cells and Myocardial Vascularization
7.4. Mast Cells and Myocardial Inflammation
8. Mast Cells as a Therapeutic Target
9. Conclusions and Future Directions
- (1)
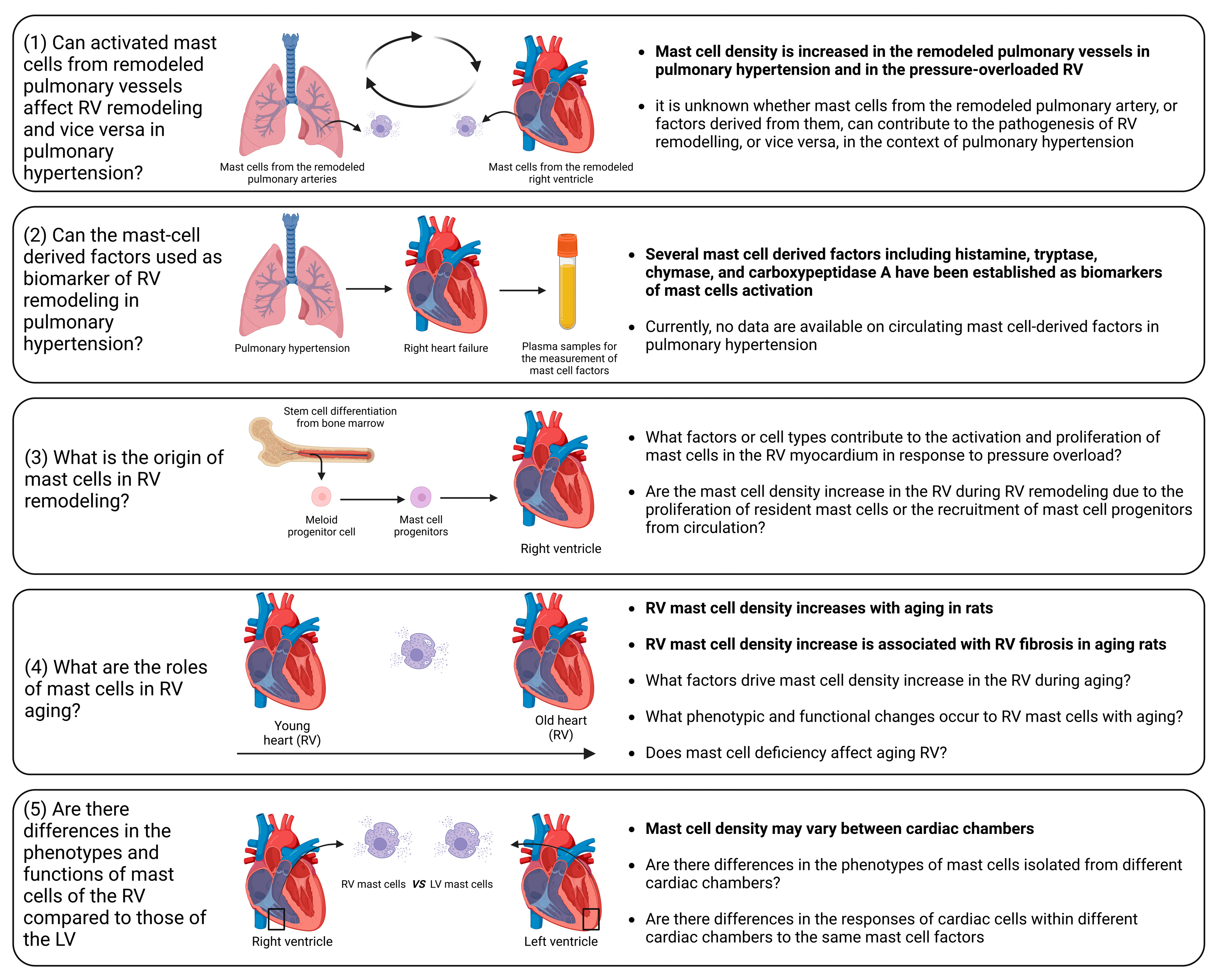
- Influence of activated mast cells from remodeled pulmonary vessels on RV remodeling and vice versa in pulmonary hypertension. It is conceivable that various factors released by activated mast cells in remodeled pulmonary arteries may be released into the circulation and transported to the RV myocardium. These factors can potentially alter the responses of the RV to pressure overload. Additionally, there is evidence of increased mast cell activation in remodeled pulmonary arteries in pulmonary hypertension [193,194,195]. Similarly, it can be postulated that the release of various factors from activated mast cells within the RV myocardium may reach the pulmonary vasculature, where they could exacerbate the remodeling processes.
- (2)
- Circulating mast-cell-derived factors as biomarkers of RV remodeling. Mediators released by activated mast cells, including histamine, tryptase, chymase, and carboxypeptidase A, can be measured in the systemic circulation and have the potential to serve as markers of mast cell activation in a number of conditions [196,197].
- (3)
- Origin of mast cells in the remodeled RV. It remains to be elucidated whether the increase in mast cell density is caused by the proliferation of resident mast cells or the recruitment of mast cell progenitors from the circulation. To address this issue, reconstitution experiments with bone marrow-derived mast cells in mast cell-deficient mice subjected to PAB can be performed.
- (4)
- Role of mast cells in RV aging. A correlation between age-related myocardial fibrosis and the density of mast cells has been previously revealed. However, there are still unanswered questions regarding mast cell-derived factors that define RV myocardial fibrosis during aging. Furthermore, which specific factors drive the increase in mast cell density in the RV during aging and what are the associated phenotypic and functional changes in RV mast cells? What are the consequences of mast cell deficiency on healthy RV aging? To date, it remains uncertain whether mast cell-deficient animals (mice or rats) maintain healthy aging of the RV.
- (5)
- What factors govern mast cell activation and proliferation in the context of RV remodeling? Although pressure overload is the main cause of mast cell activation and proliferation during cardiac remodeling, identifying the key factors that regulate this process could offer a means for scientists to prevent the onset of mast cell activation by inhibiting upstream triggers. Mast cell activation may occur due to direct mechanical strain, as previous research has indicated mast cells sense the mechanical properties of their microenvironment [198]. Another possible scenario is that cardiomyocytes may release specific mediators during the initial phases in response to pathological stimuli, promoting mast cell activation and growth. Ultimately, exploring this issue could bring us closer to understanding the mechanisms behind the disease and developing pharmacological treatments.
- (6)
- Differential impact of mast cell-derived factors on the RV and left ventricle. It is unclear which of the factors released by mast cells might have RV-specific effects in comparison to the left ventricle. It is unclear whether the effects of mast cell-derived factors differ between cardiac chambers due to compartment-specific differences in mast cell phenotypes or due to the chamber-specific phenotypes of cardiac cells. These differences in the cells targeted by mast cell factors might be partially explained by variations in receptor density or in the activated signaling pathways.
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Cohn, J.N.; Ferrari, R.; Sharpe, N. Cardiac remodeling—Concepts and clinical implications: A consensus paper from an international forum on cardiac remodeling. Behalf of an International Forum on Cardiac Remodeling. J. Am. Coll. Cardiol. 2000, 35, 569–582. [Google Scholar] [CrossRef]
- Azevedo, P.S.; Polegato, B.F.; Minicucci, M.F.; Paiva, S.A.; Zornoff, L.A. Cardiac Remodeling: Concepts, Clinical Impact, Pathophysiological Mechanisms and Pharmacologic Treatment. Arq. Bras. Cardiol. 2016, 106, 62–69. [Google Scholar] [CrossRef] [PubMed]
- Pazos-López, P.; Peteiro-Vázquez, J.; Carcía-Campos, A.; García-Bueno, L.; de Torres, J.P.; Castro-Beiras, A. The causes, consequences, and treatment of left or right heart failure. Vasc Health Risk Manag 2011, 7, 237–254. [Google Scholar] [CrossRef] [PubMed]
- Shah, A.; Gandhi, D.; Srivastava, S.; Shah, K.J.; Mansukhani, R. Heart Failure: A Class Review of Pharmacotherapy. P&T 2017, 42, 464–472. [Google Scholar]
- Iacoviello, M.; Palazzuoli, A.; Gronda, E. Recent advances in pharmacological treatment of heart failure. Eur. J. Clin. Investig. 2021, 51, e13624. [Google Scholar] [CrossRef] [PubMed]
- Ishii, H.; Amano, T.; Matsubara, T.; Murohara, T. Pharmacological intervention for prevention of left ventricular remodeling and improving prognosis in myocardial infarction. Circulation 2008, 118, 2710–2718. [Google Scholar] [CrossRef] [PubMed]
- Kim, G.H.; Uriel, N.; Burkhoff, D. Reverse remodelling and myocardial recovery in heart failure. Nat. Rev. Cardiol. 2018, 15, 83–96. [Google Scholar] [CrossRef]
- McDonagh, T.A.; Metra, M.; Adamo, M.; Gardner, R.S.; Baumbach, A.; Böhm, M.; Burri, H.; Butler, J.; Čelutkienė, J.; Chioncel, O.; et al. 2021 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure: Developed by the Task Force for the diagnosis and treatment of acute and chronic heart failure of the European Society of Cardiology (ESC). With the special contribution of the Heart Failure Association (HFA) of the ESC. Eur. J. Heart Fail. 2022, 24, 4–131. [Google Scholar] [CrossRef]
- Ameri, P.; Bertero, E.; Meliota, G.; Cheli, M.; Canepa, M.; Brunelli, C.; Balbi, M. Neurohormonal activation and pharmacological inhibition in pulmonary arterial hypertension and related right ventricular failure. Heart Fail. Rev. 2016, 21, 539–547. [Google Scholar] [CrossRef] [PubMed]
- Klinke, A.; Schubert, T.; Müller, M.; Legchenko, E.; Zelt, J.G.E.; Shimauchi, T.; Napp, L.C.; Rothman, A.M.K.; Bonnet, S.; Stewart, D.J.; et al. Emerging therapies for right ventricular dysfunction and failure. Cardiovasc Diagn Ther 2020, 10, 1735–1767. [Google Scholar] [CrossRef] [PubMed]
- Handoko, M.L.; de Man, F.S.; Allaart, C.P.; Paulus, W.J.; Westerhof, N.; Vonk-Noordegraaf, A. Perspectives on novel therapeutic strategies for right heart failure in pulmonary arterial hypertension: Lessons from the left heart. Eur. Respir. Rev. 2010, 19, 72–82. [Google Scholar] [CrossRef]
- Konstam, M.A.; Kiernan, M.S.; Bernstein, D.; Bozkurt, B.; Jacob, M.; Kapur, N.K.; Kociol, R.D.; Lewis, E.F.; Mehra, M.R.; Pagani, F.D.; et al. Evaluation and Management of Right-Sided Heart Failure: A Scientific Statement From the American Heart Association. Circulation 2018, 137, e578–e622. [Google Scholar] [CrossRef]
- Amsallem, M.; Mercier, O.; Kobayashi, Y.; Moneghetti, K.; Haddad, F. Forgotten No More: A Focused Update on the Right Ventricle in Cardiovascular Disease. JACC Heart Fail 2018, 6, 891–903. [Google Scholar] [CrossRef]
- Rubin, L.J. The adrenergic nervous system as a therapeutic target in pulmonary arterial hypertension: A cautionary tale. Eur. Respir. J. 2016, 48, 617–618. [Google Scholar] [CrossRef]
- Taverne, Y.; Sadeghi, A.; Bartelds, B.; Bogers, A.; Merkus, D. Right ventricular phenotype, function, and failure: A journey from evolution to clinics. Heart Fail. Rev. 2021, 26, 1447–1466. [Google Scholar] [CrossRef]
- Reddy, S.; Bernstein, D. Molecular Mechanisms of Right Ventricular Failure. Circulation 2015, 132, 1734–1742. [Google Scholar] [CrossRef] [PubMed]
- Haque, Z.K.; Wang, D.Z. How cardiomyocytes sense pathophysiological stresses for cardiac remodeling. Cell. Mol. Life Sci. 2017, 74, 983–1000. [Google Scholar] [CrossRef] [PubMed]
- Scarborough, E.A.; Uchida, K.; Vogel, M.; Erlitzki, N.; Iyer, M.; Phyo, S.A.; Bogush, A.; Kehat, I.; Prosser, B.L. Microtubules orchestrate local translation to enable cardiac growth. Nat. Commun. 2021, 12, 1547. [Google Scholar] [CrossRef]
- Frangogiannis, N.G. The extracellular matrix in myocardial injury, repair, and remodeling. J. Clin. Investig. 2017, 127, 1600–1612. [Google Scholar] [CrossRef] [PubMed]
- Segers, V.F.M.; Brutsaert, D.L.; De Keulenaer, G.W. Cardiac Remodeling: Endothelial Cells Have More to Say Than Just NO. Front. Physiol. 2018, 9, 382. [Google Scholar] [CrossRef]
- Kologrivova, I.; Shtatolkina, M.; Suslova, T.; Ryabov, V. Cells of the Immune System in Cardiac Remodeling: Main Players in Resolution of Inflammation and Repair After Myocardial Infarction. Front. Immunol. 2021, 12, 664457. [Google Scholar] [CrossRef]
- Sydykov, A.; Mamazhakypov, A.; Petrovic, A.; Kosanovic, D.; Sarybaev, A.S.; Weissmann, N.; Ghofrani, H.A.; Schermuly, R.T. Inflammatory Mediators Drive Adverse Right Ventricular Remodeling and Dysfunction and Serve as Potential Biomarkers. Front. Physiol. 2018, 9, 609. [Google Scholar] [CrossRef] [PubMed]
- Tello, K.; Naeije, R.; de Man, F.; Guazzi, M. Pathophysiology of the right ventricle in health and disease: An update. Cardiovasc. Res. 2023, 119, 1891–1904. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y.K.; Ma, J.S. Right ventricular failure in congenital heart disease. Korean J. Pediatr. 2013, 56, 101–106. [Google Scholar] [CrossRef] [PubMed]
- Prisco, S.Z.; Thenappan, T.; Prins, K.W. Treatment Targets for Right Ventricular Dysfunction in Pulmonary Arterial Hypertension. JACC Basic Transl. Sci. 2020, 5, 1244–1260. [Google Scholar] [CrossRef]
- Shahar, K.; Darawsha, W.; Yalonetsky, S.; Lessick, J.; Kapeliovich, M.; Dragu, R.; Mutlak, D.; Reisner, S.; Agmon, Y.; Aronson, D. Time Dependence of the Effect of Right Ventricular Dysfunction on Clinical Outcomes After Myocardial Infarction: Role of Pulmonary Hypertension. J. Am. Heart Assoc. 2016, 5. [Google Scholar] [CrossRef]
- Dini, F.L.; Pugliese, N.R.; Ameri, P.; Attanasio, U.; Badagliacca, R.; Correale, M.; Mercurio, V.; Tocchetti, C.G.; Agostoni, P.; Palazzuoli, A. Right ventricular failure in left heart disease: From pathophysiology to clinical manifestations and prognosis. Heart Fail. Rev. 2023, 28, 757–766. [Google Scholar] [CrossRef]
- Sumin, A.N.; Korok, E.V.; Sergeeva, T.Y. Impaired right ventricular filling in patients with a chronic coronary syndrome. Med. Ultrason. 2021, 23, 311–318. [Google Scholar] [CrossRef]
- Edward, J.; Banchs, J.; Parker, H.; Cornwell, W. Right ventricular function across the spectrum of health and disease. Heart 2023, 109, 349–355. [Google Scholar] [CrossRef]
- Mandoli, G.E.; De Carli, G.; Pastore, M.C.; Cameli, P.; Contorni, F.; D’Alessandro, M.; Bargagli, E.; Mondillo, S.; Cameli, M. Right cardiac involvement in lung diseases: A multimodality approach from diagnosis to prognostication. J. Intern. Med. 2021, 289, 440–449. [Google Scholar] [CrossRef]
- Mamazhakypov, A.; Sommer, N.; Assmus, B.; Tello, K.; Schermuly, R.T.; Kosanovic, D.; Sarybaev, A.S.; Weissmann, N.; Pak, O. Novel Therapeutic Targets for the Treatment of Right Ventricular Remodeling: Insights from the Pulmonary Artery Banding Model. Int. J. Environ. Res. Public Health 2021, 18, 8927. [Google Scholar] [CrossRef]
- Jabagi, H.; Mielniczuk, L.M.; Liu, P.P.; Ruel, M.; Sun, L.Y. Biomarkers in the Diagnosis, Management, and Prognostication of Perioperative Right Ventricular Failure in Cardiac Surgery—Are We There Yet? J. Clin. Med. 2019, 8, 559. [Google Scholar] [CrossRef] [PubMed]
- Pradhan, N.M.; Mullin, C.; Poor, H.D. Biomarkers and Right Ventricular Dysfunction. Crit. Care Clin. 2020, 36, 141–153. [Google Scholar] [CrossRef]
- Kret, M.; Arora, R. Pathophysiological basis of right ventricular remodeling. J. Cardiovasc. Pharmacol. Ther. 2007, 12, 5–14. [Google Scholar] [CrossRef]
- Avazmohammadi, R.; Mendiola, E.A.; Li, D.S.; Vanderslice, P.; Dixon, R.A.F.; Sacks, M.S. Interactions Between Structural Remodeling and Hypertrophy in the Right Ventricle in Response to Pulmonary Arterial Hypertension. J. Biomech. Eng. 2019, 141, 0910161–09101613. [Google Scholar] [CrossRef] [PubMed]
- Lother, A.; Kohl, P. The heterocellular heart: Identities, interactions, and implications for cardiology. Basic Res. Cardiol. 2023, 118, 30. [Google Scholar] [CrossRef]
- Sharifi Kia, D.; Kim, K.; Simon, M.A. Current Understanding of the Right Ventricle Structure and Function in Pulmonary Arterial Hypertension. Front. Physiol. 2021, 12, 641310. [Google Scholar] [CrossRef] [PubMed]
- Egemnazarov, B.; Crnkovic, S.; Nagy, B.M.; Olschewski, H.; Kwapiszewska, G. Right ventricular fibrosis and dysfunction: Actual concepts and common misconceptions. Matrix Biol. 2018. [Google Scholar] [CrossRef]
- Roe, A.T.; Frisk, M.; Louch, W.E. Targeting cardiomyocyte Ca2+ homeostasis in heart failure. Curr. Pharm. Des. 2015, 21, 431–448. [Google Scholar] [CrossRef]
- Frump, A.L.; Bonnet, S.; de Jesus Perez, V.A.; Lahm, T. Emerging role of angiogenesis in adaptive and maladaptive right ventricular remodeling in pulmonary hypertension. Am. J. Physiol. Lung Cell Mol. Physiol. 2018, 314, L443–L460. [Google Scholar] [CrossRef]
- Dupont, M.; Tang, W.H. Right ventricular afterload and the role of nitric oxide metabolism in left-sided heart failure. J. Card. Fail. 2013, 19, 712–721. [Google Scholar] [CrossRef]
- Park, J.F.; Clark, V.R.; Banerjee, S.; Hong, J.; Razee, A.; Williams, T.; Fishbein, G.; Saddic, L.; Umar, S. Transcriptomic Analysis of Right Ventricular Remodeling in Two Rat Models of Pulmonary Hypertension: Identification and Validation of Epithelial-to-Mesenchymal Transition in Human Right Ventricular Failure. Circ. Heart Fail. 2021, 14, e007058. [Google Scholar] [CrossRef]
- Imoto, K.; Okada, M.; Yamawaki, H. Expression profile of matricellular proteins in hypertrophied right ventricle of monocrotaline-induced pulmonary hypertensive rats. J. Vet. Med. Sci. 2017, 79, 1096–1102. [Google Scholar] [CrossRef] [PubMed]
- Frangogiannis, N.G. Transforming growth factor-β in myocardial disease. Nat. Rev. Cardiol. 2022, 19, 435–455. [Google Scholar] [CrossRef] [PubMed]
- Falcão-Pires, I.; Gonçalves, N.; Henriques-Coelho, T.; Moreira-Gonçalves, D.; Roncon-Albuquerque, R., Jr.; Leite-Moreira, A.F. Apelin decreases myocardial injury and improves right ventricular function in monocrotaline-induced pulmonary hypertension. Am. J. Physiol. Heart Circ. Physiol. 2009, 296, H2007–H2014. [Google Scholar] [CrossRef]
- Galli, S.J.; Tsai, M. Mast cells: Versatile regulators of inflammation, tissue remodeling, host defense and homeostasis. J. Dermatol. Sci. 2008, 49, 7–19. [Google Scholar] [CrossRef] [PubMed]
- Bruno, K.A.; Mathews, J.E.; Yang, A.L.; Frisancho, J.A.; Scott, A.J.; Greyner, H.D.; Molina, F.A.; Greenaway, M.S.; Cooper, G.M.; Bucek, A.; et al. BPA Alters Estrogen Receptor Expression in the Heart After Viral Infection Activating Cardiac Mast Cells and T Cells Leading to Perimyocarditis and Fibrosis. Front. Endocrinol. 2019, 10, 598. [Google Scholar] [CrossRef]
- da Silva, E.Z.; Jamur, M.C.; Oliver, C. Mast cell function: A new vision of an old cell. J. Histochem. Cytochem. 2014, 62, 698–738. [Google Scholar] [CrossRef]
- Plum, T.; Wang, X.; Rettel, M.; Krijgsveld, J.; Feyerabend, T.B.; Rodewald, H.R. Human Mast Cell Proteome Reveals Unique Lineage, Putative Functions, and Structural Basis for Cell Ablation. Immunity 2020, 52, 404–416.e405. [Google Scholar] [CrossRef]
- Kritikou, E.; Depuydt, M.A.C.; de Vries, M.R.; Mulder, K.E.; Govaert, A.M.; Smit, M.D.; van Duijn, J.; Foks, A.C.; Wezel, A.; Smeets, H.J.; et al. Flow Cytometry-Based Characterization of Mast Cells in Human Atherosclerosis. Cells 2019, 8, 334. [Google Scholar] [CrossRef]
- Ngkelo, A.; Richart, A.; Kirk, J.A.; Bonnin, P.; Vilar, J.; Lemitre, M.; Marck, P.; Branchereau, M.; Le Gall, S.; Renault, N.; et al. Mast cells regulate myofilament calcium sensitization and heart function after myocardial infarction. J. Exp. Med. 2016, 213, 1353–1374. [Google Scholar] [CrossRef] [PubMed]
- Sperr, W.R.; Bankl, H.C.; Mundigler, G.; Klappacher, G.; Grossschmidt, K.; Agis, H.; Simon, P.; Laufer, P.; Imhof, M.; Radaszkiewicz, T.; et al. The human cardiac mast cell: Localization, isolation, phenotype, and functional characterization. Blood 1994, 84, 3876–3884. [Google Scholar] [CrossRef] [PubMed]
- Valent, P.; Bettelheim, P. Cell surface structures on human basophils and mast cells: Biochemical and functional characterization. Adv. Immunol. 1992, 52, 333–423. [Google Scholar] [CrossRef] [PubMed]
- Martini, E.; Kunderfranco, P.; Peano, C.; Carullo, P.; Cremonesi, M.; Schorn, T.; Carriero, R.; Termanini, A.; Colombo, F.S.; Jachetti, E.; et al. Single-Cell Sequencing of Mouse Heart Immune Infiltrate in Pressure Overload-Driven Heart Failure Reveals Extent of Immune Activation. Circulation 2019, 140, 2089–2107. [Google Scholar] [CrossRef] [PubMed]
- Sridharan, G.; Shankar, A.A. Toluidine blue: A review of its chemistry and clinical utility. J. Oral Maxillofac. Pathol. 2012, 16, 251–255. [Google Scholar] [CrossRef] [PubMed]
- Krystel-Whittemore, M.; Dileepan, K.N.; Wood, J.G. Mast Cell: A Multi-Functional Master Cell. Front. Immunol. 2015, 6, 620. [Google Scholar] [CrossRef] [PubMed]
- Theoharides, T.C.; Tsilioni, I.; Ren, H. Recent advances in our understanding of mast cell activation—Or should it be mast cell mediator disorders? Expert Rev. Clin. Immunol. 2019, 15, 639–656. [Google Scholar] [CrossRef]
- Gilfillan, A.M.; Austin, S.J.; Metcalfe, D.D. Mast cell biology: Introduction and overview. Adv. Exp. Med. Biol. 2011, 716, 2–12. [Google Scholar] [CrossRef]
- Kovanen, P.T. Mast Cells as Potential Accelerators of Human Atherosclerosis-From Early to Late Lesions. Int. J. Mol. Sci. 2019, 20, 4479. [Google Scholar] [CrossRef]
- Ribatti, D. The Staining of Mast Cells: A Historical Overview. Int. Arch. Allergy Immunol. 2018, 176, 55–60. [Google Scholar] [CrossRef]
- Guimbal, S.; Cornuault, L.; Rouault, P.; Hollier, P.L.; Chapouly, C.; Bats, M.L.; Imbault, J.; Gadeau, A.P.; Couffinhal, T.; Renault, M.A. Mast Cells Are the Trigger of Small Vessel Disease and Diastolic Dysfunction in Diabetic Obese Mice. Arterioscler. Thromb. Vasc. Biol. 2021, 41, e193–e207. [Google Scholar] [CrossRef] [PubMed]
- Teodosio, C.; Mayado, A.; Sánchez-Muñoz, L.; Morgado, J.M.; Jara-Acevedo, M.; Álvarez-Twose, I.; García-Montero, A.C.; Matito, A.; Caldas, C.; Escribano, L.; et al. The immunophenotype of mast cells and its utility in the diagnostic work-up of systemic mastocytosis. J. Leukoc. Biol. 2015, 97, 49–59. [Google Scholar] [CrossRef] [PubMed]
- Moon, T.C.; Befus, A.D.; Kulka, M. Mast cell mediators: Their differential release and the secretory pathways involved. Front. Immunol. 2014, 5, 569. [Google Scholar] [CrossRef]
- Johnson, J.L.; Jackson, C.L.; Angelini, G.D.; George, S.J. Activation of matrix-degrading metalloproteinases by mast cell proteases in atherosclerotic plaques. Arterioscler. Thromb. Vasc. Biol. 1998, 18, 1707–1715. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.O.; Sommerhoff, C.P.; Paivandy, A.; Pejler, G. Mast cell chymase regulates extracellular matrix remodeling-related events in primary human small airway epithelial cells. J. Allergy Clin. Immunol. 2022, 150, 1534–1544. [Google Scholar] [CrossRef]
- Bernstein, A.; Chabot, B.; Dubreuil, P.; Reith, A.; Nocka, K.; Majumder, S.; Ray, P.; Besmer, P. The mouse W/c-kit locus. Ciba Found. Symp. 1990, 148, 158–166, discussion 166–172. [Google Scholar]
- Katz, H.R.; Austen, K.F. Mast cell deficiency, a game of kit and mouse. Immunity 2011, 35, 668–670. [Google Scholar] [CrossRef]
- Reith, A.D.; Rottapel, R.; Giddens, E.; Brady, C.; Forrester, L.; Bernstein, A. W mutant mice with mild or severe developmental defects contain distinct point mutations in the kinase domain of the c-kit receptor. Genes Dev. 1990, 4, 390–400. [Google Scholar] [CrossRef]
- Geissler, E.N.; McFarland, E.C.; Russell, E.S. Analysis of pleiotropism at the dominant white-spotting (W) locus of the house mouse: A description of ten new W alleles. Genetics 1981, 97, 337–361. [Google Scholar] [CrossRef]
- Nocka, K.; Tan, J.C.; Chiu, E.; Chu, T.Y.; Ray, P.; Traktman, P.; Besmer, P. Molecular bases of dominant negative and loss of function mutations at the murine c-kit/white spotting locus: W37, Wv, W41 and W. EMBO J. 1990, 9, 1805–1813. [Google Scholar] [CrossRef]
- Russell, E.S.; Bernstein, S.E. Blood and blood formation. Biol. Lab. Mouse 1966, 2, 351–372. [Google Scholar]
- Nagle, D.L.; Kozak, C.A.; Mano, H.; Chapman, V.M.; Bućan, M. Physical mapping of the Tec and Gabrb1 loci reveals that the Wsh mutation on mouse chromosome 5 is associated with an inversion. Hum. Mol. Genet. 1995, 4, 2073–2079. [Google Scholar] [CrossRef]
- Duttlinger, R.; Manova, K.; Chu, T.Y.; Gyssler, C.; Zelenetz, A.D.; Bachvarova, R.F.; Besmer, P. W-sash affects positive and negative elements controlling c-kit expression: Ectopic c-kit expression at sites of kit-ligand expression affects melanogenesis. Development 1993, 118, 705–717. [Google Scholar] [CrossRef] [PubMed]
- Grimbaldeston, M.A.; Chen, C.C.; Piliponsky, A.M.; Tsai, M.; Tam, S.Y.; Galli, S.J. Mast cell-deficient W-sash c-kit mutant Kit W-sh/W-sh mice as a model for investigating mast cell biology in vivo. Am. J. Pathol. 2005, 167, 835–848. [Google Scholar] [CrossRef]
- Nigrovic, P.A.; Gray, D.H.; Jones, T.; Hallgren, J.; Kuo, F.C.; Chaletzky, B.; Gurish, M.; Mathis, D.; Benoist, C.; Lee, D.M. Genetic inversion in mast cell-deficient (Wsh) mice interrupts corin and manifests as hematopoietic and cardiac aberrancy. Am. J. Pathol. 2008, 173, 1693–1701. [Google Scholar] [CrossRef]
- Kitamura, Y.; Go, S.; Hatanaka, K. Decrease of mast cells in W/Wv mice and their increase by bone marrow transplantation. Blood 1978, 52, 447–452. [Google Scholar] [CrossRef] [PubMed]
- Tsai, M.; Valent, P.; Galli, S.J. KIT as a master regulator of the mast cell lineage. J. Allergy Clin. Immunol. 2022, 149, 1845–1854. [Google Scholar] [CrossRef]
- Reber, L.L.; Marichal, T.; Galli, S.J. New models for analyzing mast cell functions in vivo. Trends Immunol. 2012, 33, 613–625. [Google Scholar] [CrossRef]
- Hayashi, S.; Kunisada, T.; Ogawa, M.; Yamaguchi, K.; Nishikawa, S. Exon skipping by mutation of an authentic splice site of c-kit gene in W/W mouse. Nucleic Acids Res. 1991, 19, 1267–1271. [Google Scholar] [CrossRef]
- Puddington, L.; Olson, S.; Lefrançois, L. Interactions between stem cell factor and c-Kit are required for intestinal immune system homeostasis. Immunity 1994, 1, 733–739. [Google Scholar] [CrossRef]
- Huizinga, J.D.; Thuneberg, L.; Klüppel, M.; Malysz, J.; Mikkelsen, H.B.; Bernstein, A. W/kit gene required for interstitial cells of Cajal and for intestinal pacemaker activity. Nature 1995, 373, 347–349. [Google Scholar] [CrossRef] [PubMed]
- Galli, S.J.; Arizono, N.; Murakami, T.; Dvorak, A.M.; Fox, J.G. Development of large numbers of mast cells at sites of idiopathic chronic dermatitis in genetically mast cell-deficient WBB6F1-W/Wv mice. Blood 1987, 69, 1661–1666. [Google Scholar] [CrossRef]
- Bhattacharya, K.; Farwell, K.; Huang, M.; Kempuraj, D.; Donelan, J.; Papaliodis, D.; Vasiadi, M.; Theoharides, T.C. Mast cell deficient W/Wv mice have lower serum IL-6 and less cardiac tissue necrosis than their normal littermates following myocardial ischemia-reperfusion. Int. J. Immunopathol. Pharmacol. 2007, 20, 69–74. [Google Scholar] [CrossRef]
- Shao, Z.; Nazari, M.; Guo, L.; Li, S.H.; Sun, J.; Liu, S.M.; Yuan, H.P.; Weisel, R.D.; Li, R.K. The cardiac repair benefits of inflammation do not persist: Evidence from mast cell implantation. J. Cell. Mol. Med. 2015, 19, 2751–2762. [Google Scholar] [CrossRef]
- Hara, M.; Ono, K.; Hwang, M.W.; Iwasaki, A.; Okada, M.; Nakatani, K.; Sasayama, S.; Matsumori, A. Evidence for a role of mast cells in the evolution to congestive heart failure. J. Exp. Med. 2002, 195, 375–381. [Google Scholar] [CrossRef]
- Liao, C.H.; Akazawa, H.; Tamagawa, M.; Ito, K.; Yasuda, N.; Kudo, Y.; Yamamoto, R.; Ozasa, Y.; Fujimoto, M.; Wang, P.; et al. Cardiac mast cells cause atrial fibrillation through PDGF-A-mediated fibrosis in pressure-overloaded mouse hearts. J. Clin. Investig. 2010, 120, 242–253. [Google Scholar] [CrossRef]
- Sydykov, A.; Luitel, H.; Mamazhakypov, A.; Wygrecka, M.; Pradhan, K.; Pak, O.; Petrovic, A.; Kojonazarov, B.; Weissmann, N.; Seeger, W.; et al. Genetic Deficiency and Pharmacological Stabilization of Mast Cells Ameliorate Pressure Overload-Induced Maladaptive Right Ventricular Remodeling in Mice. Int. J. Mol. Sci. 2020, 21, 9099. [Google Scholar] [CrossRef] [PubMed]
- Behrends, D.A.; Cheng, L.; Sullivan, M.B.; Wang, M.H.; Roby, G.B.; Zayed, N.; Gao, C.; Henderson, J.E.; Martineau, P.A. Defective bone repair in mast cell deficient mice with c-Kit loss of function. Eur Cell Mater 2014, 28, 209–221, discussion 221–222. [Google Scholar] [CrossRef] [PubMed]
- Widiapradja, A.; Manteufel, E.J.; Dehlin, H.M.; Pena, J.; Goldspink, P.H.; Sharma, A.; Kolb, L.L.; Imig, J.D.; Janicki, J.S.; Lu, B.; et al. Regulation of Cardiac Mast Cell Maturation and Function by the Neurokinin-1 Receptor in the Fibrotic Heart. Sci. Rep. 2019, 9, 11004. [Google Scholar] [CrossRef]
- Buckley, C.L.; Stokes, A.J. Corin-deficient W-sh mice poorly tolerate increased cardiac afterload. Regul. Pept. 2011, 172, 44–50. [Google Scholar] [CrossRef]
- Feyerabend, T.B.; Weiser, A.; Tietz, A.; Stassen, M.; Harris, N.; Kopf, M.; Radermacher, P.; Möller, P.; Benoist, C.; Mathis, D.; et al. Cre-mediated cell ablation contests mast cell contribution in models of antibody- and T cell-mediated autoimmunity. Immunity 2011, 35, 832–844. [Google Scholar] [CrossRef]
- Tchougounova, E.; Pejler, G.; Abrink, M. The chymase, mouse mast cell protease 4, constitutes the major chymotrypsin-like activity in peritoneum and ear tissue. A role for mouse mast cell protease 4 in thrombin regulation and fibronectin turnover. J. Exp. Med. 2003, 198, 423–431. [Google Scholar] [CrossRef] [PubMed]
- Younan, G.; Suber, F.; Xing, W.; Shi, T.; Kunori, Y.; Abrink, M.; Pejler, G.; Schlenner, S.M.; Rodewald, H.R.; Moore, F.D., Jr.; et al. The inflammatory response after an epidermal burn depends on the activities of mouse mast cell proteases 4 and 5. J. Immunol. 2010, 185, 7681–7690. [Google Scholar] [CrossRef] [PubMed]
- Houde, M.; Schwertani, A.; Touil, H.; Desbiens, L.; Sarrhini, O.; Lecomte, R.; Lepage, M.; Gagnon, H.; Takai, S.; Pejler, G.; et al. Mouse Mast Cell Protease 4 Deletion Protects Heart Function and Survival After Permanent Myocardial Infarction. Front. Pharmacol. 2018, 9, 868. [Google Scholar] [CrossRef] [PubMed]
- Tejada, T.; Tan, L.; Torres, R.A.; Calvert, J.W.; Lambert, J.P.; Zaidi, M.; Husain, M.; Berce, M.D.; Naib, H.; Pejler, G.; et al. IGF-1 degradation by mouse mast cell protease 4 promotes cell death and adverse cardiac remodeling days after a myocardial infarction. Proc. Natl. Acad. Sci. USA 2016, 113, 6949–6954. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Liu, C.L.; Fang, W.; Zhang, X.; Yang, C.; Li, J.; Liu, J.; Sukhova, G.K.; Gurish, M.F.; Libby, P.; et al. Deficiency of mouse mast cell protease 4 mitigates cardiac dysfunctions in mice after myocardium infarction. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 1170–1181. [Google Scholar] [CrossRef]
- Litviňuková, M.; Talavera-López, C.; Maatz, H.; Reichart, D.; Worth, C.L.; Lindberg, E.L.; Kanda, M.; Polanski, K.; Heinig, M.; Lee, M.; et al. Cells of the adult human heart. Nature 2020, 588, 466–472. [Google Scholar] [CrossRef]
- Cohen, C.D.; Rousseau, S.T.; Bermea, K.C.; Bhalodia, A.; Lovell, J.P.; Zita, M.D.; Čiháková, D.; Adamo, L. Myocardial Immune Cells: The Basis of Cardiac Immunology. J. Immunol. 2023, 210, 1198–1207. [Google Scholar] [CrossRef]
- Marone, G.; de Crescenzo, G.; Adt, M.; Patella, V.; Arbustini, E.; Genovese, A. Immunological characterization and functional importance of human heart mast cells. Immunopharmacology 1995, 31, 1–18. [Google Scholar] [CrossRef]
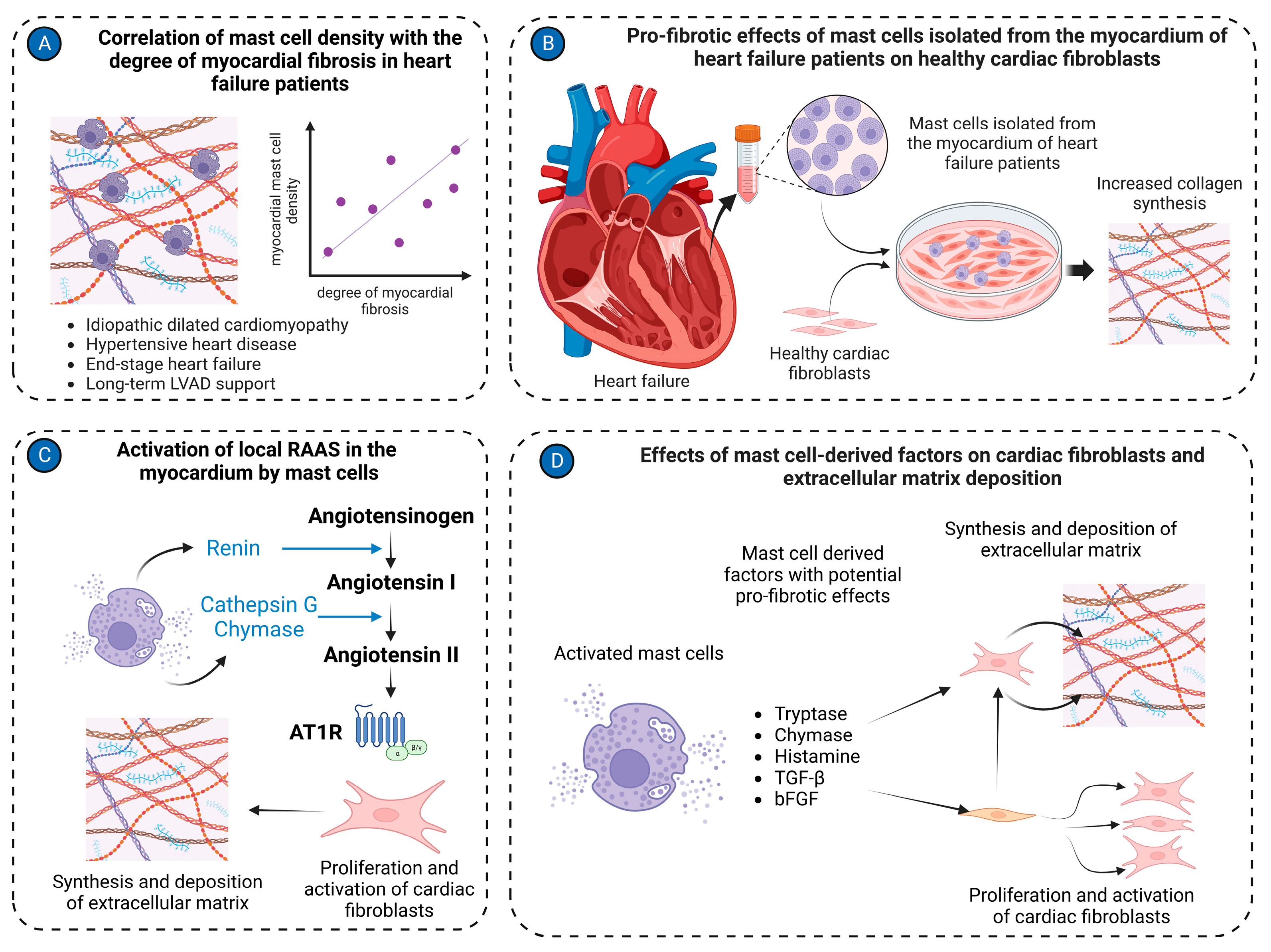
- Silver, R.B.; Reid, A.C.; Mackins, C.J.; Askwith, T.; Schaefer, U.; Herzlinger, D.; Levi, R. Mast cells: A unique source of renin. Proc. Natl. Acad. Sci. USA 2004, 101, 13607–13612. [Google Scholar] [CrossRef] [PubMed]
- Levick, S.P.; McLarty, J.L.; Murray, D.B.; Freeman, R.M.; Carver, W.E.; Brower, G.L. Cardiac mast cells mediate left ventricular fibrosis in the hypertensive rat heart. Hypertension 2009, 53, 1041–1047. [Google Scholar] [CrossRef]
- Gersch, C.; Dewald, O.; Zoerlein, M.; Michael, L.H.; Entman, M.L.; Frangogiannis, N.G. Mast cells and macrophages in normal C57/BL/6 mice. Histochem. Cell Biol. 2002, 118, 41–49. [Google Scholar] [CrossRef]
- Ingason, A.B.; Mechmet, F.; Atacho, D.A.M.; Steingrímsson, E.; Petersen, P.H. Distribution of mast cells within the mouse heart and its dependency on Mitf. Mol. Immunol. 2019, 105, 9–15. [Google Scholar] [CrossRef]
- Engels, W.; Reiters, P.H.; Daemen, M.J.; Smits, J.F.; van der Vusse, G.J. Transmural changes in mast cell density in rat heart after infarct induction in vivo. J. Pathol. 1995, 177, 423–429. [Google Scholar] [CrossRef] [PubMed]
- Stamenov, N.; Kotov, G.; Iliev, A.; Landzhov, B.; Kirkov, V.; Stanchev, S. Mast cells and basic fibroblast growth factor in physiological aging of rat heart and kidney. Biotech. Histochem. 2022, 97, 504–518. [Google Scholar] [CrossRef] [PubMed]
- Panizo, A.; Mindan, F.J.; Galindo, M.F.; Cenarruzabeitia, E.; Hernandez, M.; Diez, J. Are mast cells involved in hypertensive heart disease? J. Hypertens. 1995, 13, 1201–1208. [Google Scholar] [CrossRef] [PubMed]
- Rakusan, K.; Sarkar, K.; Turek, Z.; Wicker, P. Mast cells in the rat heart during normal growth and in cardiac hypertrophy. Circ. Res. 1990, 66, 511–516. [Google Scholar] [CrossRef] [PubMed]
- Frangogiannis, N.G.; Burns, A.R.; Michael, L.H.; Entman, M.L. Histochemical and morphological characteristics of canine cardiac mast cells. Histochem. J. 1999, 31, 221–229. [Google Scholar] [CrossRef]
- Dai, S.; Ogle, C.W. Ventricular histamine concentrations and mast cell counts in the rat heart during acute ischaemia. Agents Actions 1990, 29, 138–143. [Google Scholar] [CrossRef]
- Kwon, J.S.; Kim, Y.S.; Cho, A.S.; Cho, H.H.; Kim, J.S.; Hong, M.H.; Jeong, S.Y.; Jeong, M.H.; Cho, J.G.; Park, J.C.; et al. The novel role of mast cells in the microenvironment of acute myocardial infarction. J. Mol. Cell. Cardiol. 2011, 50, 814–825. [Google Scholar] [CrossRef]
- Kotov, G.; Landzhov, B.; Stamenov, N.; Stanchev, S.; Iliev, A. Changes in the number of mast cells, expression of fibroblast growth factor-2 and extent of interstitial fibrosis in established and advanced hypertensive heart disease. Ann Anat 2020, 232, 151564. [Google Scholar] [CrossRef] [PubMed]
- Akgul, A.; Youker, K.A.; Noon, G.P.; Loebe, M. Quantitative changes in mast cell populations after left ventricular assist device implantation. ASAIO J. 2005, 51, 275–280. [Google Scholar] [CrossRef] [PubMed]
- Patella, V.; de Crescenzo, G.; Ciccarelli, A.; Marinò, I.; Adt, M.; Marone, G. Human heart mast cells: A definitive case of mast cell heterogeneity. Int. Arch. Allergy Immunol. 1995, 106, 386–393. [Google Scholar] [CrossRef]
- Jin, J.; Jiang, Y.; Chakrabarti, S.; Su, Z. Cardiac Mast Cells: A Two-Head Regulator in Cardiac Homeostasis and Pathogenesis Following Injury. Front. Immunol. 2022, 13, 963444. [Google Scholar] [CrossRef]
- Kennedy, R.H.; Hauer-Jensen, M.; Joseph, J. Cardiac function in hearts isolated from a rat model deficient in mast cells. Am. J. Physiol. Heart Circ. Physiol. 2005, 288, H632–H637. [Google Scholar] [CrossRef] [PubMed]
- Gorr, M.W.; Sriram, K.; Chinn, A.M.; Muthusamy, A.; Insel, P.A. Transcriptomic profiles reveal differences between the right and left ventricle in normoxia and hypoxia. Physiol. Rep. 2020, 8, e14344. [Google Scholar] [CrossRef]
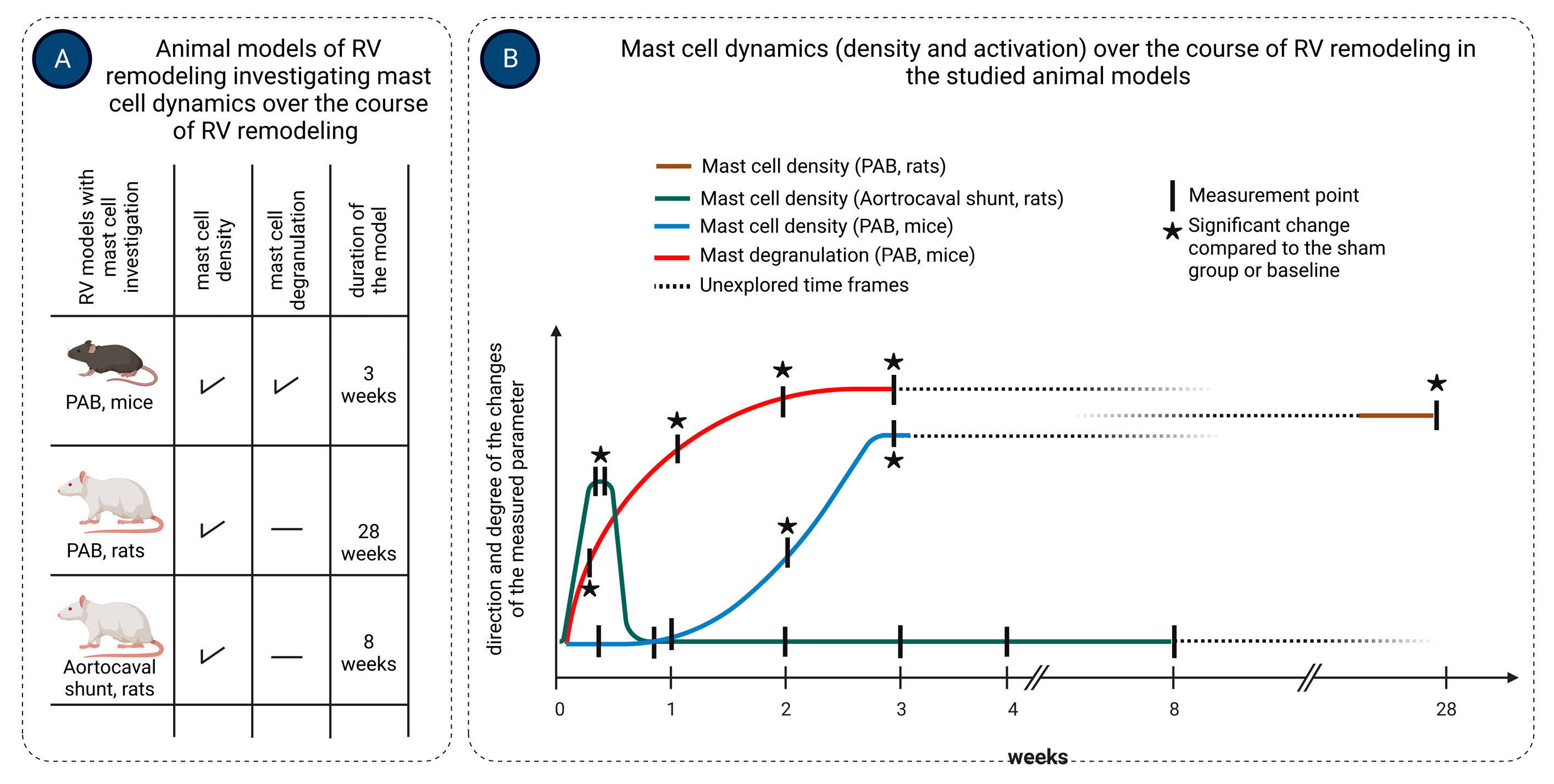
- Luitel, H.; Sydykov, A.; Schymura, Y.; Mamazhakypov, A.; Janssen, W.; Pradhan, K.; Wietelmann, A.; Kosanovic, D.; Dahal, B.K.; Weissmann, N.; et al. Pressure overload leads to an increased accumulation and activity of mast cells in the right ventricle. Physiol. Rep. 2017, 5, e13146. [Google Scholar] [CrossRef]
- Brower, G.L.; Chancey, A.L.; Thanigaraj, S.; Matsubara, B.B.; Janicki, J.S. Cause and effect relationship between myocardial mast cell number and matrix metalloproteinase activity. Am. J. Physiol. Heart Circ. Physiol. 2002, 283, H518–H525. [Google Scholar] [CrossRef]
- Olivetti, G.; Lagrasta, C.; Ricci, R.; Sonnenblick, E.H.; Capasso, J.M.; Anversa, P. Long-term pressure-induced cardiac hypertrophy: Capillary and mast cell proliferation. Am. J. Physiol. 1989, 257, H1766–H1772. [Google Scholar] [CrossRef]
- Aharinejad, S.; Schraufnagel, D.E.; Böck, P.; MacKay, C.A.; Larson, E.K.; Miksovsky, A.; Marks, S.C. Spontaneously hypertensive rats develop pulmonary hypertension and hypertrophy of pulmonary venous sphincters. Am. J. Pathol. 1996, 148, 281–290. [Google Scholar]
- Janssens, S.P.; Thompson, B.T.; Spence, C.R.; Hales, C.A. Functional and structural changes with hypoxia in pulmonary circulation of spontaneously hypertensive rats. J. Appl. Physiol. 1994, 77, 1101–1107. [Google Scholar] [CrossRef]
- Sun, M.; Chen, M.; Dawood, F.; Zurawska, U.; Li, J.Y.; Parker, T.; Kassiri, Z.; Kirshenbaum, L.A.; Arnold, M.; Khokha, R.; et al. Tumor necrosis factor-alpha mediates cardiac remodeling and ventricular dysfunction after pressure overload state. Circulation 2007, 115, 1398–1407. [Google Scholar] [CrossRef]
- Fang, G.; Li, Y.; Yuan, J.; Cao, W.; Song, S.; Chen, L.; Wang, Y.; Wang, Q. Cadherin-11-Interleukin-6 Signaling between Cardiac Fibroblast and Cardiomyocyte Promotes Ventricular Remodeling in a Mouse Pressure Overload-Induced Heart Failure Model. Int. J. Mol. Sci. 2023, 24, 6549. [Google Scholar] [CrossRef] [PubMed]
- Joseph, J.; Kennedy, R.H.; Devi, S.; Wang, J.; Joseph, L.; Hauer-Jensen, M. Protective role of mast cells in homocysteine-induced cardiac remodeling. Am. J. Physiol. Heart Circ. Physiol. 2005, 288, H2541–H2545. [Google Scholar] [CrossRef] [PubMed]
- Cildir, G.; Pant, H.; Lopez, A.F.; Tergaonkar, V. The transcriptional program, functional heterogeneity, and clinical targeting of mast cells. J. Exp. Med. 2017, 214, 2491–2506. [Google Scholar] [CrossRef] [PubMed]
- Scandiuzzi, L.; Beghdadi, W.; Daugas, E.; Abrink, M.; Tiwari, N.; Brochetta, C.; Claver, J.; Arouche, N.; Zang, X.; Pretolani, M.; et al. Mouse mast cell protease-4 deteriorates renal function by contributing to inflammation and fibrosis in immune complex-mediated glomerulonephritis. J. Immunol. 2010, 185, 624–633. [Google Scholar] [CrossRef] [PubMed]
- Aldi, S.; Takano, K.; Tomita, K.; Koda, K.; Chan, N.Y.; Marino, A.; Salazar-Rodriguez, M.; Thurmond, R.L.; Levi, R. Histamine H4-receptors inhibit mast cell renin release in ischemia/reperfusion via protein kinase C ε-dependent aldehyde dehydrogenase type-2 activation. J. Pharmacol. Exp. Ther. 2014, 349, 508–517. [Google Scholar] [CrossRef]
- Aldi, S.; Marino, A.; Tomita, K.; Corti, F.; Anand, R.; Olson, K.E.; Marcus, A.J.; Levi, R. E-NTPDase1/CD39 modulates renin release from heart mast cells during ischemia/reperfusion: A novel cardioprotective role. FASEB J. 2015, 29, 61–69. [Google Scholar] [CrossRef]
- Akgul, A.; Skrabal, C.A.; Thompson, L.O.; Loebe, M.; Lafuente, J.A.; Noon, G.P.; Youker, K.A. Role of mast cells and their mediators in failing myocardium under mechanical ventricular support. J. Heart Lung Transplant. 2004, 23, 709–715. [Google Scholar] [CrossRef]
- He, A.; Fang, W.; Zhao, K.; Wang, Y.; Li, J.; Yang, C.; Benadjaoud, F.; Shi, G.P. Mast cell-deficiency protects mice from streptozotocin-induced diabetic cardiomyopathy. Transl. Res. 2019, 208, 1–14. [Google Scholar] [CrossRef]
- Zeng, Z.; Shen, L.; Li, X.; Luo, T.; Wei, X.; Zhang, J.; Cao, S.; Huang, X.; Fukushima, Y.; Bin, J.; et al. Disruption of histamine H2 receptor slows heart failure progression through reducing myocardial apoptosis and fibrosis. Clin. Sci. 2014, 127, 435–448. [Google Scholar] [CrossRef]
- Hara, M.; Matsumori, A.; Ono, K.; Kido, H.; Hwang, M.W.; Miyamoto, T.; Iwasaki, A.; Okada, M.; Nakatani, K.; Sasayama, S. Mast cells cause apoptosis of cardiomyocytes and proliferation of other intramyocardial cells in vitro. Circulation 1999, 100, 1443–1449. [Google Scholar] [CrossRef] [PubMed]
- Powell, P.C.; Wei, C.C.; Fu, L.; Pat, B.; Bradley, W.E.; Collawn, J.F.; Dell’Italia, L.J. Chymase uptake by cardiomyocytes results in myosin degradation in cardiac volume overload. Heliyon 2019, 5, e01397. [Google Scholar] [CrossRef]
- Kuzmin, V.S.; Malykhina, I.A.; Pustovit, K.B.; Ivanova, A.D.; Kuniewicz, M.; Walocha, J.; Atkinson, A.; Aminu, A.J.; Dobrzynski, H. Inflammatory degranulation of the cardiac resident mast cells suppresses the pacemaking and affects activation pattern in the sinoatrial node. Transl. Res. Anat. 2022, 26, 100170. [Google Scholar] [CrossRef]
- Mackins, C.J.; Kano, S.; Seyedi, N.; Schäfer, U.; Reid, A.C.; Machida, T.; Silver, R.B.; Levi, R. Cardiac mast cell-derived renin promotes local angiotensin formation, norepinephrine release, and arrhythmias in ischemia/reperfusion. J. Clin. Investig. 2006, 116, 1063–1070. [Google Scholar] [CrossRef] [PubMed]
- Marino, A.; Sakamoto, T.; Robador, P.A.; Tomita, K.; Levi, R. S1P receptor 1-Mediated Anti-Renin-Angiotensin System Cardioprotection: Pivotal Role of Mast Cell Aldehyde Dehydrogenase Type 2. J. Pharmacol. Exp. Ther. 2017, 362, 230–242. [Google Scholar] [CrossRef] [PubMed]
- Marino, A.; Sakamoto, T.; Tang, X.H.; Gudas, L.J.; Levi, R. A Retinoic Acid β(2)-Receptor Agonist Exerts Cardioprotective Effects. J. Pharmacol. Exp. Ther. 2018, 366, 314–321. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.; Xiong, Y.; Li, X.; Yang, Y. Cardiac Fibrosis: Cellular Effectors, Molecular Pathways, and Exosomal Roles. Front. Cardiovasc. Med. 2021, 8, 715258. [Google Scholar] [CrossRef]
- Frangogiannis, N.G. Cardiac fibrosis. Cardiovasc. Res. 2021, 117, 1450–1488. [Google Scholar] [CrossRef]
- Varricchi, G.; Marone, G.; Kovanen, P.T. Cardiac Mast Cells: Underappreciated Immune Cells in Cardiovascular Homeostasis and Disease. Trends Immunol. 2020, 41, 734–746. [Google Scholar] [CrossRef]
- Levick, S.P.; Melendez, G.C.; Plante, E.; McLarty, J.L.; Brower, G.L.; Janicki, J.S. Cardiac mast cells: The centrepiece in adverse myocardial remodelling. Cardiovasc. Res. 2011, 89, 12–19. [Google Scholar] [CrossRef]
- Legere, S.A.; Haidl, I.D.; Legare, J.F.; Marshall, J.S. Mast Cells in Cardiac Fibrosis: New Insights Suggest Opportunities for Intervention. Front. Immunol. 2019, 10, 580. [Google Scholar] [CrossRef]
- Batlle, M.; Pérez-Villa, F.; Lázaro, A.; Garcia-Pras, E.; Ramirez, J.; Ortiz, J.; Orús, J.; Roqué, M.; Heras, M.; Roig, E. Correlation between mast cell density and myocardial fibrosis in congestive heart failure patients. Transplant. Proc. 2007, 39, 2347–2349. [Google Scholar] [CrossRef]
- Juliano, G.R.; Skaf, M.F.; Ramalho, L.S.; Juliano, G.R.; Torquato, B.G.S.; Oliveira, M.S.; Oliveira, F.A.; Espíndula, A.P.; Cavellani, C.L.; Teixeira, V.P.A.; et al. Analysis of mast cells and myocardial fibrosis in autopsied patients with hypertensive heart disease. Rev. Port. Cardiol. (Engl. Ed.) 2020, 39, 89–96. [Google Scholar] [CrossRef] [PubMed]
- Skrabal, C.A.; Thompson, L.O.; Southard, R.E.; Joyce, D.L.; Noon, G.P.; Loebe, M.; Youker, K.A. Interaction between isolated human myocardial mast cells and cultured fibroblasts. J. Surg. Res. 2004, 118, 66–70. [Google Scholar] [CrossRef] [PubMed]
- Palaniyandi, S.S.; Inagaki, K.; Mochly-Rosen, D. Mast cells and epsilonPKC: A role in cardiac remodeling in hypertension-induced heart failure. J. Mol. Cell. Cardiol. 2008, 45, 779–786. [Google Scholar] [CrossRef] [PubMed]
- Shiota, N.; Rysa, J.; Kovanen, P.T.; Ruskoaho, H.; Kokkonen, J.O.; Lindstedt, K.A. A role for cardiac mast cells in the pathogenesis of hypertensive heart disease. J. Hypertens. 2003, 21, 1935–1944. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Jubair, S.; Janicki, J.S. Estrogen inhibits mast cell chymase release to prevent pressure overload-induced adverse cardiac remodeling. Hypertension 2015, 65, 328–334. [Google Scholar] [CrossRef]
- Zhao, X.Y.; Zhao, L.Y.; Zheng, Q.S.; Su, J.L.; Guan, H.; Shang, F.J.; Niu, X.L.; He, Y.P.; Lu, X.L. Chymase induces profibrotic response via transforming growth factor-beta 1/Smad activation in rat cardiac fibroblasts. Mol. Cell. Biochem. 2008, 310, 159–166. [Google Scholar] [CrossRef]
- Takai, S.; Jin, D.; Sakaguchi, M.; Katayama, S.; Muramatsu, M.; Sakaguchi, M.; Matsumura, E.; Kim, S.; Miyazaki, M. A novel chymase inhibitor, 4-[1-([bis-(4-methyl-phenyl)-methyl]-carbamoyl)3-(2-ethoxy-benzyl)-4-oxo-azetidine-2-yloxy]-benzoic acid (BCEAB), suppressed cardiac fibrosis in cardiomyopathic hamsters. J. Pharmacol. Exp. Ther. 2003, 305, 17–23. [Google Scholar] [CrossRef]
- Batlle, M.; Roig, E.; Perez-Villa, F.; Lario, S.; Cejudo-Martin, P.; Garcia-Pras, E.; Ortiz, J.; Roque, M.; Orus, J.; Rigol, M.; et al. Increased expression of the renin-angiotensin system and mast cell density but not of angiotensin-converting enzyme II in late stages of human heart failure. J. Heart Lung Transplant. 2006, 25, 1117–1125. [Google Scholar] [CrossRef]
- Schnee, J.M.; Hsueh, W.A. Angiotensin II, adhesion, and cardiac fibrosis. Cardiovasc. Res. 2000, 46, 264–268. [Google Scholar] [CrossRef]
- Jahanyar, J.; Youker, K.A.; Loebe, M.; Assad-Kottner, C.; Koerner, M.M.; Torre-Amione, G.; Noon, G.P. Mast cell-derived cathepsin g: A possible role in the adverse remodeling of the failing human heart. J. Surg. Res. 2007, 140, 199–203. [Google Scholar] [CrossRef] [PubMed]
- Jin, D.; Takai, S.; Sakaguchi, M.; Okamoto, Y.; Muramatsu, M.; Miyazaki, M. An antiarrhythmic effect of a chymase inhibitor after myocardial infarction. J. Pharmacol. Exp. Ther. 2004, 309, 490–497. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, T.; Wada, A.; Tsutamoto, T.; Ohnishi, M.; Isono, T.; Kinoshita, M. Chymase inhibition prevents cardiac fibrosis and improves diastolic dysfunction in the progression of heart failure. Circulation 2003, 107, 2555–2558. [Google Scholar] [CrossRef] [PubMed]
- Kawano, H.; Do, Y.S.; Kawano, Y.; Starnes, V.; Barr, M.; Law, R.E.; Hsueh, W.A. Angiotensin II has multiple profibrotic effects in human cardiac fibroblasts. Circulation 2000, 101, 1130–1137. [Google Scholar] [CrossRef] [PubMed]
- Crabos, M.; Roth, M.; Hahn, A.W.; Erne, P. Characterization of angiotensin II receptors in cultured adult rat cardiac fibroblasts. Coupling to signaling systems and gene expression. J. Clin. Investig. 1994, 93, 2372–2378. [Google Scholar] [CrossRef]
- McLarty, J.L.; Melendez, G.C.; Brower, G.L.; Janicki, J.S.; Levick, S.P. Tryptase/Protease-activated receptor 2 interactions induce selective mitogen-activated protein kinase signaling and collagen synthesis by cardiac fibroblasts. Hypertension 2011, 58, 264–270. [Google Scholar] [CrossRef]
- Chancey, A.L.; Brower, G.L.; Janicki, J.S. Cardiac mast cell-mediated activation of gelatinase and alteration of ventricular diastolic function. Am. J. Physiol. Heart Circ. Physiol. 2002, 282, H2152–H2158. [Google Scholar] [CrossRef]
- Murray, D.B.; Gardner, J.D.; Brower, G.L.; Janicki, J.S. Endothelin-1 mediates cardiac mast cell degranulation, matrix metalloproteinase activation, and myocardial remodeling in rats. Am. J. Physiol. Heart Circ. Physiol. 2004, 287, H2295–H2299. [Google Scholar] [CrossRef]
- Meléndez, G.C.; Li, J.; Law, B.A.; Janicki, J.S.; Supowit, S.C.; Levick, S.P. Substance P induces adverse myocardial remodelling via a mechanism involving cardiac mast cells. Cardiovasc. Res. 2011, 92, 420–429. [Google Scholar] [CrossRef]
- Hans, C.P.; Feng, Y.; Naura, A.S.; Troxclair, D.; Zerfaoui, M.; Siddiqui, D.; Jihang, J.; Kim, H.; Kaye, A.D.; Matrougui, K.; et al. Opposing roles of PARP-1 in MMP-9 and TIMP-2 expression and mast cell degranulation in dyslipidemic dilated cardiomyopathy. Cardiovasc. Pathol. 2011, 20, e57–e68. [Google Scholar] [CrossRef]
- de Almeida, A.; Mustin, D.; Forman, M.F.; Brower, G.L.; Janicki, J.S.; Carver, W. Effects of mast cells on the behavior of isolated heart fibroblasts: Modulation of collagen remodeling and gene expression. J. Cell. Physiol. 2002, 191, 51–59. [Google Scholar] [CrossRef] [PubMed]
- Levick, S.P.; Gardner, J.D.; Holland, M.; Hauer-Jensen, M.; Janicki, J.S.; Brower, G.L. Protection from adverse myocardial remodeling secondary to chronic volume overload in mast cell deficient rats. J. Mol. Cell. Cardiol. 2008, 45, 56–61. [Google Scholar] [CrossRef]
- Luxán, G.; Dimmeler, S. The vasculature: A therapeutic target in heart failure? Cardiovasc. Res. 2022, 118, 53–64. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; da Silva, J.; Alencar, A.; Zapata-Sudo, G.; Lin, M.R.; Sun, X.; Ahmad, S.; Ferrario, C.M.; Groban, L. Mast Cell Inhibition Attenuates Cardiac Remodeling and Diastolic Dysfunction in Middle-aged, Ovariectomized Fischer 344 × Brown Norway Rats. J. Cardiovasc. Pharmacol. 2016, 68, 49–57. [Google Scholar] [CrossRef] [PubMed]
- Dona, M.S.I.; Hsu, I.; Meuth, A.I.; Brown, S.M.; Bailey, C.A.; Aragonez, C.G.; Russell, J.J.; Krstevski, C.; Aroor, A.R.; Chandrasekar, B.; et al. Multi-omic analysis of the cardiac cellulome defines a vascular contribution to cardiac diastolic dysfunction in obese female mice. Basic Res. Cardiol. 2023, 118, 11. [Google Scholar] [CrossRef] [PubMed]
- Neumann, J.; Kirchhefer, U.; Dhein, S.; Hofmann, B.; Gergs, U. The Roles of Cardiovascular H(2)-Histamine Receptors Under Normal and Pathophysiological Conditions. Front. Pharmacol. 2021, 12, 732842. [Google Scholar] [CrossRef]
- Leary, P.J.; Tedford, R.J.; Bluemke, D.A.; Bristow, M.R.; Heckbert, S.R.; Kawut, S.M.; Krieger, E.V.; Lima, J.A.; Masri, C.S.; Ralph, D.D.; et al. Histamine H2 Receptor Antagonists, Left Ventricular Morphology, and Heart Failure Risk: The MESA Study. J. Am. Coll. Cardiol. 2016, 67, 1544–1552. [Google Scholar] [CrossRef]
- Deliargyris, E.N.; Upadhya, B.; Sane, D.C.; Dehmer, G.J.; Pye, J.; Smith, S.C., Jr.; Boucher, W.S.; Theoharides, T.C. Mast cell tryptase: A new biomarker in patients with stable coronary artery disease. Atherosclerosis 2005, 178, 381–386. [Google Scholar] [CrossRef]
- Heikkilä, H.M.; Lätti, S.; Leskinen, M.J.; Hakala, J.K.; Kovanen, P.T.; Lindstedt, K.A. Activated mast cells induce endothelial cell apoptosis by a combined action of chymase and tumor necrosis factor-alpha. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 309–314. [Google Scholar] [CrossRef]
- Lätti, S.; Leskinen, M.; Shiota, N.; Wang, Y.; Kovanen, P.T.; Lindstedt, K.A. Mast cell-mediated apoptosis of endothelial cells in vitro: A paracrine mechanism involving TNF-alpha-mediated down-regulation of bcl-2 expression. J. Cell. Physiol. 2003, 195, 130–138. [Google Scholar] [CrossRef] [PubMed]
- Kupreishvili, K.; Fuijkschot, W.W.; Vonk, A.B.; Smulders, Y.M.; Stooker, W.; Van Hinsbergh, V.W.; Niessen, H.W.; Krijnen, P.A. Mast cells are increased in the media of coronary lesions in patients with myocardial infarction and may favor atherosclerotic plaque instability. J. Cardiol. 2017, 69, 548–554. [Google Scholar] [CrossRef]
- Laine, P.; Kaartinen, M.; Penttilä, A.; Panula, P.; Paavonen, T.; Kovanen, P.T. Association between myocardial infarction and the mast cells in the adventitia of the infarct-related coronary artery. Circulation 1999, 99, 361–369. [Google Scholar] [CrossRef]
- Somasundaram, P.; Ren, G.; Nagar, H.; Kraemer, D.; Mendoza, L.; Michael, L.H.; Caughey, G.H.; Entman, M.L.; Frangogiannis, N.G. Mast cell tryptase may modulate endothelial cell phenotype in healing myocardial infarcts. J. Pathol. 2005, 205, 102–111. [Google Scholar] [CrossRef] [PubMed]
- de Souza Junior, D.A.; Mazucato, V.M.; Santana, A.C.; Oliver, C.; Jamur, M.C. Mast Cells Interact with Endothelial Cells to Accelerate In Vitro Angiogenesis. Int. J. Mol. Sci. 2017, 18, 2674. [Google Scholar] [CrossRef]
- Theoharides, T.C.; Alysandratos, K.D.; Angelidou, A.; Delivanis, D.A.; Sismanopoulos, N.; Zhang, B.; Asadi, S.; Vasiadi, M.; Weng, Z.; Miniati, A.; et al. Mast cells and inflammation. Biochim. Biophys. Acta 2012, 1822, 21–33. [Google Scholar] [CrossRef]
- Frangogiannis, N.G.; Lindsey, M.L.; Michael, L.H.; Youker, K.A.; Bressler, R.B.; Mendoza, L.H.; Spengler, R.N.; Smith, C.W.; Entman, M.L. Resident cardiac mast cells degranulate and release preformed TNF-alpha, initiating the cytokine cascade in experimental canine myocardial ischemia/reperfusion. Circulation 1998, 98, 699–710. [Google Scholar] [CrossRef] [PubMed]
- Gilles, S.; Zahler, S.; Welsch, U.; Sommerhoff, C.P.; Becker, B.F. Release of TNF-alpha during myocardial reperfusion depends on oxidative stress and is prevented by mast cell stabilizers. Cardiovasc. Res. 2003, 60, 608–616. [Google Scholar] [CrossRef]
- Paivandy, A.; Pejler, G. Novel Strategies to Target Mast Cells in Disease. J. Innate Immun. 2021, 1–17. [Google Scholar] [CrossRef]
- Toshner, M.; Spiekerkoetter, E.; Bogaard, H.; Hansmann, G.; Nikkho, S.; Prins, K.W. Repurposing of medications for pulmonary arterial hypertension. Pulm Circ 2020, 10, 2045894020941494. [Google Scholar] [CrossRef]
- Cha, Y.; Erez, T.; Reynolds, I.J.; Kumar, D.; Ross, J.; Koytiger, G.; Kusko, R.; Zeskind, B.; Risso, S.; Kagan, E.; et al. Drug repurposing from the perspective of pharmaceutical companies. Br. J. Pharmacol. 2018, 175, 168–180. [Google Scholar] [CrossRef]
- Ahmad, S.; Ferrario, C.M. Chymase inhibitors for the treatment of cardiac diseases: A patent review (2010-2018). Expert Opin. Ther. Pat. 2018, 28, 755–764. [Google Scholar] [CrossRef]
- Kanemitsu, H.; Takai, S.; Tsuneyoshi, H.; Nishina, T.; Yoshikawa, K.; Miyazaki, M.; Ikeda, T.; Komeda, M. Chymase inhibition prevents cardiac fibrosis and dysfunction after myocardial infarction in rats. Hypertens. Res. 2006, 29, 57–64. [Google Scholar] [CrossRef]
- Jin, D.; Takai, S.; Yamada, M.; Sakaguchi, M.; Kamoshita, K.; Ishida, K.; Sukenaga, Y.; Miyazaki, M. Impact of chymase inhibitor on cardiac function and survival after myocardial infarction. Cardiovasc. Res. 2003, 60, 413–420. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Oyamada, S.; Bianchi, C.; Takai, S.; Chu, L.M.; Sellke, F.W. Chymase inhibition reduces infarction and matrix metalloproteinase-9 activation and attenuates inflammation and fibrosis after acute myocardial ischemia/reperfusion. J. Pharmacol. Exp. Ther. 2011, 339, 143–151. [Google Scholar] [CrossRef] [PubMed]
- Wei, C.C.; Hase, N.; Inoue, Y.; Bradley, E.W.; Yahiro, E.; Li, M.; Naqvi, N.; Powell, P.C.; Shi, K.; Takahashi, Y.; et al. Mast cell chymase limits the cardiac efficacy of Ang I-converting enzyme inhibitor therapy in rodents. J. Clin. Investig. 2010, 120, 1229–1239. [Google Scholar] [CrossRef]
- Takahama, H.; Asanuma, H.; Sanada, S.; Fujita, M.; Sasaki, H.; Wakeno, M.; Kim, J.; Asakura, M.; Takashima, S.; Minamino, T.; et al. A histamine H2 receptor blocker ameliorates development of heart failure in dogs independently of β-adrenergic receptor blockade. Basic Res. Cardiol. 2010, 105, 787–794. [Google Scholar] [CrossRef]
- Leary, P.J.; Hess, E.; Barón, A.E.; Branch, K.R.; Choudhary, G.; Hough, C.L.; Maron, B.A.; Ralph, D.D.; Ryan, J.J.; Tedford, R.J.; et al. H2 Receptor Antagonist Use and Mortality in Pulmonary Hypertension: Insight from the VA-CART Program. Am. J. Respir. Crit. Care Med. 2018, 197, 1638–1641. [Google Scholar] [CrossRef]
- Leary, P.J.; Barr, R.G.; Bluemke, D.A.; Bristow, M.R.; Kronmal, R.A.; Lima, J.A.; Ralph, D.D.; Ventetuolo, C.E.; Kawut, S.M. H2 receptor antagonists and right ventricular morphology: The MESA right ventricle study. Ann Am Thorac Soc 2014, 11, 1379–1386. [Google Scholar] [CrossRef]
- Zhang, J.; Cai, W.K.; Zhang, Z.; Wang, P.; Lin, X.Q.; Feng, J.; Fu, S.C.; He, G.H. Cardioprotective effect of histamine H2 antagonists in congestive heart failure: A systematic review and meta-analysis. Medicine 2018, 97, e0409. [Google Scholar] [CrossRef]
- Mukai, K.; Tsai, M.; Saito, H.; Galli, S.J. Mast cells as sources of cytokines, chemokines, and growth factors. Immunol. Rev. 2018, 282, 121–150. [Google Scholar] [CrossRef]
- Farha, S.; Sharp, J.; Asosingh, K.; Park, M.; Comhair, S.A.; Tang, W.H.; Thomas, J.; Farver, C.; Hsieh, F.; Loyd, J.E.; et al. Mast cell number, phenotype, and function in human pulmonary arterial hypertension. Pulm Circ 2012, 2, 220–228. [Google Scholar] [CrossRef]
- Hoffmann, J.; Yin, J.; Kukucka, M.; Yin, N.; Saarikko, I.; Sterner-Kock, A.; Fujii, H.; Leong-Poi, H.; Kuppe, H.; Schermuly, R.T.; et al. Mast cells promote lung vascular remodelling in pulmonary hypertension. Eur. Respir. J. 2011, 37, 1400–1410. [Google Scholar] [CrossRef]
- Bartelds, B.; van Loon, R.L.E.; Mohaupt, S.; Wijnberg, H.; Dickinson, M.G.; Boersma, B.; Takens, J.; van Albada, M.; Berger, R.M.F. Mast cell inhibition improves pulmonary vascular remodeling in pulmonary hypertension. Chest 2012, 141, 651–660. [Google Scholar] [CrossRef]
- Metcalfe, D.D.; Pawankar, R.; Ackerman, S.J.; Akin, C.; Clayton, F.; Falcone, F.H.; Gleich, G.J.; Irani, A.M.; Johansson, M.W.; Klion, A.D.; et al. Biomarkers of the involvement of mast cells, basophils and eosinophils in asthma and allergic diseases. World Allergy Organ J. 2016, 9, 7. [Google Scholar] [CrossRef]
- Kabashima, K.; Nakashima, C.; Nonomura, Y.; Otsuka, A.; Cardamone, C.; Parente, R.; De Feo, G.; Triggiani, M. Biomarkers for evaluation of mast cell and basophil activation. Immunol. Rev. 2018, 282, 114–120. [Google Scholar] [CrossRef]
- Hu, K.K.; Bruce, M.A.; Butte, M.J. Spatiotemporally and mechanically controlled triggering of mast cells using atomic force microscopy. Immunol. Res. 2014, 58, 211–217. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mouse Lines | Generation and Genetic Characteristics | Off-Target Phenotypes | LV Remodeling Models | RV Remodeling Models |
|---|---|---|---|---|
| KitW/W-v |
| PAB-induced RV remodeling [87] | ||
| KitW-sh/W-sh | A spontaneous inversion mutation in the transcriptional regulatory elements upstream of the c-KIT transcription start site on mouse chromosome 5 [74] | Not assessed | ||
| Cpa3Cre/+ | 60% reduction in spleen basophils numbers [91] | LAD-occlusion-induced myocardial infarction [51] | Not assessed | |
| mMCP4 knockout | Targeted inactivation of mMCP-4 gene [92] | Increased tryptase activity in peritoneal mast cells [93] | LAD-occlusion-induced myocardial infarction [94,95,96] | Not assessed |
| Animal Model | Mast Cells in the RV | Mast Cells in the LV | Main Conclusion | References |
|---|---|---|---|---|
| Healthy C57BL/6J mice | Mast cell density 2.1 ± 0.25 cells/mm2 | Mast cell density 1.09 ± 0.09 cells/mm2 | Mast cell density is significantly higher in the RV as compared to the LV | Ingason et al. [103] |
| Healthy mongrel dogs | Mast cell density 6.53 ± 1.04 cells/mm2 | Mast cell density 7.82 ± 1.16 cells/mm2 | Mast cells are equally distributed between ventricles | Frangogiannis et al. [108] |
| Healthy young (6-month-old) and aging (12-month-old) Wistar rats | Significant increase in mast cell density in 12-month-old rats compared to 6-month-old rats Lower mast cell density in the RV than in the LV | Significant increase in mast cell density in the LV of 12-month-old rats compared to 6-month-old rats Greater mast cell density in the LV than in the RV | Increase in mast cell density in the myocardium with aging | Stamenov et al. [105] |
| Wistar rats (3-month-old) raised at sea level or simulated high altitude (3500 m) | No significant effect of altitude on mast cell density | Higher mast cell density in high altitude rats | Higher mast cell density in the RV than in the LV both at sea level and high altitude | Rakusan et al. [107] |
| Acute LAD occlusion in Sprague Dawley rats (2, 5, and 10 min) | Increase in RV histamine concentration after 2 min of LAD occlusion No change in the RV mast cell density after LAD occlusion No effects of mast cell stabilization | Decrease in the LV histamine concentration after 2 min of LAD occlusion No change in the LV mast cell density after LAD occlusion No effects of mast cell stabilization | Changes in myocardial histamine concentrations during acute myocardial ischemia are not related to mast cells | Dai et al. [109] |
| LAD occlusion (1 h) induced ischemia-reperfusion cardiomyopathy in rats | No significant changes in mast cell density following LAD | Increase in mast cell density in the infarct region of the LV at 1 day and 21 days after MI induction | Cardioprotective role of mast cell granules in MI via the prolonged survival of cardiomyocytes and the induction of angiogenesis | Kwon et al. [110] |
| Normotensive Wistar-Kyoto rats and SHR | Higher mast cell density in SHR | Higher mast cell density in SHR | Higher mast cell density in SHR and in the LV than in the RV independent of strain | Panizo et al. [106] |
| SHR with established hypertension and cardiac hypertrophy (6-month-old) and advanced or late-stage hypertension and cardiac hypertrophy (12-month-old) | Higher mast cell density in 12-month-old than 6-month-old SHR | Higher mast cell density in 12-month-old than 6-month-old SHR | Lower mean values for mast cell markers in the RV than the LV, irrespective of the age group of SHR | Kotov et al. [111] |
| Heart tissues from donor hearts and from patients with end-stage cardiomyopathy at the time of LVAD implantation and at the time of LVAD removal | No differences in mast cell density in RV compared to the LV | Higher mast cell density in cardiomyopathy than in donor hearts and lower than in LVAD-supported hearts Significant correlation between mast cell density and collagen in patients before LVAD implantation | Increase in mast cell density in cardiomyopathy A secondary increase in mast cell density due to mechanical support with LVAD and decrease in myocardial fibrosis | Akgul et al. [112] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mamazhakypov, A.; Maripov, A.; Sarybaev, A.S.; Schermuly, R.T.; Sydykov, A. Mast Cells in Cardiac Remodeling: Focus on the Right Ventricle. J. Cardiovasc. Dev. Dis. 2024, 11, 54. https://doi.org/10.3390/jcdd11020054
Mamazhakypov A, Maripov A, Sarybaev AS, Schermuly RT, Sydykov A. Mast Cells in Cardiac Remodeling: Focus on the Right Ventricle. Journal of Cardiovascular Development and Disease. 2024; 11(2):54. https://doi.org/10.3390/jcdd11020054
Chicago/Turabian StyleMamazhakypov, Argen, Abdirashit Maripov, Akpay S. Sarybaev, Ralph Theo Schermuly, and Akylbek Sydykov. 2024. "Mast Cells in Cardiac Remodeling: Focus on the Right Ventricle" Journal of Cardiovascular Development and Disease 11, no. 2: 54. https://doi.org/10.3390/jcdd11020054
APA StyleMamazhakypov, A., Maripov, A., Sarybaev, A. S., Schermuly, R. T., & Sydykov, A. (2024). Mast Cells in Cardiac Remodeling: Focus on the Right Ventricle. Journal of Cardiovascular Development and Disease, 11(2), 54. https://doi.org/10.3390/jcdd11020054