p-21 Activated Kinase as a Molecular Target for Chemoprevention in Diabetes †


Abstract
1. Introduction
2. Methods
2.1. Molecular Analysis of PAK Signaling Pathways and Their Involvement in Response to Anti-Diabetic Drugs
2.2. Identification of PAK Interacting Partners
2.3. Clinical Study Inclusion/Exclusion Criteria and Mini-Meta-Analysis
3. Results
3.1. PAK Signaling Is Associated with Diabetes and Cancer
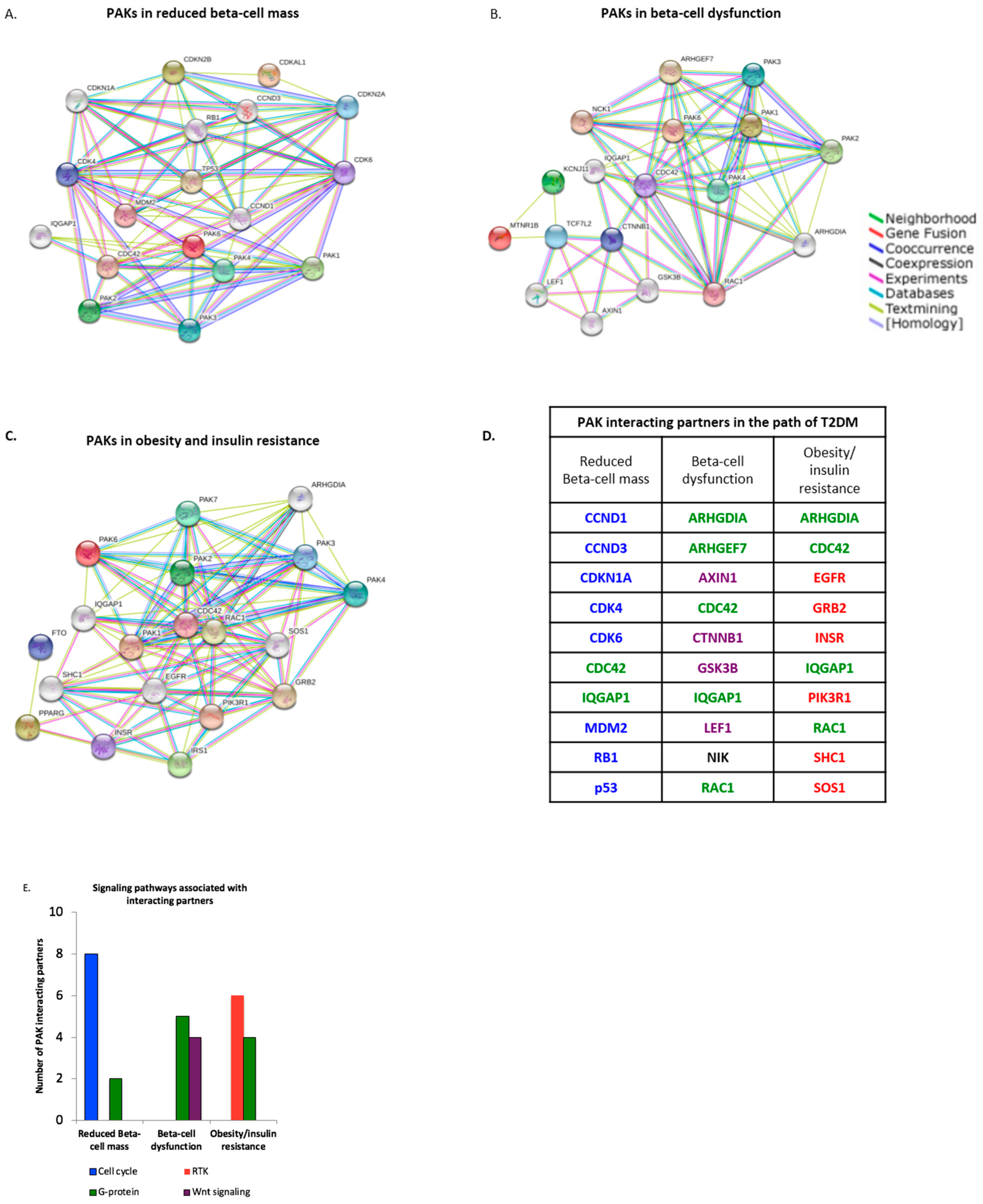
3.2. PAK Interacting Partners Are Associated with the Pathogenesis of T2DM
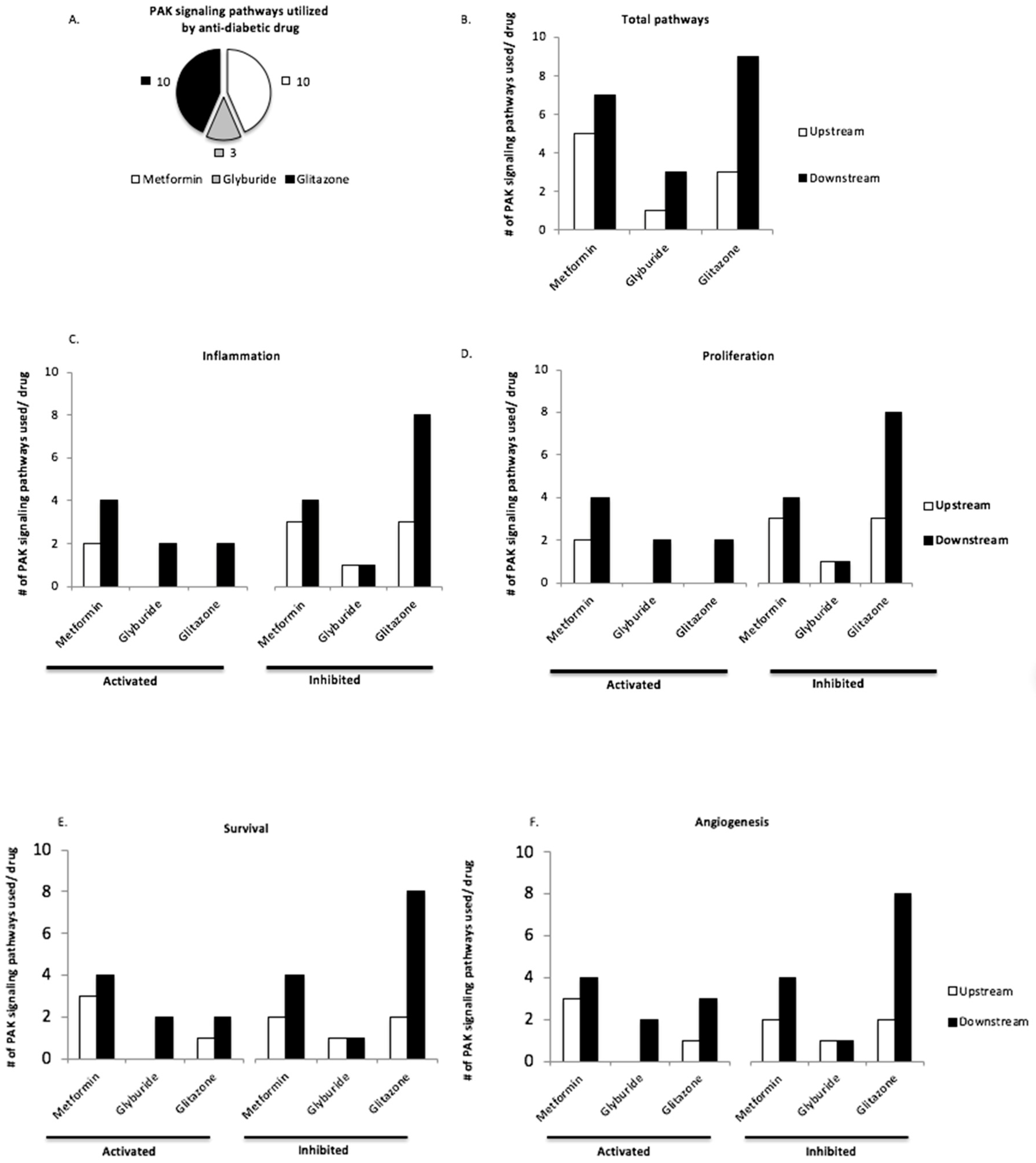
3.3. Upstream and Downstream PAK Signaling Pathways Are Utilized by Anti-Diabetic Drugs Pioglitazone and Metformin
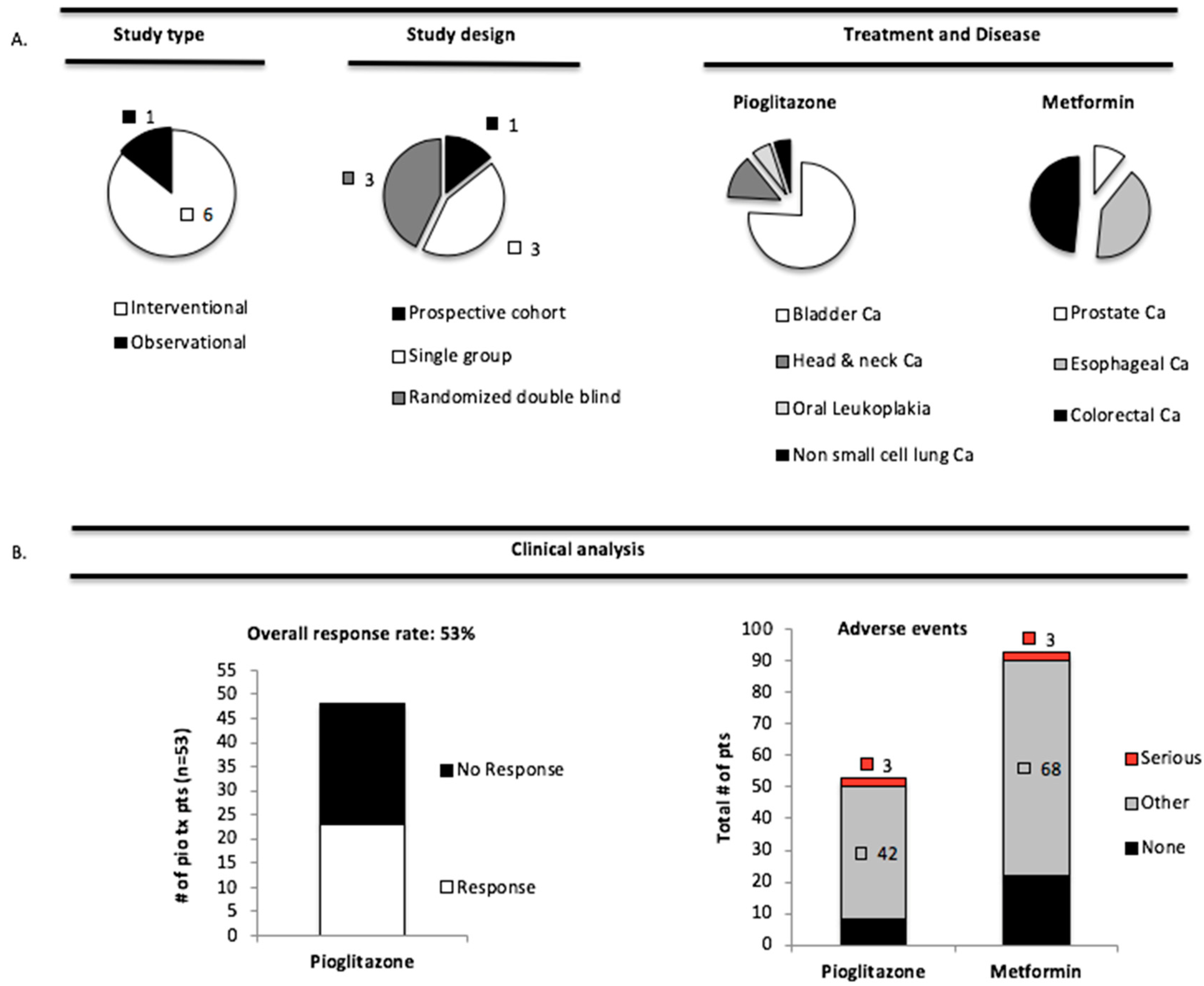
3.4. Pioglitazone and Metformin Have Therapeutic Limitations in Cancer Patients with PAK Overexpression
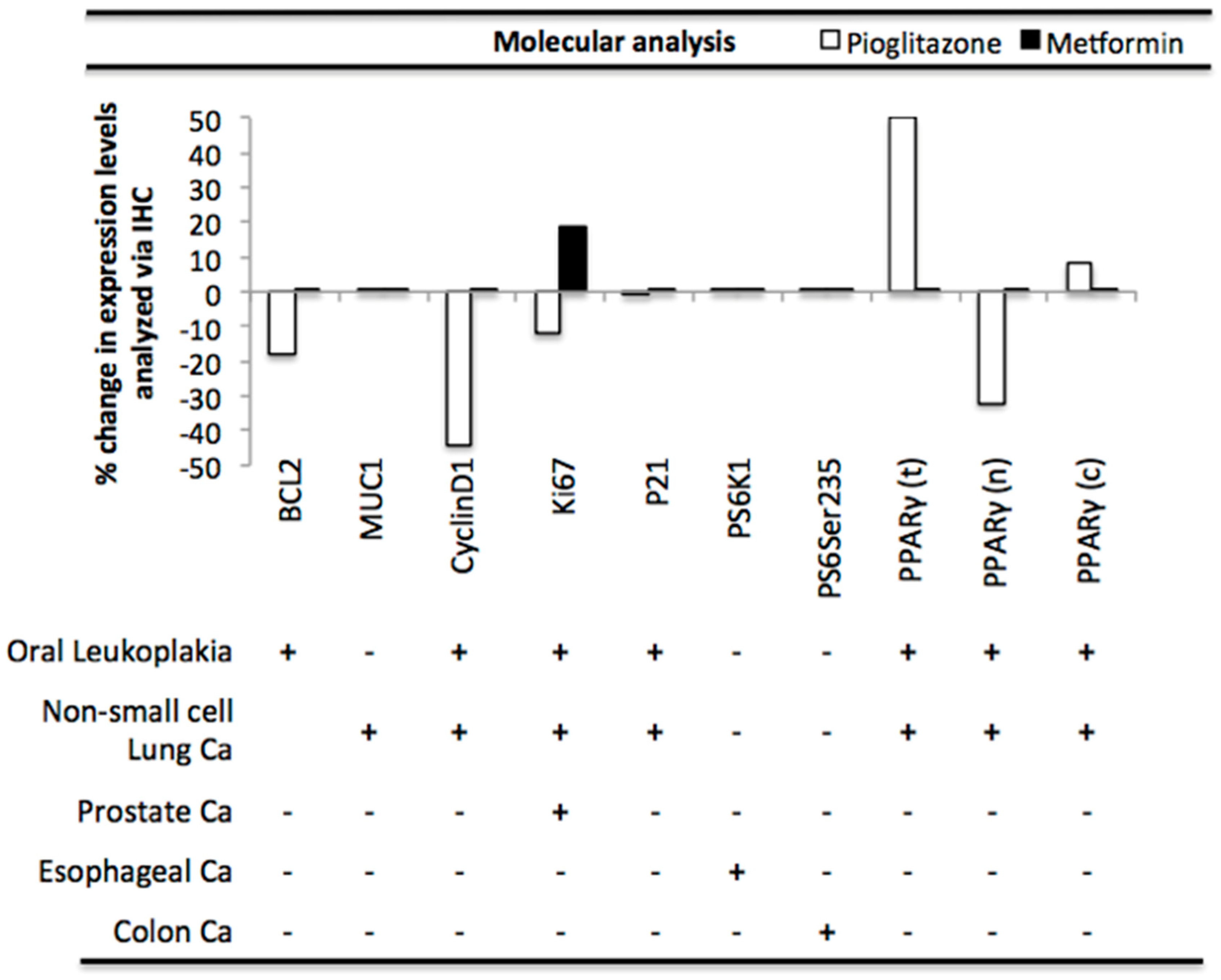
3.5. Pioglitazone and Metformin Alter Biomarkers Downstream of PAK in Human Disease
4. Discussion
4.1. Future Directions
4.2. Concluding Remarks
Supplementary Materials
Disclosure
Funding
Conflicts of Interest
References
- McCarthy, M.I. Genomics, type 2 diabetes, and obesity. N. Engl. J. Med. 2010, 363, 2339–2350. [Google Scholar] [CrossRef] [PubMed]
- Chang, S.-C.; Yang, W.-C.V. Hyperglycemia, tumorigenesis, and chronic inflammation. Crit. Rev. Oncol./Hematol. 2016, 108, 146–153. [Google Scholar] [CrossRef] [PubMed]
- Dammann, K.; Khare, V.; Gasche, C. Tracing PAKs from GI inflammation to cancer. Gut 2014, 63, 1173–1184. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Franceschini, A.; Wyder, S.; Forslund, K.; Heller, D.; Huerta-Cepas, J.; Simonovic, M.; Roth, A.; Santos, A.; Tsafou, K.P.; et al. STRING v10: Protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2015, 43, D447–D452. [Google Scholar] [CrossRef] [PubMed]
- Campregher, C.; Schmid, G.; Ferk, F.; Knasmuller, S.; Khare, V.; Kortum, B.; Dammann, K.; Lang, M.; Scharl, T.; Spittler, A.; et al. MSH3-deficiency initiates EMAST without oncogenic transformation of human colon epithelial cells. PLoS ONE 2012, 7, e50541. [Google Scholar] [CrossRef] [PubMed]
- Kong, D.; Dagon, Y.; Campbell, J.N.; Guo, Y.; Yang, Z.; Yi, X.; Aryal, P.; Wellenstein, K.; Kahn, B.B.; Sabatini, B.L.; et al. A Postsynaptic AMPK-->p21-Activated Kinase Pathway Drives Fasting-Induced Synaptic Plasticity in AgRP Neurons. Neuron 2016, 91, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Dodeller, F.; Schulze-Koops, H. The p38 mitogen-activated protein kinase signaling cascade in CD4 T cells. Arthritis Res. Ther. 2006, 8, 205. [Google Scholar] [CrossRef] [PubMed]
- Menard, R.E.; Mattingly, R.R. Cell surface receptors activate p21-activated kinase 1 via multiple Ras and PI3-kinase-dependent pathways. Cell. Signal. 2003, 15, 1099–1109. [Google Scholar] [CrossRef]
- Huynh, N.; Liu, K.H.; Baldwin, G.S.; He, H. P21-activated kinase 1 stimulates colon cancer cell growth and migration/invasion via ERK- and AKT-dependent pathways. Biochim. Biophys. Acta 2010, 1803, 1106–1113. [Google Scholar] [CrossRef] [PubMed]
- Gu, S.; Kounenidakis, M.; Schmidt, E.-M.; Deshpande, D.; Alkahtani, S.; Alarifi, S.; Föller, M.; Alevizopoulos, K.; Lang, F.; Stournaras, C. Rapid activation of FAK/mTOR/p70S6K/PAK1-signaling controls the early testosterone-induced actin reorganization in colon cancer cells. Cell. Signal. 2013, 25, 66–73. [Google Scholar] [CrossRef] [PubMed]
- Ishida, H.; Li, K.; Yi, M.; Lemon, S.M. p21-activated kinase 1 is activated through the mammalian target of rapamycin/p70 S6 kinase pathway and regulates the replication of hepatitis C virus in human hepatoma cells. J. Biol. Chem. 2007, 282, 11836–11848. [Google Scholar] [CrossRef] [PubMed]
- Khare, V.; Dammann, K.; Asboth, M.; Krnjic, A.; Jambrich, M.; Gasche, C. Overexpression of PAK1 promotes cell survival in inflammatory bowel diseases and colitis-associated cancer. Inflamm. Bowel Dis. 2015, 21, 287–296. [Google Scholar] [CrossRef] [PubMed]
- Ijuin, T.; Takenawa, T. Regulation of Insulin Signaling by the Phosphatidylinositol 3,4,5-Triphosphate Phosphatase SKIP through the Scaffolding Function of Pak1. Mol. Cell. Biol. 2012, 32, 3570–3584. [Google Scholar] [CrossRef] [PubMed]
- Dammann, K.; Khare, V.; Lang, M.; Claudel, T.; Harpain, F.; Granofszky, N.; Evstatiev, R.; Williams, J.M.; Pritchard, D.M.; Watson, A.; et al. PAK1 modulates a PPARγ/NF-κB cascade in intestinal inflammation. Biochim. Biophys. Acta (BBA) Mol. Cell Res. 2015, 1853, 2349–2360. [Google Scholar] [CrossRef] [PubMed]
- Shin, Y.J.; Kim, E.H.; Roy, A.; Kim, J.-H. Evidence for a novel mechanism of the PAK1 interaction with the Rho-GTPases Cdc42 and Rac. PLoS ONE 2013, 8, e71495. [Google Scholar] [CrossRef] [PubMed]
- DeSantiago, J.; Bare, D.J.; Xiao, L.; Ke, Y.; Solaro, R.J.; Banach, K. p21-Activated kinase1 (Pak1) is a negative regulator of NADPH-oxidase 2 in ventricular myocytes. J. Mol. Cell. Cardiol. 2014, 67, 77–85. [Google Scholar] [CrossRef] [PubMed]
- Khare, V.; Lyakhovich, A.; Dammann, K.; Lang, M.; Borgmann, M.; Tichy, B.; Pospisilova, S.; Luciani, G.; Campregher, C.; Evstatiev, R.; et al. Mesalamine modulates intercellular adhesion through inhibition of p-21 activated kinase-1. Biochem. Pharmacol. 2013, 85, 234–244. [Google Scholar] [CrossRef] [PubMed]
- Zhu, G.; Wang, Y.; Huang, B.; Liang, J.; Ding, Y.; Xu, A.; Wu, W. A Rac1/PAK1 cascade controls beta-catenin activation in colon cancer cells. Oncogene 2012, 31, 1001–1012. [Google Scholar] [CrossRef] [PubMed]
- Park, M.-H.; Kim, D.-J.; You, S.-T.; Lee, C.-S.; Kim, H.K.; Park, S.M.; Shin, E.Y.; Kim, E.G. Phosphorylation of beta-catenin at serine 663 regulates its transcriptional activity. Biochem. Biophys. Res. Commun. 2012, 419, 543–549. [Google Scholar] [CrossRef] [PubMed]
- Viollet, B.; Lantier, L.; Devin-Leclerc, J.; Hébrard, S.; Amouyal, C.; Mounier, R.; Foretz, M.; Andreelli, F. Targeting the AMPK pathway for the treatment of Type 2 diabetes. Front. Biosci. 2009, 14, 3380–3400. [Google Scholar] [CrossRef]
- Sozen, B.; Ozturk, S.; Yaba, A.; Demir, N. The p38 MAPK signalling pathway is required for glucose metabolism, lineage specification and embryo survival during mouse preimplantation development. Mech. Dev. 2015, 138, 375–398. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Thompson, B.J.; Hietakangas, V.; Cohen, S.M. MAPK/ERK signaling regulates insulin sensitivity to control glucose metabolism in Drosophila. PLoS Genet. 2011, 7, e1002429. [Google Scholar] [CrossRef] [PubMed]
- Altomare, D.A.; Khaled, A.R. Homeostasis and the Importance for a Balance Between AKT/mTOR Activity and Intracellular Signaling. Curr. Med. Chem. 2012, 19, 3748–3762. [Google Scholar] [CrossRef] [PubMed]
- Mauro, C.; Leow, S.C.; Anso, E.; Rocha, S.; Thotakura, A.K.; Tornatore, L.; Moretti, M.; De Smaele, E.; Beg, A.A.; Tergaonkar, V.; et al. NF-kappaB controls energy homeostasis and metabolic adaptation by upregulating mitochondrial respiration. Nat. Cell Biol. 2011, 13, 1272–1279. [Google Scholar] [CrossRef] [PubMed]
- Chiang, Y.A.; Jin, T. p21-Activated protein kinases and their emerging roles in glucose homeostasis. Am. J. Physiol. Endocrinol. Metab. 2014, 306, E707–E722. [Google Scholar] [CrossRef] [PubMed]
- Sylow, L.; Kleinert, M.; Pehmøller, C.; Prats, C.; Chiu, T.T.; Klip, A.; Richter, E.A.; Jensen, T.E. Akt and Rac1 signaling are jointly required for insulin-stimulated glucose uptake in skeletal muscle and downregulated in insulin resistance. Cell. Signal. 2014, 26, 323–331. [Google Scholar] [CrossRef] [PubMed]
- Picard, F.; Auwerx, J. PPAR(gamma) and glucose homeostasis. Annu. Rev. Nutr. 2002, 22, 167–197. [Google Scholar] [CrossRef] [PubMed]
- Gautam, S.; Ishrat, N.; Singh, R.; Narender, T.; Srivastava, A.K. Aegeline from Aegle marmelos stimulates glucose transport via Akt and Rac1 signaling, and contributes to a cytoskeletal rearrangement through PI3K/Rac1. Eur. J. Pharmacol. 2015, 762, 419–429. [Google Scholar] [CrossRef] [PubMed]
- Ahn, M.; Yoder, S.M.; Wang, Z.; Oh, E.; Ramalingam, L.; Tunduguru, R.; Thurmond, D.C. The p21-activated kinase (PAK1) is involved in diet-induced beta cell mass expansion and survival in mice and human islets. Diabetologia 2016, 59, 2145–2155. [Google Scholar] [CrossRef] [PubMed]
- Manchester, J.; Kong, X.; Lowry, O.H.; Lawrence, J.C. Ras signaling in the activation of glucose transport by insulin. Proc. Natl. Acad. Sci. USA 1994, 91, 4644–4648. [Google Scholar] [CrossRef] [PubMed]
- Elghazi, L.; Gould, A.P.; Weiss, A.J.; Barker, D.J.; Callaghan, J.; Opland, D.; Myers, M.; Cras-Méneur, C.; Bernal-Mizrachi, E. Importance of beta-Catenin in glucose and energy homeostasis. Sci. Rep. 2012, 2, 693. [Google Scholar] [CrossRef] [PubMed]
- Salminen, A.; Hyttinen, J.M.T.; Kaarniranta, K. AMP-activated protein kinase inhibits NF-κB signaling and inflammation: Impact on healthspan and lifespan. J. Mol. Med. 2011, 89, 667–676. [Google Scholar] [CrossRef] [PubMed]
- Motoshima, H.; Goldstein, B.J.; Igata, M.; Araki, E. AMPK and cell proliferation—AMPK as a therapeutic target for atherosclerosis and cancer. J. Physiol. 2006, 574 Pt 1, 63–71. [Google Scholar] [CrossRef]
- Domenech, E.; Maestre, C.; Esteban-Martinez, L.; Partida, D.; Pascual, R.; Fernandez-Miranda, G.; Seco, E.; Campos-Olivas, R.; Pérez, M.; Megias, D.; et al. AMPK and PFKFB3 mediate glycolysis and survival in response to mitophagy during mitotic arrest. Nat. Cell Biol. 2015, 17, 1304–1316. [Google Scholar] [CrossRef] [PubMed]
- Ouchi, N.; Shibata, R.; Walsh, K. AMP-activated protein kinase signaling stimulates VEGF expression and angiogenesis in skeletal muscle. Circ. Res. 2005, 96, 838–846. [Google Scholar] [CrossRef] [PubMed]
- Cuadrado, A.; Nebreda, A.R. Mechanisms and functions of p38 MAPK signalling. Biochem. J. 2010, 429, 403–417. [Google Scholar] [CrossRef] [PubMed]
- Karar, J.; Maity, A. PI3K/AKT/mTOR Pathway in Angiogenesis. Front. Mol. Neurosci. 2011, 4, 51. [Google Scholar] [CrossRef] [PubMed]
- Dammann, K.; Khare, V.; Harpain, F.; Lang, M.; Kurtovic, A.; Mesteri, I.; Evstatiev, R.; Gasche, C. PAK1 promotes intestinal tumor initiation. Cancer Prev. Res. 2015, 8, 1093–1101. [Google Scholar] [CrossRef] [PubMed]
- Salomone, S. Pleiotropic Effects of Glitazones: A Double Edge Sword? Front. Pharmacol. 2011, 2, 14. [Google Scholar] [CrossRef] [PubMed]
- Kotlinowski, J.; Jozkowicz, A. PPAR Gamma and Angiogenesis: Endothelial Cells Perspective. J. Diabetes Res. 2016, 2016, 8492353. [Google Scholar] [CrossRef] [PubMed]
- Liou, G.-Y.; Storz, P. Reactive oxygen species in cancer. Free Radic. Res. 2010, 44, 479–496. [Google Scholar] [CrossRef] [PubMed]
- Devery, A.M.; Wadekar, R.; Bokobza, S.M.; Weber, A.M.; Jiang, Y.; Ryan, A.J. Vascular endothelial growth factor directly stimulates tumour cell proliferation in non-small cell lung cancer. Int. J. Oncol. 2015, 47, 849–856. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Lee, C.; Tang, Z.; Zhang, F.; Arjunan, P.; Li, Y.; Hou, X.; Kumar, A.; Dong, L. VEGF-B: A survival, or an angiogenic factor? Cell Adhes. Migr. 2009, 3, 322–327. [Google Scholar] [CrossRef]
- You, G.Y.; Lee, J.O.; Kim, J.H.; Kim, N.; Lee, S.K.; Moon, J.W.; Jie, S.; Lee, H.J.; Kim, S.J.; Park, S.H.; et al. Tiam-1, a GEF for Rac1, plays a critical role in metformin-mediated glucose uptake in C2C12 cells. Cell. Signal. 2013, 25, 2558–2565. [Google Scholar] [CrossRef] [PubMed]
- Coletta, D.K.; Sriwijitkamol, A.; Wajcberg, E.; Tantiwong, P.; Li, M.; Prentki, M.; Madiraju, M.; Jenkinson, C.P.; Cersosimo, E.; Musi, N.; et al. Pioglitazone stimulates AMP-activated protein kinase signalling and increases the expression of genes involved in adiponectin signalling, mitochondrial function and fat oxidation in human skeletal muscle in vivo: A randomised trial. Diabetologia 2009, 52, 723–732. [Google Scholar] [CrossRef] [PubMed]
- Wu, N.; Gu, C.; Gu, H.; Hu, H.; Han, Y.; Li, Q. Metformin induces apoptosis of lung cancer cells through activating JNK/p38 MAPK pathway and GADD153. Neoplasma 2011, 58, 482–490. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Al-Sammarraie, N.; DiPette, D.J.; Singh, U.S. Metformin impairs Rho GTPase signaling to induce apoptosis in neuroblastoma cells and inhibits growth of tumors in the xenograft mouse model of neuroblastoma. Oncotarget 2014, 5, 11709–11722. [Google Scholar] [CrossRef] [PubMed]
- Qian, X.; Li, J.; Ding, J.; Wang, Z.; Duan, L.; Hu, G. Glibenclamide exerts an antitumor activity through reactive oxygen species-c-jun NH2-terminal kinase pathway in human gastric cancer cell line MGC-803. Biochem. Pharmacol. 2008, 76, 1705–1715. [Google Scholar] [CrossRef] [PubMed]
- Okami, N.; Narasimhan, P.; Yoshioka, H.; Sakata, H.; Kim, G.S.; Jung, J.E.; Maier, C.M.; Chan, P.H. Prevention of JNK phosphorylation as a mechanism for rosiglitazone in neuroprotection after transient cerebral ischemia: Activation of dual specificity phosphatase. J. Cereb. Blood Flow Metab. 2013, 33, 106–114. [Google Scholar] [CrossRef] [PubMed]
- Bolden, A.; Bernard, L.; Jones, D.; Akinyeke, T.; Stewart, L.V. The PPAR gamma agonist troglitazone regulates erk 1/2 phosphorylation via a PPARγ-independent, MEK-dependent pathway in human prostate cancer cells. PPAR Res. 2012, 2012. [Google Scholar] [CrossRef] [PubMed]
- Nair, V.; Sreevalsan, S.; Basha, R.; Abdelrahim, M.; Abudayyeh, A.; Hoffman, A.R.; Safe, S. Mechanism of metformin-dependent inhibition of mammalian target of rapamycin (mTOR) and Ras activity in pancreatic cancer: Role of specificity protein (Sp) transcription factors. J. Biol. Chem. 2014, 289, 27692–27701. [Google Scholar] [CrossRef] [PubMed]
- San, Y.-Z.; Liu, Y.; Zhang, Y.; Shi, P.-P.; Zhu, Y.-L. Peroxisome proliferator-activated receptor-gamma agonist inhibits the mammalian target of rapamycin signaling pathway and has a protective effect in a rat model of status epilepticus. Mol. Med. Rep. 2015, 12, 1877–1883. [Google Scholar] [CrossRef] [PubMed]
- Blanchard, P.-G.; Festuccia, W.T.; Houde, V.P.; St-Pierre, P.; Brule, S.; Turcotte, V.; Côté, M.; Bellmann, K.; Marette, A.; Deshaies, Y. Major involvement of mTOR in the PPARgamma-induced stimulation of adipose tissue lipid uptake and fat accretion. J. Lipid Res. 2012, 53, 1117–1125. [Google Scholar] [CrossRef] [PubMed]
- Isoda, K.; Young, J.L.; Zirlik, A.; MacFarlane, L.A.; Tsuboi, N.; Gerdes, N.; Schonbeck, U.; Libby, P. Metformin inhibits proinflammatory responses and nuclear factor-kappaB in human vascular wall cells. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 611–617. [Google Scholar] [CrossRef] [PubMed]
- Hou, Y.; Moreau, F.; Chadee, K. PPARgamma is an E3 ligase that induces the degradation of NFkappaB/p65. Nat. Commun. 2012, 3, 1300. [Google Scholar] [CrossRef] [PubMed]
- Yuan, X.; Zhang, Z.; Gong, K.; Zhao, P.; Qin, J.; Liu, N. Inhibition of reactive oxygen species/extracellular signal-regulated kinases pathway by pioglitazone attenuates advanced glycation end products-induced proliferation of vascular smooth muscle cells in rats. Biol. Pharm. Bull. 2011, 34, 618–623. [Google Scholar] [CrossRef] [PubMed]
- Dallaglio, K.; Bruno, A.; Cantelmo, A.R.; Esposito, A.I.; Ruggiero, L.; Orecchioni, S.; Calleri, A.; Bertolini, F.; Pfeffer, U.; Noonan, D.M.; et al. Paradoxic effects of metformin on endothelial cells and angiogenesis. Carcinogenesis 2014, 35, 1055–1066. [Google Scholar] [CrossRef] [PubMed]
- Ersoy, C.; Kiyici, S.; Budak, F.; Oral, B.; Guclu, M.; Duran, C.; Selimoglu, H.; Erturk, E.; Tuncel, E.; Imamoglu, S. The effect of metformin treatment on VEGF and PAI-1 levels in obese type 2 diabetic patients. Diabetes Res. Clin. Pract. 2008, 81, 56–60. [Google Scholar] [CrossRef] [PubMed]
- Lu, D.; Carson, D.A. Repression of beta-catenin signaling by PPAR gamma ligands. Eur. J. Pharmacol. 2010, 636, 198–202. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.-S.; Chou, F.-S.; Bloomston, M.; Vonau, M.S.; Saji, M.; Espinosa, A.; Pinzone, J.J. Thiazolidinediones downregulate Wnt/beta-catenin signaling via multiple mechanisms in breast cancer cells. J. Surg. Res. 2009, 153, 210–216. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.; Li, D.-Q. PAKs in Human Cancer Progression: From Inception to Cancer Therapeutic to Future Oncobiology. Adv. Cancer Res. 2016, 130, 137–209. [Google Scholar] [CrossRef] [PubMed]
- Donath, M.Y.; Shoelson, S.E. Type 2 diabetes as an inflammatory disease. Nat. Rev. Immunol. 2011, 11, 98–107. [Google Scholar] [CrossRef] [PubMed]
- Bosi, E. Metformin—The gold standard in type 2 diabetes: What does the evidence tell us? Diabetes Obes. Metab. 2009, 11 (Suppl. 2), 3–8. [Google Scholar] [CrossRef] [PubMed]
- Hubsman, M.W.; Volinsky, N.; Manser, E.; Yablonski, D.; Aronheim, A. Autophosphorylation-dependent degradation of Pak1, triggered by the Rho-family GTPase, Chp. Biochem. J. 2007, 404, 487–497. [Google Scholar] [CrossRef]
- Li, R.; Debreceni, B.; Jia, B.; Gao, Y.; Tigyi, G.; Zheng, Y. Localization of the PAK1-, WASP-, and IQGAP1-specifying regions of Cdc42. J. Biol. Chem. 1999, 274, 29648–29654. [Google Scholar] [CrossRef] [PubMed]
- Drews, G.; Krippeit-Drews, P.; Dufer, M. Oxidative stress and beta-cell dysfunction. Pflugers Arch. Eur. J. Physiol. 2010, 460, 703–718. [Google Scholar] [CrossRef] [PubMed]
- Essers, M.A.G.; de Vries-Smits, L.M.M.; Barker, N.; Polderman, P.E.; Burgering, B.M.T.; Korswagen, H.C. Functional interaction between beta-catenin and FOXO in oxidative stress signaling. Science 2005, 308, 1181–1184. [Google Scholar] [CrossRef] [PubMed]
- Mazumdar, A.; Kumar, R. Estrogen regulation of Pak1 and FKHR pathways in breast cancer cells. FEBS Lett. 2003, 535, 6–10. [Google Scholar] [CrossRef]
- Li, X.; Cheng, K.K.Y.; Liu, Z.; Yang, J.-K.; Wang, B.; Jiang, X.; Zhou, Y.; Hallenborg, P.; Hoo, R.L.; Lam, K.S.; et al. The MDM2-p53-pyruvate carboxylase signalling axis couples mitochondrial metabolism to glucose-stimulated insulin secretion in pancreatic beta-cells. Nat. Commun. 2016, 7, 11740. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Li, Y.; Gu, H.; Zhu, G.; Li, J.; Cao, L.; Li, F. P21-Activated kinase 6 (PAK6) inhibits prostate cancer growth via phosphorylation of androgen receptor and tumorigenic E3 ligase murine double minute-2 (MDM2). J. Biol. Chem. 2013, 288, 3359–3369. [Google Scholar] [CrossRef] [PubMed]
- Murray, B.W.; Guo, C.; Piraino, J.; Westwick, J.K.; Zhang, C.; Lamerdin, J.; Dagostino, E.; Knighton, D.; Loi, C.M.; Zager, M.; et al. Small-molecule p21-activated kinase inhibitor PF-3758309 is a potent inhibitor of oncogenic signaling and tumor growth. Proc. Natl. Acad. Sci. USA 2010, 107, 9446–9451. [Google Scholar] [CrossRef] [PubMed]
- Park, S.-Y.; Lee, J.H.; Ha, M.; Nam, J.-W.; Kim, V.N. miR-29 miRNAs activate p53 by targeting p85 alpha and CDC42. Nat. Struct. Mol. Biol. 2009, 16, 23–29. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Nikulenkov, F.; Zawacka-Pankau, J.; Li, H.; Gabdoulline, R.; Xu, J.; Eriksson, S.; Hedström, E.; Issaeva, N.; Kel, A.; et al. ROS-dependent activation of JNK converts p53 into an efficient inhibitor of oncogenes leading to robust apoptosis. Cell Death Differ. 2014, 21, 612–623. [Google Scholar] [CrossRef] [PubMed]
- Parvathy, M.; Sreeja, S.; Kumar, R.; Pillai, M.R. Potential role of p21 Activated Kinase 1 (PAK1) in the invasion and motility of oral cancer cells. BMC Cancer 2016, 16 (Suppl. 1), 293. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, S.; Dong, Q.-Z.; Jiang, G.-Y.; Han, Y.; Wang, L.; Wang, E.-H. The P21-activated kinase expression pattern is different in non-small cell lung cancer and affects lung cancer cell sensitivity to epidermal growth factor receptor tyrosine kinase inhibitors. Med. Oncol. 2016, 33, 22. [Google Scholar] [CrossRef] [PubMed]
- Gan, J.; Zhang, Y.; Ke, X.; Tan, C.; Ren, H.; Dong, H.; Jiang, J.; Chen, S.; Zhuang, Y.; Zhang, H. Dysregulation of PAK1 Is Associated with DNA Damage and Is of Prognostic Importance in Primary Esophageal Small Cell Carcinoma. Int. J. Mol. Sci. 2015, 16, 12035–12050. [Google Scholar] [CrossRef] [PubMed]
- Ito, M.; Nishiyama, H.; Kawanishi, H.; Matsui, S.; Guilford, P.; Reeve, A.; Ogawa, O. P21-activated kinase 1: A new molecular marker for intravesical recurrence after transurethral resection of bladder cancer. J. Urol. 2007, 178 Pt 1, 1073–1079. [Google Scholar] [CrossRef] [PubMed]
- Song, B.; Wang, W.; Zheng, Y.; Yang, J.; Xu, Z. P21-activated kinase 1 and 4 were associated with colorectal cancer metastasis and infiltration. J. Surg. Res. 2015, 196, 130–135. [Google Scholar] [CrossRef] [PubMed]
- Girnun, G.D.; Smith, W.M.; Drori, S.; Sarraf, P.; Mueller, E.; Eng, C.; Nambiar, P.; Rosenberg, D.W.; Bronson, R.T.; Edelmann, W.; et al. APC-dependent suppression of colon carcinogenesis by PPARgamma. Proc. Natl. Acad. Sci. USA 2002, 99, 13771–13776. [Google Scholar] [CrossRef] [PubMed]
- Grommes, C.; Karlo, J.C.; Caprariello, A.; Blankenship, D.; Dechant, A.; Landreth, G.E. The PPARgamma agonist pioglitazone crosses the blood-brain barrier and reduces tumor growth in a human xenograft model. Cancer Chemother. Pharmacol. 2013, 71, 929–936. [Google Scholar] [CrossRef] [PubMed]
- Burotto, M.; Szabo, E. PPARγ in Head and Neck Cancer Prevention. Oral Oncol. 2014, 50, 924–929. [Google Scholar] [CrossRef] [PubMed]
- Lewis, J.D.; Habel, L.A.; Quesenberry, C.P.; Strom, B.L.; Peng, T.; Hedderson, M.M.; Ehrlich, S.F.; Mamtani, R.; Bilker, W.; Vaughn, D.J.; et al. Pioglitazone Use and Risk of Bladder Cancer and Other Common Cancers in Persons With Diabetes. JAMA 2015, 314, 265–277. [Google Scholar] [CrossRef] [PubMed]
- Majima, T.; Komatsu, Y.; Doi, K.; Shigemoto, M.; Takagi, C.; Fukao, A.; Corners, J.; Nakao, K. Safety and efficacy of low-dose pioglitazone (7.5 mg/day) vs. standard-dose pioglitazone (15 mg/day) in Japanese women with type 2 diabetes mellitus. Endocr. J. 2006, 53, 325–330. [Google Scholar] [CrossRef] [PubMed]
- Bradley, M.C.; Ferrara, A.; Achacoso, N.; Ehrlich, S.F.; Quesenberry, C.P.J.; Habel, L.A. A Cohort Study of Metformin and Colorectal Cancer Risk among Patients with Diabetes Mellitus. Cancer Epidemiol. Biomark. Prev. 2018, 27, 525–530. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Hu, D.; Chen, H.; Shi, G.; Fetahu, I.S.; Wu, F.; Rabidou, K.; Fang, R.; Tan, L.; Xu, S.; et al. Glucose-regulated phosphorylation of TET2 by AMPK reveals a pathway linking diabetes to cancer. Nature 2018, 559, 637–641. [Google Scholar] [CrossRef] [PubMed]
- Drew, D.A.; Cao, Y.; Chan, A.T. Aspirin and colorectal cancer: The promise of precision chemoprevention. Nat. Rev. Cancer 2016, 16, 173–186. [Google Scholar] [CrossRef] [PubMed]
- Lyakhovich, A.; Gasche, C. Systematic review: Molecular chemoprevention of colorectal malignancy by mesalazine. Aliment. Pharmacol. Ther. 2010, 31, 202–209. [Google Scholar] [CrossRef] [PubMed]
- Karagozian, R.; Burakoff, R. The role of mesalamine in the treatment of ulcerative colitis. Ther. Clin. Risk Manag. 2007, 3, 893–903. [Google Scholar] [PubMed]
- Rousseaux, C.; Lefebvre, B.; Dubuquoy, L.; Lefebvre, P.; Romano, O.; Auwerx, J.; Metzger, D.; Wahli, W.; Desvergne, B.; Naccari, G.C.; et al. Intestinal antiinflammatory effect of 5-aminosalicylic acid is dependent on peroxisome proliferator-activated receptor-gamma. J. Exp. Med. 2005, 201, 1205–1215. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Upstream | Downstream | ||
|---|---|---|---|
| AMPK | [6] | MAPK-p38/JNK | [7] |
| RAS | [8] | MAPK-ERK | [9] |
| mTOR | [10,11] | mTOR | [12] |
| PI3k/AKT | [13] | PI3k/AKT | [9,12,14] |
| RAC1 | [15] | NF-kB | [14] |
| CDC42 | [15] | p-PAK1 | [14] |
| PPARy | [14] | ||
| ROS | [16] | ||
| VEGF | [9] | ||
| Wnt/B-catenin | [17,18,19] | ||
| PAK Signaling Pathways in Glucose Homeostasis | |
|---|---|
| Reference | |
| AMPK | [6,20] |
| MAPK-p38/JNK | [21] |
| MAPK-ERK | [22] |
| mTOR | [23] |
| NF-kB | [24] |
| p-PAK1 | [25] |
| PI3K/AKT | [26] |
| PPARy | [27] |
| RAC1/CDC42 | [28,29] |
| RAS | [30] |
| Wnt/Beta-catenin | [31] |
| Role of PAK Signaling Pathways in Disease | |||||
|---|---|---|---|---|---|
| Pathway | Inflammation | Proliferation | Survival | Angiogenesis | Reference |
| AMPK | ↓ | ↓ | ↑ | ↑ | [32,33,34,35] |
| MAPK-p38/JNK | ↑ | ↑ | ↑ | ↑ | [36] |
| MAPK-ERK | ↑ | ↑ | ↑ | ↑ | [9] |
| mTOR | ↑ | ↑ | ↑ | ↑ | [37] |
| NF-kB | ↑ | ↑ | ↑ | ↑ | [14] |
| p-PAK1 | ↑ | ↑ | ↑ | ↑ | [38] |
| PI3k/AKT | ↑ | ↑ | ↑ | ↑ | [37] |
| PPARy | ↓ | ↓ | ↓ | ↑↓ | [39,40] |
| RAC1/CDC42 | ↑ | ↑ | ↑ | ↑ | [15,18,26,28] |
| ROS | ↑ | ↑↓ | ↑↓ | ↑↓ | [16,41] |
| VEGF | ↑ | ↑ | ↑ | ↑ | [42,43] |
| Wnt/B-catenin | ↑ | ↑ | ↑ | ↑ | [17,18,19,38] |
| PAK Signaling Pathways Utilized by Anti-diabetic Drugs | ||||
|---|---|---|---|---|
| Pathway | Metformin | Glyburide | Glitazone | Citation |
| AMPK | ↑ | - | ↑ | [44,45] |
| MAPK-p38/JNK | ↑ | ↑ | ↓ | [46,47,48,49] |
| MAPK-ERK | ↓ | - | ↑↓ | [47,50] |
| mTOR | ↓ | - | ↓ | [51,52,53] |
| NF-kB | ↓ | - | ↓ | [14,38,54,55] |
| p-PAK1 | ↑ | - | - | [44] |
| PI3K/AKT | ↑ | ↓ | ↓ | [37,48] |
| PPAR-γ | - | - | ↑ | [27] |
| RAC1/CDC42 | ↑ | - | - | [47] |
| RAS | ↓ | - | - | [51] |
| ROS | - | ↑ | ↓ | [56] |
| VEGF | ↑↓ | - | ↑↓ | [57,58] |
| Wnt/ B-catenin | - | - | ↓ | [59,60] |
| Summary of Clinical Study Designs | |||||||
|---|---|---|---|---|---|---|---|
| Study Type | Design | Condition | Intervention | Primary Outcome Measure | Number of Patients Treatment (tx); Control (con) | Duration | Identifier |
| Observational | Prospective cohort | Diabetes Bladder Ca | Pioglitazone | Incident Diagnoses | Tx: 34,181 Con: 158,918 | 10 years | NCT01637935 |
| Interventional | Single group; prevention | Head/Neck Ca; Oral Leukoplakia | Pioglitazone | Overall response | Tx: 21 Con: 0 | 12 weeks | NCT00099021 |
| Interventional | Randomized; double blind; treatment | Oral Leukoplakia | Pioglitazone | Overall response | Tx: 27 Con: 25 | 24 weeks | NCT00951379 |
| Interventional | Single group; Treatment | Non Small cell lung Ca | Pioglitazone | % change Ki67 IHC | Tx: 6 Con: 0 | 14–42 days | NCT01342770 |
| Interventional | Randomized; double blind; treatment | Prostate adenocarcinoma | Metformin | % change Ki67 IHC | Tx: 10 Con: 10 | 4–12 weeks | NCT01433913 |
| Interventional | Randomized; double blind; prevention | Barrett Esophagus; Esophageal Ca | Metformin | % change pS6K1 IHC | Tx: 38 Con: 36 | 3 months | NCT01447927 |
| Interventional | Single group; prevention | Adenomatous polyp; CRC; obesity | Metformin | % change S6-serine235 | Tx: 45 Con: 0 | 12 weeks | NCT01312467 |
| Biomarker Analysis in Cancer Patients Treated with Pioglitazone or Metformin | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Disease | Drug | Marker analyzed via immunohistochemistry (IHC) | Study ID | ||||||||||
| Apoptosis | Cell cycle control | PI3K/mTOR | PPARy | ||||||||||
| pioglitazone | metformin | BCL2 | MUC1 | CyclinD1 | Ki67 | P21 | PS6K1 | PS6ser235 | Total | Nuclear | Cytoplasm | ||
| Oral leukoplakia | + | - | ↓ ↓ | - | ↓ ↓ | ↓ | ↑ | - | - | - | ↓ ↓ ↓ | ↑ | NCT0095 1379 |
| Non-small cell lung Ca | + | - | - | 0 | ↓ ↓ ↓ ↓ | ↓ ↓ | ↓ | - | - | ↑ ↑ ↑ ↑ | - | - | NCT0134 2770 |
| Prostate Ca | - | + | - | - | - | ↑ ↑ | - | - | - | - | - | - | NCT0143 3913 |
| Esophageal Ca | - | + | - | - | - | - | - | ↑ | - | - | - | - | NCT0144 7927 |
| Colon Ca | - | + | - | - | - | - | - | - | 0 | - | - | - | NCT0131 2467 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dammann, K.; Khare, V.; Coleman, C.; Berdel, H.; Gasche, C. p-21 Activated Kinase as a Molecular Target for Chemoprevention in Diabetes. Geriatrics 2018, 3, 73. https://doi.org/10.3390/geriatrics3040073
Dammann K, Khare V, Coleman C, Berdel H, Gasche C. p-21 Activated Kinase as a Molecular Target for Chemoprevention in Diabetes. Geriatrics. 2018; 3(4):73. https://doi.org/10.3390/geriatrics3040073
Chicago/Turabian StyleDammann, Kyle, Vineeta Khare, Clyde Coleman, Henrik Berdel, and Christoph Gasche. 2018. "p-21 Activated Kinase as a Molecular Target for Chemoprevention in Diabetes" Geriatrics 3, no. 4: 73. https://doi.org/10.3390/geriatrics3040073
APA StyleDammann, K., Khare, V., Coleman, C., Berdel, H., & Gasche, C. (2018). p-21 Activated Kinase as a Molecular Target for Chemoprevention in Diabetes. Geriatrics, 3(4), 73. https://doi.org/10.3390/geriatrics3040073