
Development of Liver-on-Chip Integrating a Hydroscaffold Mimicking the Liver’s Extracellular Matrix
 , , , , ,
, , , , ,
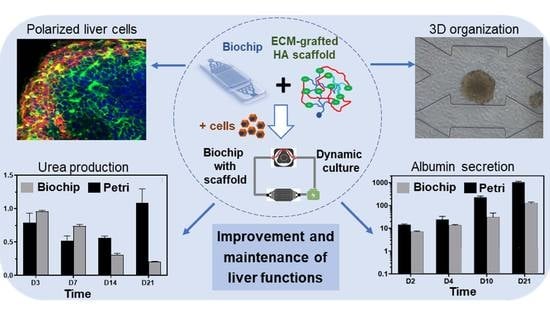
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Biochip Fabrication
2.2. Hydroscaffold Preparation
2.3. IDCCM Fluidic Platform
2.4. Cell Culture
2.5. Cell Viability
2.6. Immunostaining Assays
2.7. Albumin and Urea Measurements
2.8. Flow Control and Pressure Drop Measurement
2.9. Scanning Electron Microscopy (SEM)
2.10. Statistical Analysis
3. Results
3.1. Integration of the Hydroscaffold into the Biochip
3.2. Cell Culture in Biochip Containing the Hydroscaffold
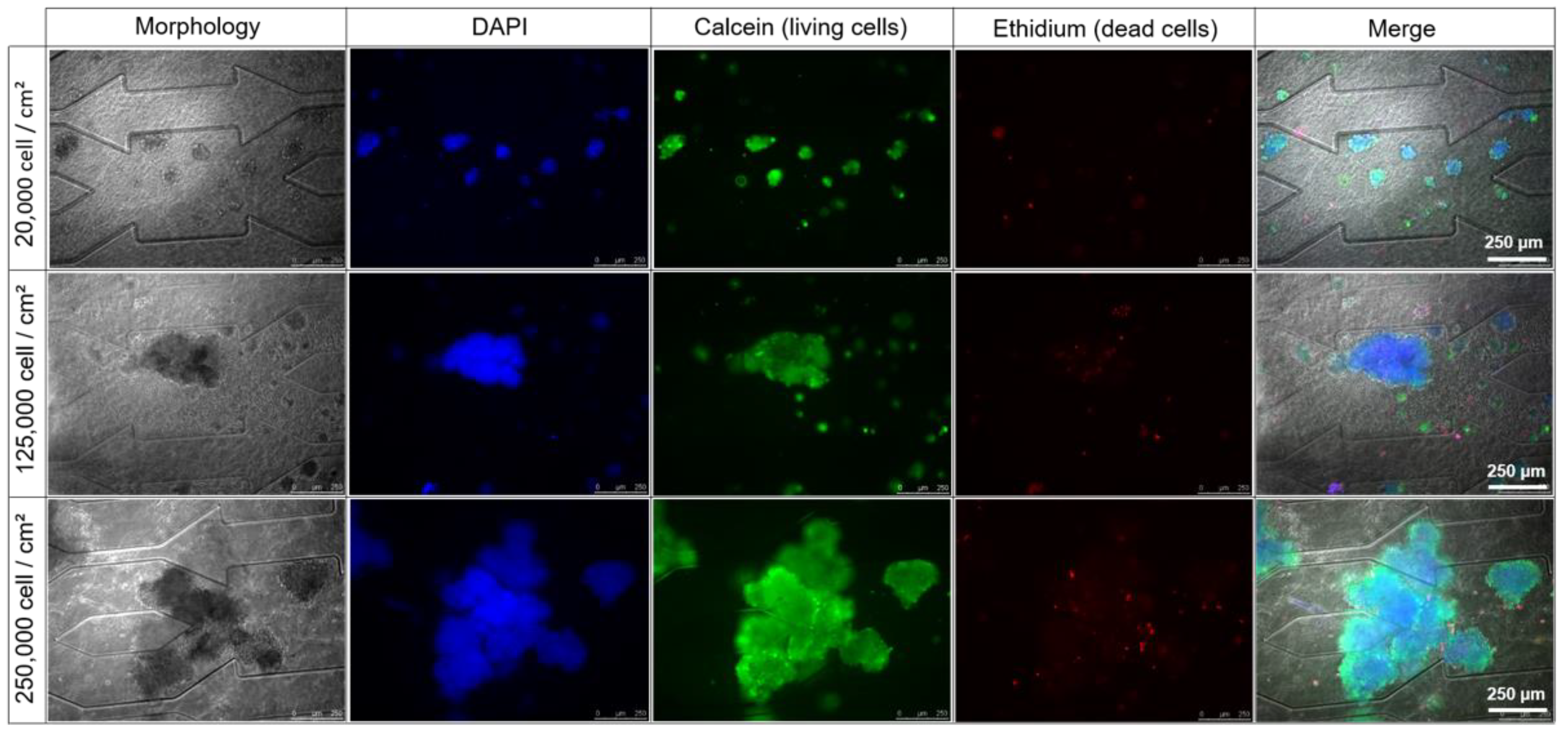
3.2.1. Effect of Cell Seeding Density: Morphology
3.2.2. Cell Viability and Functionality
3.3. Long-Term Cell Culture in a Biochip Containing the Hydroscaffold
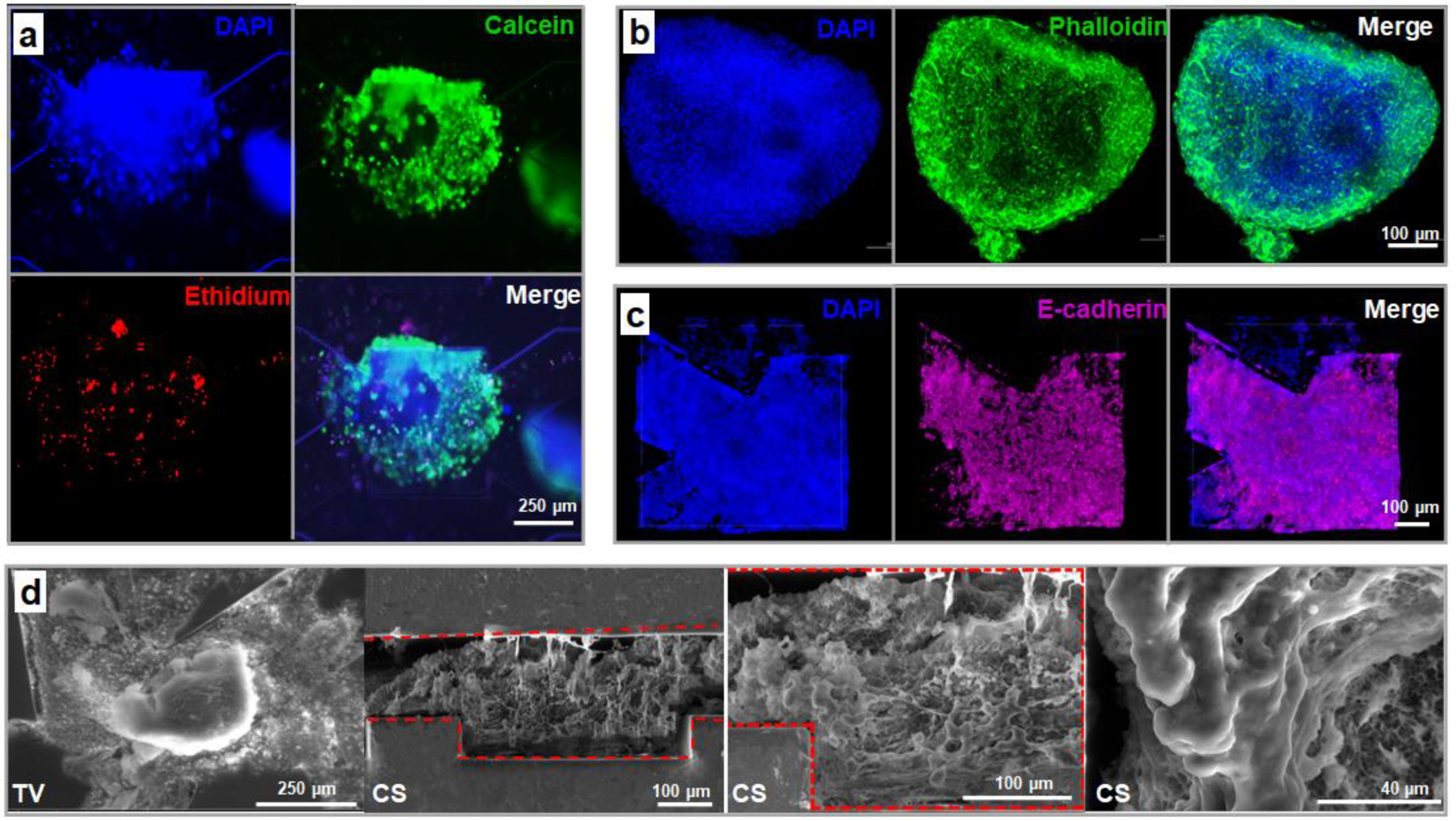
3.3.1. Cell Proliferation and Spheroid Formation
3.3.2. Spheroid Morphology and Integrity
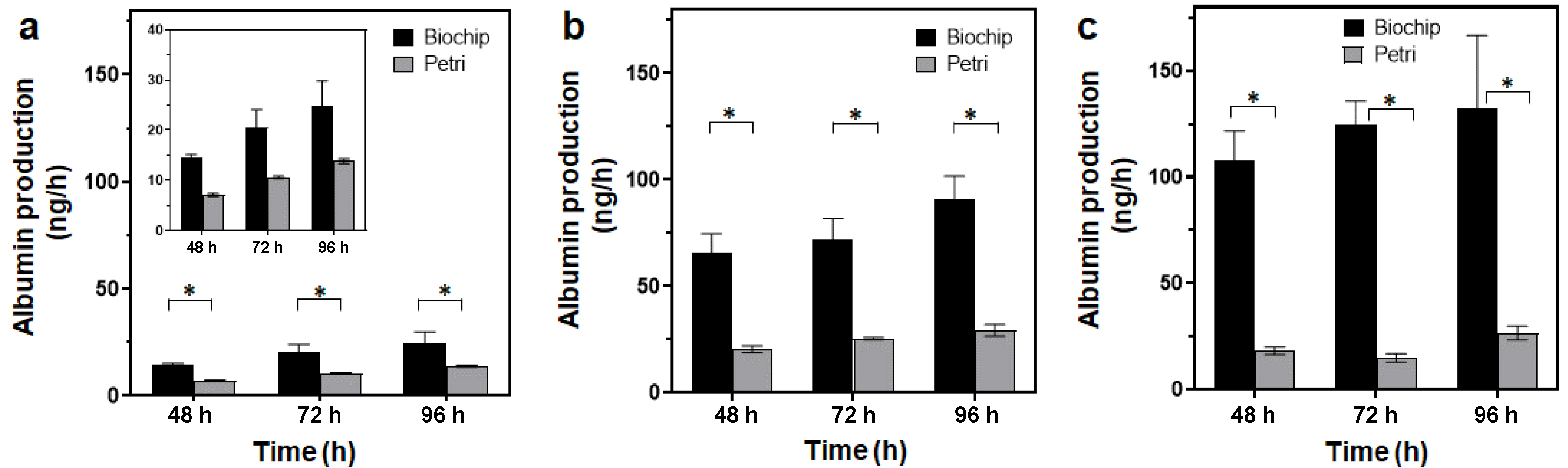
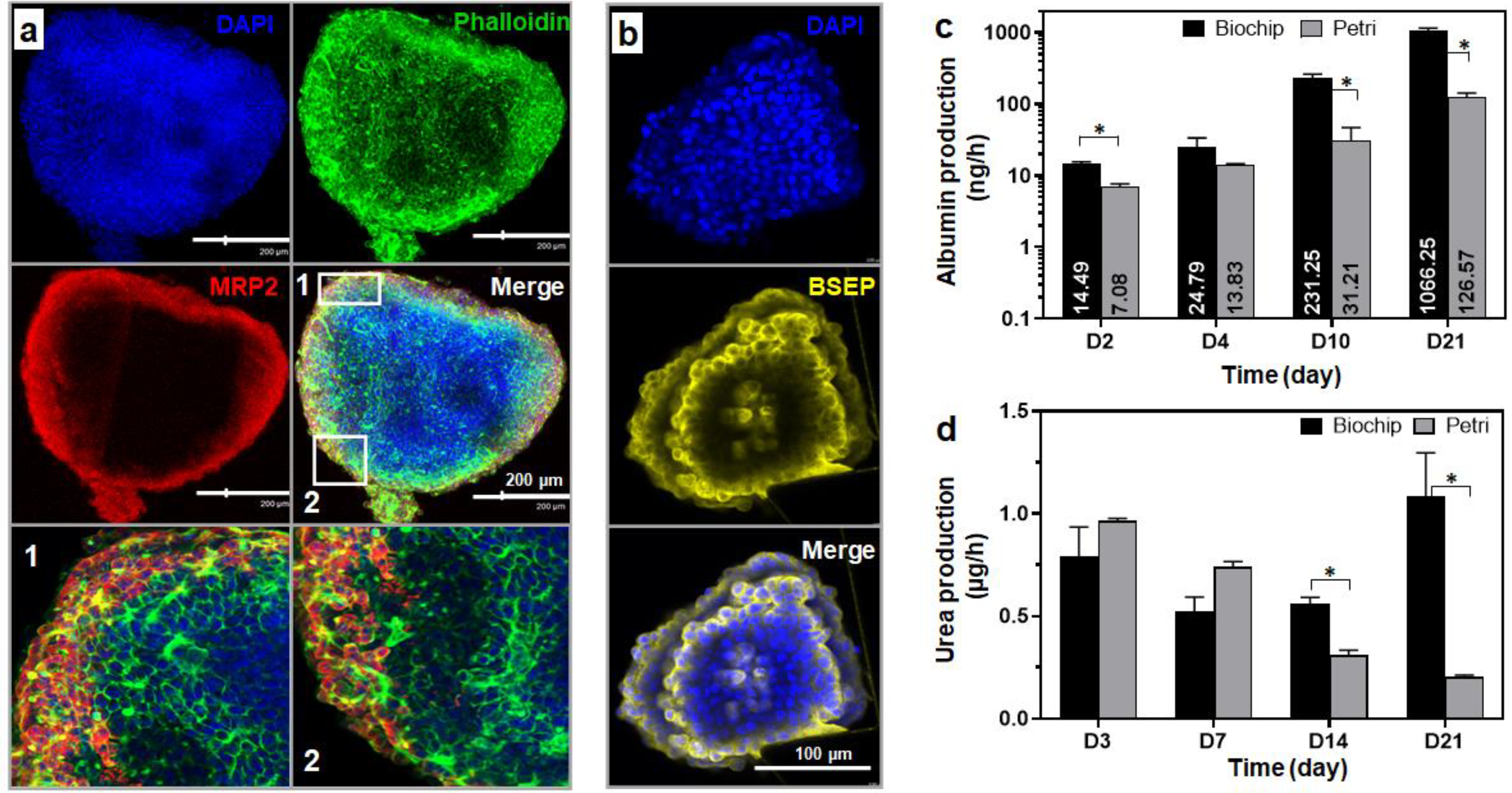
3.3.3. Spheroid Functionality
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hughes, J.P.; Rees, S.; Kalindjian, S.B.; Philpott, K.L. Principles of early drug discovery. Br. J. Pharmacol. 2011, 162, 1239–1249. [Google Scholar] [CrossRef]
- Khetani, S.R.; Berger, D.R.; Ballinger, K.R.; Davidson, M.D.; Lin, C.; Ware, B.R. Microengineered liver tissues for drug testing. J. Lab. Autom. 2015, 20, 216–250. [Google Scholar] [CrossRef]
- Freyer, N.; Knöspel, F.; Strahl, N.; Amini, L.; Schrade, P.; Bachmann, S.; Damm, G.; Seehofer, D.; Jacobs, F.; Monshouwer, M.; et al. Hepatic differentiation of human induced pluripotent stem cells in a perfused three-dimensional multicompartment bioreactor. Biores. Open Access 2016, 5, 235–248. [Google Scholar] [CrossRef]
- Sivaraman, A.; Leach, J.K.; Townsend, S.; Iida, T.; Hogan, B.J.; Stolz, D.B.; Fry, R.; Samson, L.D.; Tannenbaum, S.R.; Griffith, L.G. A microscale in vitro physiological model of the liver: Predictive screens for drug metabolism and enzyme induction. Curr. Drug. Metab. 2005, 6, 569–591. [Google Scholar] [CrossRef]
- Mann, D.A. Human induced pluripotent stem cell-derived hepatocytes for toxicology testing. Expert Opin. Drug Metab. Toxicol. 2015, 11, 1–5. [Google Scholar] [CrossRef]
- Merlier, F.; Jellali, R.; Leclerc, E. Online hepatic rat metabolism by coupling liver biochip and mass spectrometry. Analyst 2017, 142, 3747–3757. [Google Scholar] [CrossRef]
- Bell, C.C.; Lauschke, V.M.; Vorrink, S.U.; Palmgren, H.; Duffin, R.; Andersson, T.B.; Ingelman-Sundberg, M. Transcriptional, functional, and mechanistic comparisons of stem cell-derived hepatocytes, HepaRG cells, and three-dimensional human hepatocyte spheroids as predictive in vitro systems for drug-induced liver injury. Drug Metab. Dispos. 2017, 45, 419–429. [Google Scholar] [CrossRef]
- Polidoro, M.A.; Ferrari, E.; Marzorati, S.; Lleo, A.; Rasponi, M. Experimental liver models: From cell culture techniques to microfluidic organs-on-chip. Liver Int. 2021, 41, 1744–1761. [Google Scholar] [CrossRef]
- Ghabril, M.; Chalasani, N.; Björnsson, E. Drug-induced liver injury: A clinical update. Curr. Opin. Gastroenterol. 2010, 26, 222–226. [Google Scholar] [CrossRef]
- Bernal, W.; Wendon, J. 2013. Acute liver failure. N. Engl. J. Med. 2013, 369, 2525–2534. [Google Scholar] [CrossRef]
- Donato, M.T.; Tolosa, L. Stem-cell derived hepatocyte-like cells for the assessment of drug-induced liver injury. Differentiation 2019, 106, 15–22. [Google Scholar] [CrossRef]
- Kuna, L.; Bozic, I.; Kizivat, T.; Bojanic, K.; Mrso, M.; Kralj, E.; Smolic, R.; Wu, G.Y.; Smolic, M. Models of drug induced liver injury (DILI)—Current issues and future perspectives. Curr. Drug Metab. 2018, 19, 830–838. [Google Scholar] [CrossRef]
- Chen, M.; Bisgin, H.; Tong, L.; Hong, H.; Fang, H.; Borlak, J.; Tong, W. Toward predictive models for drug-induced liver injury in humans: Are we there yet? Biomark. Med. 2014, 8, 201–213. [Google Scholar] [CrossRef]
- Segovia-Zafra, A.; Di Zeo-Sánchez, D.; López-Gómez, C.; Pérez-Valdés, Z.; García-Fuentes, E.; Andrade, R.; Lucena, I.; Villanueva-Paz, M. Preclinical models of idiosyncratic drug-induced liver injury (iDILI): Moving towards prediction. Acta Pharm. Sin. B 2021, 11, 3685–3726. [Google Scholar] [CrossRef]
- Bhatia, S.N.; Ingber, D.E. Microfluidic organs-on-chips. Nat. Biotechnol. 2014, 32, 760–772. [Google Scholar] [CrossRef]
- Chen, X.; Zhang, Y.S.; Zhang, X.; Liu, C. Organ-on-a-chip platforms for accelerating the evaluation of nanomedicine. Bioact. Mater. 2021, 6, 1012–1027. [Google Scholar] [CrossRef]
- Fang, Y.; Eglen, R.M. Three-dimensional cell cultures in drug discovery and development. SLAS Discov. 2017, 22, 456–472. [Google Scholar] [CrossRef]
- Gómez-Lechón, M.J.; Tolosa, L.; Conde, I.; Donato, M.T. Competency of different cell models to predict human hepatotoxic drugs. Expert Opin. Drug Metab. Toxicol. 2014, 10, 1553–1568. [Google Scholar] [CrossRef]
- Lauschke, V.M.; Hendriks, D.F.; Bell, C.C.; Andersson, T.B.; Ingelman-Sundberg, M. Novel 3D culture systems for studies of human liver function and assessments of the hepatotoxicity of drugs and drug candidates. Chem. Res. Toxicol. 2016, 29, 1936–1955. [Google Scholar] [CrossRef]
- Ruoß, M.; Vosough, M.; Königsrainer, A.; Nadalin, S.; Wagner, S.; Sajadian, S.; Huber, D.; Heydari, Z.; Ehnert, S.; Hengstler, J.G.; et al. Towards improved hepatocyte cultures: Progress and limitations. Food Chem. Toxicol. 2020, 138, 111188. [Google Scholar] [CrossRef]
- Foster, A.J.; Chouhan, B.; Regan, S.L.; Rollison, H.; Amberntsson, S.; Andersson, L.C.; Srivastava, A.; Darnell, M.; Cairns, J.; Lazic, S.E.; et al. Integrated in vitro models for hepatic safety and metabolism: Evaluation of a human liver-chip and liver spheroid. Arch. Toxicol. 2019, 93, 1021–1037. [Google Scholar] [CrossRef]
- Ingber, D.E. Is it time for reviewer 3 to request human organ chip experiments instead of animal validation studies? Adv. Sci. 2020, 7, 2002030. [Google Scholar] [CrossRef]
- Essaouiba, A.; Okitsu, T.; Jellali, R.; Shinohara, M.; Danoy, M.; Tauran, Y.; Legallais, C.; Sakai, Y.; Leclerc, E. Microwell-based pancreas-on-chip model enhances genes expression and functionality of rat islets of Langerhans. Mol. Cell. Endocrinol. 2020, 514, 110892. [Google Scholar] [CrossRef]
- Godoy, P.; Hewitt, N.J.; Albrecht, U.; Andersen, M.E.; Ansari, N.; Bhattacharya, S.; Bode, J.G.; Bolleyn, J.; Borner, C.; Böttger, J.; et al. Recent advances in 2D and 3D in vitro systems using primary hepatocytes, alternative hepatocyte sources and non-parenchymal liver cells and their use in investigating mechanisms of hepatotoxicity, cell signaling and ADME. Arch. Toxicol. 2013, 87, 1315–1530. [Google Scholar] [CrossRef]
- Cui, X.; Hartanto, Y.; Zhang, H. Advances in multicellular spheroids formation. J. R. Soc. Interface 2017, 14, 20160877. [Google Scholar] [CrossRef]
- Fu, J.J.; Lv, X.H.; Wang, L.X.; He, X.; Li, Y.; Yu, L.; Li, C.M. Cutting and bonding parafilm® to fast prototyping flexible hanging drop chips for 3D spheroid cultures. Cell. Mol. Bioeng. 2020, 14, 187–199. [Google Scholar] [CrossRef]
- Prot, J.M.; Videau, O.; Brochot, C.; Legallais, C.; Bénech, H.; Leclerc, E. A cocktail of metabolic probes demonstrates the relevance of primary human hepatocyte cultures in a microfluidic biochip for pharmaceutical drug screening. Int. J. Pharm. 2011, 408, 67–75. [Google Scholar] [CrossRef]
- Jellali, R.; Bricks, T.; Jacques, S.; Fleury, M.J.; Paullier, P.; Merlier, F.; Leclerc, E. Long-term human primary hepatocyte cultures in a microfluidic liver biochip show maintenance of mRNA levels and higher drug metabolism compared with Petri cultures. Biopharm. Drug Dispos. 2016, 37, 264–275. [Google Scholar] [CrossRef]
- Choucha-Snouber, L.; Aninat, C.; Grsicom, L.; Madalinski, G.; Brochot, C.; Poleni, P.E.; Razan, F.; Guillouzo, C.G.; Legallais, C.; Corlu, A.; et al. Investigation of ifosfamide nephrotoxicity induced in a liver–kidney co-culture biochip. Biotechnol. Bioeng. 2013, 110, 597–608. [Google Scholar] [CrossRef]
- Leclerc, E.; Hamon, J.; Claude, I.; Jellali, R.; Naudot, M.; Bois, F. Investigation of acetaminophen toxicity in HepG2/C3a microscale cultures using a system biology model of glutathione depletion. Cell Biol. Toxicol. 2015, 31, 173–185. [Google Scholar] [CrossRef]
- Jellali, R.; Gilard, F.; Pandolfi, V.; Legendre, A.; Fleury, M.-J.; Paullier, P.; Legallais, C.; Leclerc, E. Metabolomics-on-a-chip approach to study hepatotoxicity of DDT, permethrin and their mixtures. J. Appl. Toxicol. 2018, 38, 1121–1134. [Google Scholar] [CrossRef]
- Danoy, M.; Tauran, Y.; Poulain, S.; Jellali, R.; Bruce, J.; Leduc, M.; Le Gall, M.; Gilard, F.; Kido, T.; Arakawa, H.; et al. Multi-omics analysis of hiPSCs-derived HLCs matured on-chip revealed patterns typical of liver regeneration. Biotechnol. Bioeng. 2021, 118, 3716–3732. [Google Scholar] [CrossRef]
- Boulais, L.; Jellali, R.; Pereira, U.; Leclerc, E.; Bencherif, S.A.; Legallais, C. Cryogel-integrated biochip for liver tissue engineering. ACS Appl. Bio. Mater. 2021, 4, 5617–5626. [Google Scholar] [CrossRef]
- Baudoin, R.; Alberto, G.; Paullier, P.; Legallais, C.; Leclerc, E. Parallelized microfluidic biochips in multi well plate applied to liver tissue engineering. Sens. Actuators B Chem. 2012, 173, 919–926. [Google Scholar] [CrossRef]
- Jellali, R.; Paullier, P.; Fleury, M.-J.; Leclerc, E. Liver and kidney cells cultures in a new perfluoropolyether biochip. Sens. Actuators B Chem. 2016, 229, 396–407. [Google Scholar] [CrossRef]
- Souguir, Z.; Vidal, G.; Demange, E.; Louis, F. Method for producing hydrogel from modified hyaluronic acid and type I collagen. W.O. Patent 2016166479-A1, 20 October 2016. [Google Scholar]
- Baudoin, R.; Griscom, L.; Prot, J.M.; Legallais, C.; Leclerc, E. Behaviour of HepG2/C3a cell culture in a microfluidic bioreactor. Biochem. Eng. J. 2011, 53, 172–181. [Google Scholar] [CrossRef]
- Gaskell, H.; Sharma, P.; Colley, H.E.; Murdoch, C.; Williams, D.P.; Webb, S.D. Characterization of a functional C3A liver spheroid model. Toxicol. Res. 2016, 5, 1053–1065. [Google Scholar] [CrossRef]
- Wrzesinski, K.; Magnone, M.C.; Hansen, L.V.; Kruse, M.E.; Bergauer, T.; Bobadilla, M.; Gubler, M.; Mizrahi, J.; Zhang, K.; Andreasen, C.M.; et al. HepG2/C3A 3D spheroids exhibit stable physiological functionality for at least 24 days after recovering from trypsinisation. Toxicol. Res. 2013, 2, 163–172. [Google Scholar] [CrossRef]
- Ramaiahgari, S.C.; den Braver, M.W.; Herpers, B.; Terpstra, V.; Commandeur, J.N.M.; van de Water, B.; Price, L.S. A 3D in vitro model of differentiated HepG2 cell spheroids with improved liver-like properties for repeated dose high-throughput toxicity studies. Arch. Toxicol. 2014, 88, 1083–1095. [Google Scholar] [CrossRef]
- Moscato, S.; Ronca, F.; Campani, D.; Danti, S. Poly(vinyl alcohol)/gelatin hydrogels cultured with HepG2 cells as a 3D model of hepatocellular carcinoma: A morphological study. J. Funct. Biomater. 2015, 6, 16–32. [Google Scholar] [CrossRef] [Green Version]
- Danoy, M.; Poulain, S.; Lereau-Bernier, M.; Kato, S.; Scheidecker, B.; Kido, T.; Miyajima, A.; Sakai, Y.; Plessy, C.; Leclerc, E. Characterisation of the liver zonation like transcriptomic patterns in HLCs derived from hiPCs in a microfluidic biochip environment. Biotechnol. Prog. 2020, 36, e3013. [Google Scholar] [CrossRef]
- Matsumoto, S.; Rizki Safitir, A.; Danoy, M.; Maekawa, T.; Kinoshita, H.; Shinohara, M.; Sakai, Y.; Fujii, T.; Leclerc, E. Investigation of the hepatic respiration and liver zonation on rat hepatocytes using an integrated oxygen biosensor in a microscale device. Biotechnol. Prog. 2019, 35, e2854. [Google Scholar] [CrossRef]
- Allen, J.W.; Khetani, S.R.; Bhatia, S.N. In vitro zonation and toxicity in a hepatocyte bioreactor. Toxicol. Sci. 2004, 84, 110–119. [Google Scholar] [CrossRef]
- Lee, P.J.; Hung, P.J.; Lee, L.P. An artificial liver sinusoid with a microfluidic endothelial-like barrier for primary hepatocyte culture. Biotechnol. Bioeng. 2007, 97, 1340–1346. [Google Scholar] [CrossRef]
- Baudoin, R.; Corlu, A.; Griscom, L.; Legallais, C.; Leclerc, E. Trends in the development of microfluidic cell biochips for in vitro hepatotoxicity. Toxicol. Vitr. 2007, 21, 535–544. [Google Scholar] [CrossRef]
- Yu, F.; Deng, R.; Tong, W.H.; Huan, L.; Way, N.C.; IslamBadhan, A.; Iliescu, C.; Yu, H. A perfusion incubator liver chip for 3D cell culture with application on chronic hepatotoxicity testing. Sci. Rep. 2017, 7, 14528. [Google Scholar] [CrossRef]
- Gissen, P.; Arias, I.M. Structural and functional hepatocyte polarity and liver disease. J. Hepatol. 2015, 63, 1023–1037. [Google Scholar] [CrossRef]
- Bual, R.P.; Ijima, H. Intact extracellular matrix component promotes maintenance of liver-specific functions and larger aggregates formation of primary rat hepatocytes. Regen. Ther. 2019, 11, 258–268. [Google Scholar] [CrossRef] [PubMed]
- Andez, A.M.-H.; Amenta, P.S. The extracellular matrix in hepatic regeneration. FASEB J. 1995, 9, 1401–1410. [Google Scholar] [CrossRef]
- Arriazu, E.; de Galarreta, M.R.; Cubero, F.J.; Varela-Rey, M.; de Obanos, M.P.P.; Leung, T.M.; Lopategi, A.; Benedicto, A.; Abraham-Enachescu, I.; Nieto, N. Extracellular matrix and liver disease. Antioxid. Redox Signal. 2014, 21, 1078–1097. [Google Scholar] [CrossRef] [Green Version]
- Ijima, H.; Nakamura, S.; Bual, R.P.; Yoshida, K. Liver-specific extracellular matrix hydrogel promotes liver-specific functions of hepatocytes in vitro and survival of transplanted hepatocytes in vivo. J. Biosci. Bioeng. 2019, 128, 365–372. [Google Scholar] [CrossRef]
- Gressner, A.M.; Krull, N.; Bachem, M.G. Regulation of proteoglycan expression in fibrotic liver and cultured fat-storing cells. Pathol. Res. Pract. 1994, 190, 864–882. [Google Scholar] [CrossRef]
- Liu, J.; Abate, W.; Xu, J.; Corry, D.; Kaul, B.; Jackson, S.K. Three-dimensional spheroid cultures of A549 and HepG2 cells exhibit different lipopolysaccharide (LPS) receptor expression and LPS-induced cytokine response compared with monolayer cultures. Innate Immun. 2011, 17, 245–255. [Google Scholar]
- Deng, J.; Zhang, X.; Chen, Z.; Luo, Y.; Lu, Y.; Liu, T.; Wu, Z.; Jin, Y.; Zhao, W.; Lin, B. A cell lines derived microfluidic liver model for investigation of hepatotoxicity induced by drug-drug interaction. Biomicrofluidics 2019, 13, 024101. [Google Scholar] [CrossRef]
- Boulais, L. Cryogel-Integrated Hepatic Cell Culture Microchips for Liver Tissue Engineering. Ph.D. Thesis, Université de Technologie de Compiègne, Compiègne, France, 2020. [Google Scholar]
- Baudoin, R.; Prot, J.M.; Legallais, C.; Leclerc, E. Microfluidic Biochip for In Vitro Toxicity Analysis; ESAO: Compiègne, France, 2009. [Google Scholar] [CrossRef]
- Pauty, J.; Usuba, R.; Cheng, I.G.; Hespel, L.; Takahashi, H.; Kato, K.; Kobayashi, M.; Nakajima, H.; Lee, E.; Yger, F.; et al. A vascular endothelial growth factor-dependent sprouting angiogenesis assay based on an in vitro human blood vessel model for the study of anti-angiogenic drugs. EBioMedicine 2018, 27, 225–236. [Google Scholar] [CrossRef]
- Pettinato, G.; Lehoux, S.; Ramanathan, R.; Salem, M.M.; He, L.-X.; Muse, O.; Flaumenhaft, R.; Thompson, M.T.; Rouse, E.A.; Cummings, R.D.; et al. Generation of fully functional hepatocyte-like organoids from human induced pluripotent stem cells mixed with Endothelial Cells. Sci. Rep. 2019, 9, 8920. [Google Scholar] [CrossRef]
- Melchiorri, A.J.; Bracaglia, L.G.; Kimerer, L.K.; Hibino, N.; Fisher, J.P. In vitro endothelialization of biodegradable vascular grafts via endothelial progenitor cell seeding and maturation in a tubular perfusion system bioreactor. Tissue Eng. Part C Methods 2016, 22, 663–670. [Google Scholar] [CrossRef]
- Messelmani, T.; Morisseau, L.; Sakai, Y.; Legallais, C.; Le Goff, A.; Leclerc, E.; Jellali, R. Liver organ-on-chip models for toxicity studies and risk assessment. Lab Chip 2022, 22, 2423–2450. [Google Scholar] [CrossRef] [PubMed]
- Štampar, M.; Breznik, B.; Filipič, M.; Žegura, B. Characterization of In Vitro 3D Cell Model Developed from Human Hepatocellular Carcinoma (HepG2) Cell Line. Cells 2020, 9, 2557. [Google Scholar] [CrossRef]
- Freyer, N.; Greuel, S.; Knöspel, F.; Gerstmann, F.; Storch, L.; Damm, G.; Seehofer, D.; Harris, J.F.; Iyer, R.; Schubert, F.; et al. Microscale 3D Liver Bioreactor for In Vitro Hepatotoxicity Testing under Perfusion Conditions. Bioengineering 2018, 5, 24. [Google Scholar] [CrossRef] [PubMed] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Seeded Cells | 48 h | 96 h | |
|---|---|---|---|
| Biochip + hydroscaffold | 1.25 × 105 cells/cm2 | 66 ± 15 | 91 ± 18 |
| 2.5 × 105 cells/cm2 | 108 ± 13 | 132 ± 34 | |
| Petri + hydroscaffold | 1.25 × 105 cells/cm2 | 20.5 ± 2.55 | 29.5 ± 5 |
| 2.5 × 105 cells/cm2 | 18 ± 1.8 | 26.5 ± 3.2 | |
| Biochip ** | 1.25 × 105 cells/cm2 | 95 ± 5 | 90 ± 40 |
| 2.5 × 105 cells/cm2 | 118 ± 25 | 190 ± 85 | |
| Biochip + alginate cryogel * | 2.5 × 105 cells/cm2 | 88 ± 25 | 135 ± 60 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Messelmani, T.; Le Goff, A.; Souguir, Z.; Maes, V.; Roudaut, M.; Vandenhaute, E.; Maubon, N.; Legallais, C.; Leclerc, E.; Jellali, R. Development of Liver-on-Chip Integrating a Hydroscaffold Mimicking the Liver’s Extracellular Matrix. Bioengineering 2022, 9, 443. https://doi.org/10.3390/bioengineering9090443
Messelmani T, Le Goff A, Souguir Z, Maes V, Roudaut M, Vandenhaute E, Maubon N, Legallais C, Leclerc E, Jellali R. Development of Liver-on-Chip Integrating a Hydroscaffold Mimicking the Liver’s Extracellular Matrix. Bioengineering. 2022; 9(9):443. https://doi.org/10.3390/bioengineering9090443
Chicago/Turabian StyleMesselmani, Taha, Anne Le Goff, Zied Souguir, Victoria Maes, Méryl Roudaut, Elodie Vandenhaute, Nathalie Maubon, Cécile Legallais, Eric Leclerc, and Rachid Jellali. 2022. "Development of Liver-on-Chip Integrating a Hydroscaffold Mimicking the Liver’s Extracellular Matrix" Bioengineering 9, no. 9: 443. https://doi.org/10.3390/bioengineering9090443