
Wool Keratin-Based Nanofibres—In Vitro Validation

 ,
,  ,
,  ,
,  ,
, 

Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. NFs-Membrane Production
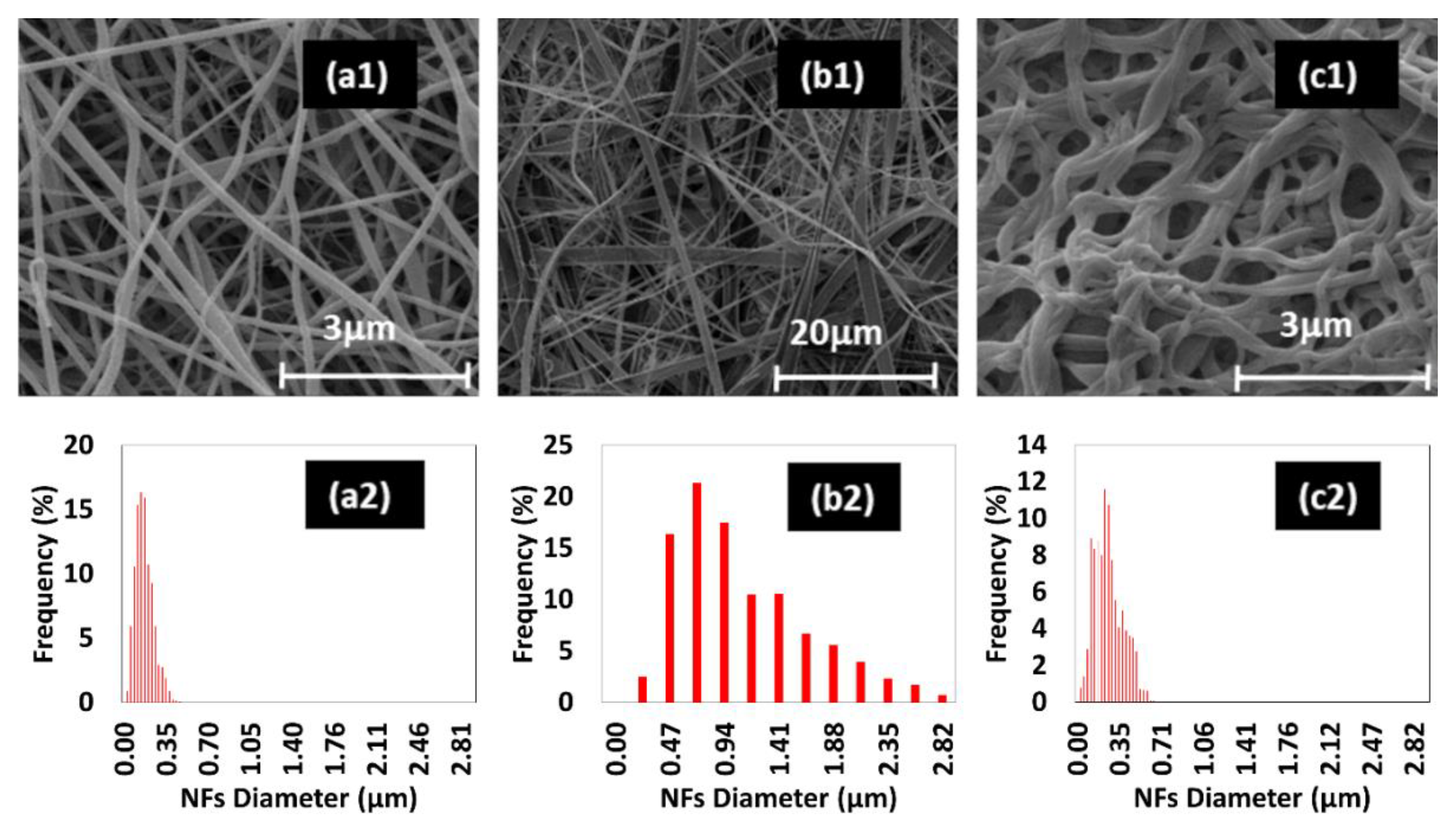
2.3. Morphological Characterization (SEM)
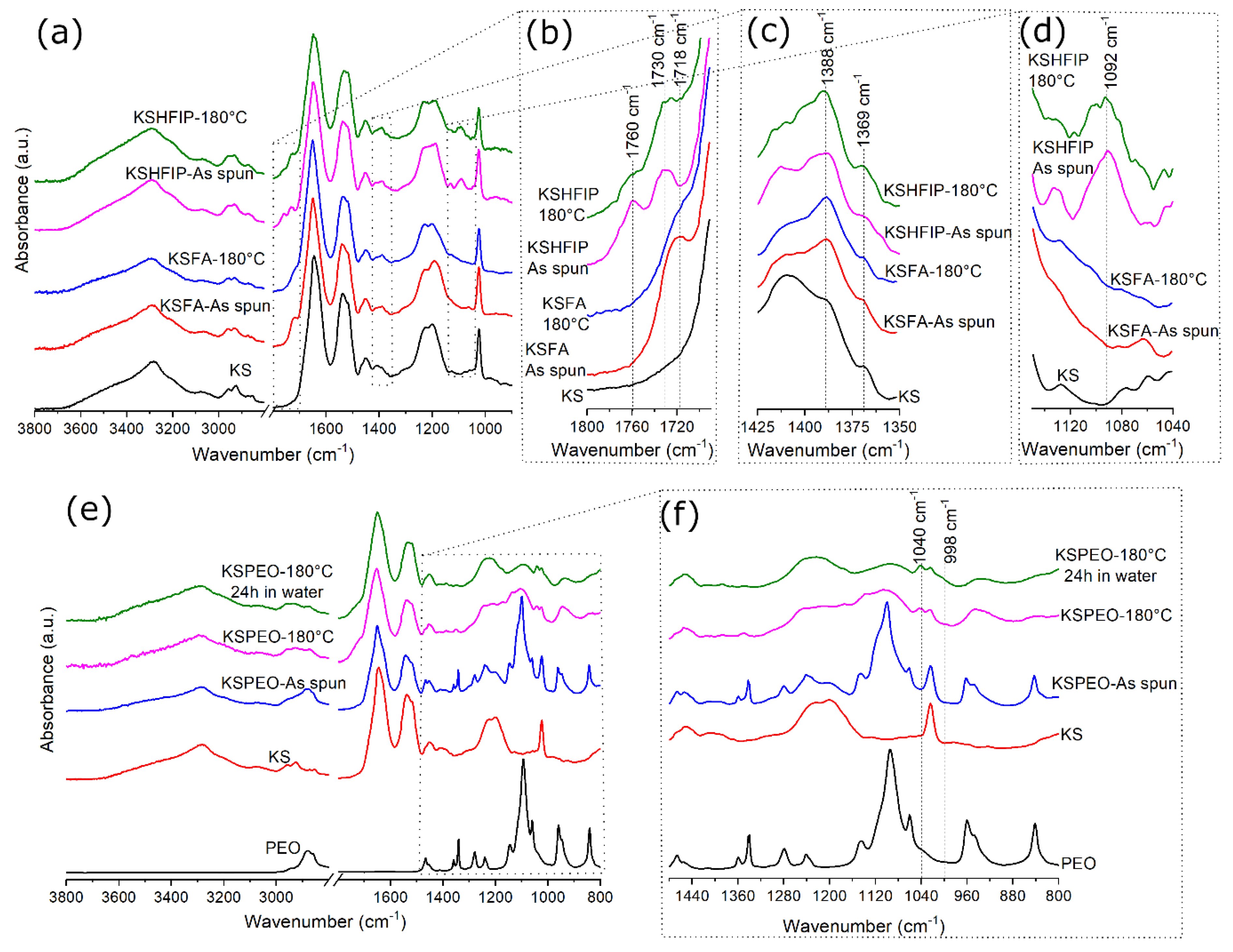
2.4. FTIR Analyses
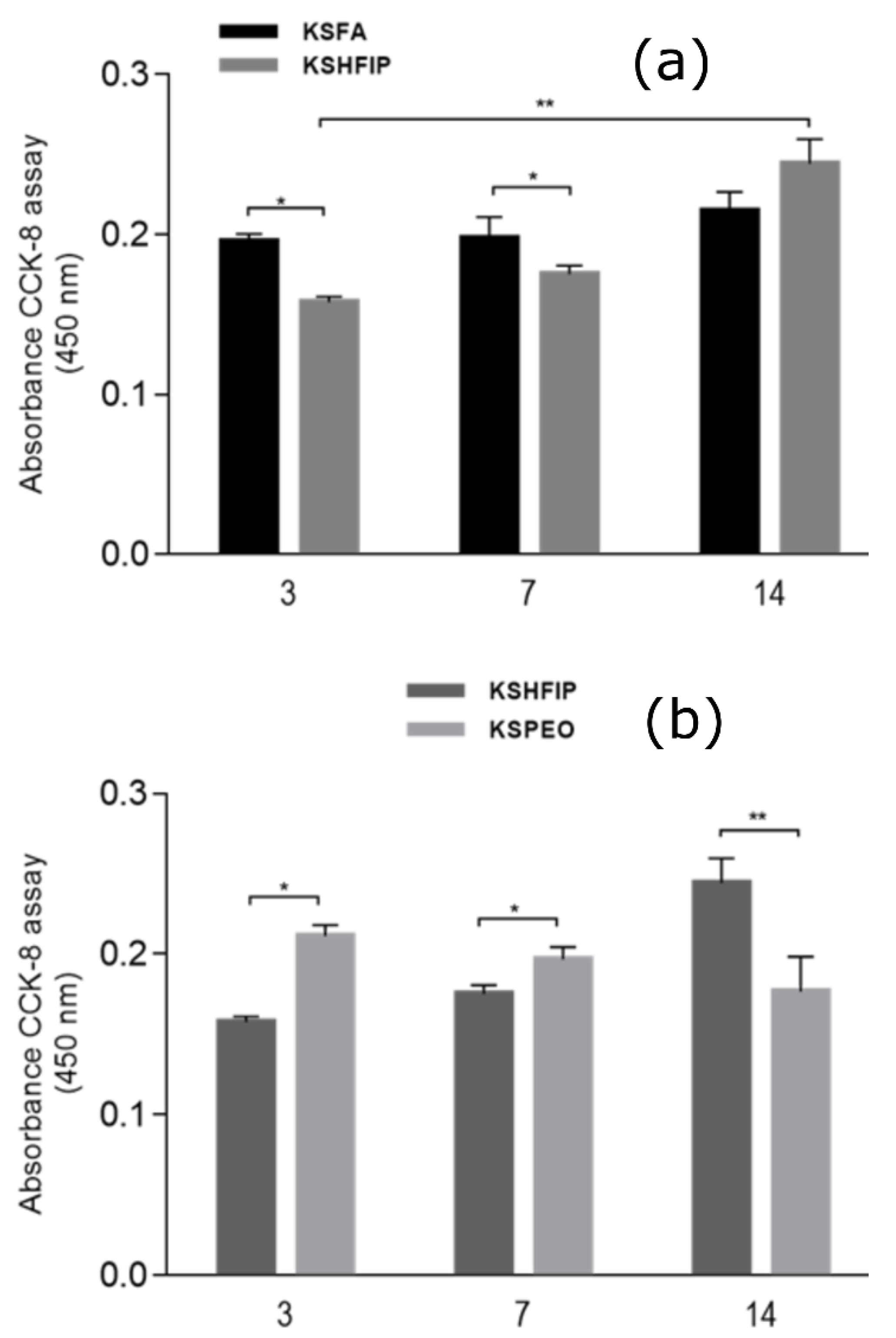
2.5. Cell Culture Tests
3. Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ferraris, S.; Guarino, V.; Cochis, A.; Varesano, A.; Cruz Maya, I.; Vineis, C.; Rimondini, L.; Spriano, S. Aligned keratin submicrometric-fibers for fibroblasts guidance onto nanogrooved titanium surfaces for transmucosal implants. Mater. Lett. 2018, 229, 1–4. [Google Scholar] [CrossRef]
- Sanchez Ramirez, D.O.; Carletto, R.A.; Truffa Giachet, F. Keratin Processing. In Kerain as a Protein Biopolymer; Sharma, S., Kumar, A., Eds.; Springer: Cham, Switzerland, 2019; pp. 77–121. ISBN 978-3-030-02900-5. [Google Scholar]
- Cochis, A.; Ferraris, S.; Sorrentino, R.; Azzimonti, B.; Novara, C.; Geobaldo, F.; Truffa Giachet, F.; Vineis, C.; Varesano, A.; Sayed Abdelgeliel, A.; et al. Silver-doped keratin nanofibers preserve a titanium surface from biofilm contamination and favor soft-tissue healing. J. Mater. Chem. B 2017, 5, 8366–8377. [Google Scholar] [CrossRef] [PubMed]
- Aldana, A.A.; Abraham, G.A. Current advances in electrospun gelatin-based scaffolds for tissue engineering applications. Int. J. Pharm. 2017, 523, 441–453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cruz-Maya, I.; Varesano, A.; Vineis, C.; Guarino, V. Comparative study on protein-rich electrospun fibers for in vitro applications. Polymers 2020, 12, 1671. [Google Scholar] [CrossRef]
- Ferraris, S.; Spriano, S.; Scalia, A.C.; Cochis, A.; Rimondini, L.; Cruz-Maya, I.; Guarino, V.; Varesano, A.; Vineis, C. Topographical and biomechanical guidance of electrospun fibers for biomedical applications. Polymers 2020, 12, 2896. [Google Scholar] [CrossRef] [PubMed]
- Pires, L.R.; Guarino, V.; Oliveira, M.J.; Ribeiro, C.C.; Barbosa, M.A.; Ambrosio, L.; Pêgo, A.P. Ibuprofen-loaded poly(trimethylene carbonate-co-ε-caprolactone) electrospun fibres for nerve regeneration. J. Tissue Eng. Regen. Med. 2016, 10, E154–E166. [Google Scholar] [CrossRef] [Green Version]
- Tayi, A.S.; Pashuck, E.T.; Newcomb, C.J.; McClendon, M.T.; Stupp, S.I. Electrospinning bioactive supramolecular polymers from water. Biomacromolecules 2014, 15, 1323–1327. [Google Scholar] [CrossRef] [PubMed]
- Grodowska, K.; Parczewski, A. Organic solvents in the pharmaceutical industry. Acta Pol. Pharm. Drug Res. 2010, 67, 3–12. [Google Scholar]
- Wilk, S.; Benko, A. Advances in fabricating the electrospun biopolymer-based biomaterials. J. Funct. Biomater. 2021, 12, 26. [Google Scholar] [CrossRef]
- Xiao, J.; Shi, C.; Zheng, H.; Shi, Z.; Jiang, D.; Li, Y.; Huang, Q. Kafirin Protein Based Electrospun Fibers with Tunable Mechanical Property, Wettability, and Release Profile. J. Agric. Food Chem. 2016, 64, 3226–3233. [Google Scholar] [CrossRef]
- Cruz-Maya, I.; Guarino, V.; Almaguer-Flores, A.; Alvarez-Perez, M.A.; Varesano, A.; Vineis, C. Highly polydisperse keratin rich nanofibers: Scaffold design and in vitro characterization. J. Biomed. Mater. Res. Part A 2019, 107, 1803–1813. [Google Scholar] [CrossRef] [PubMed]
- Khaw, Y.Y.; Chee, C.Y.; Gan, S.N.; Singh, R.; Ghazali, N.N.N.; Liu, N.-S. Poly(lactic acid) composite films reinforced with microcrystalline cellulose and keratin from chicken feather fiber in 1-butyl-3-methylimidazolium chloride. J. Appl. Polym. Sci. 2019, 136, 47642. [Google Scholar] [CrossRef]
- Elmowafy, E.; Abdal-Hay, A.; Skouras, A.; Tiboni, M.; Casettari, L.; Guarino, V. Polyhydroxyalkanoate (PHA): Applications in drug delivery and tissue engineering. Expert Rev. Med. Devices 2019, 16, 467–482. [Google Scholar] [CrossRef] [PubMed]
- Guarino, V.; Cirillo, V.; Ambrosio, L. Bicomponent electrospun scaffolds to design extracellular matrix tissue analogs. Expert Rev. Med. Devices 2015, 13, 83–102. [Google Scholar] [CrossRef] [PubMed]
- Varesano, A.; Vineis, C.; Tonetti, C.; Sanchez Ramirez, D.O.; Mazzuchetti, G. Chemical and physical modifications of electrospun keratin nanofibers induced by heating treatments. J. Appl. Polym. Sci. 2014, 131, 40532. [Google Scholar] [CrossRef]
- Sanchez Ramirez, D.O.; Cruz-Maya, I.; Vineis, C.; Tonetti, C.; Varesano, A.; Guarino, V. Design of Asymmetric Nanofibers-membranes based on Polyvinylalcohol and Wool-keratin for Wound Healing Applications. J. Funct. Biomater. J. 2021, in press. [Google Scholar]
- Varesano, A.; Vineis, C.; Tonetti, C.; Sanchez Ramirez, D.O.; Mazzuchetti, G.; Ortelli, S.; Blosi, M.; Costa, A.L. Multifunctional hybrid nanocomposite nanofibers produced by colloid electrospinning from water solutions. Curr. Nanosci. 2015, 11, 41–48. [Google Scholar] [CrossRef]
- Van De Weert, M.; Haris, P.I.; Hennink, W.E.; Crommelin, D.J.A. Fourier transform infrared spectrometric analysis of protein conformation: Effect of sampling method and stress factors. Anal. Biochem. 2001, 297, 160–169. [Google Scholar] [CrossRef] [PubMed]
- Cobb, J.S.; Zai-Rose, V.; Correia, J.J.; Janorkar, A.V. FT-IR Spectroscopic Analysis of the Secondary Structures Present during the Desiccation Induced Aggregation of Elastin-Like Polypeptide on Silica. ACS Omega 2020, 5, 8403–8413. [Google Scholar] [CrossRef]
- Troullier, A.; Reinstädler, D.; Dupont, Y.; Naumann, D.; Forge, V. Transient non-native secondary structures during the refolding of α- lactalbumin detected by infrared spectroscopy. Nat. Struct. Biol. 2000, 7, 78–86. [Google Scholar] [CrossRef]
- Tian, F.; Middaugh, C.R.; Offerdahl, T.; Munson, E.; Sane, S.; Rytting, J.H. Spectroscopic evaluation of the stabilization of humanized monoclonal antibodies in amino acid formulations. Int. J. Pharm. 2007, 335, 20–31. [Google Scholar] [CrossRef]
- Baird, G.; Farrell, C.; Cheung, J.; Semple, A.; Blue, J.; Ahl, P.L. FTIR Spectroscopy Detects Intermolecular β-Sheet Formation Above the High Temperature Tm for Two Monoclonal Antibodies. Protein J. 2020, 39, 318–327. [Google Scholar] [CrossRef]
- Belton, D.J.; Plowright, R.; Kaplan, D.L.; Perry, C.C. A robust spectroscopic method for the determination of protein conformational composition—Application to the annealing of silk. Acta Biomater. 2018, 73, 355–364. [Google Scholar] [CrossRef] [Green Version]
- DeFlores, L.P.; Ganim, Z.; Nicodemus, R.A.; Tokmakoff, A. Amide I′−II′ 2D IR Spectroscopy Provides Enhanced Protein Secondary Structural Sensitivity. J. Am. Chem. Soc. 2009, 131, 3385–3391. [Google Scholar] [CrossRef] [PubMed]
- Cardamone, J.M. Investigating the microstructure of keratin extracted from wool: Peptide sequence (MALDI-TOF/TOF) and protein conformation (FTIR). J. Mol. Struct. 2010, 969, 97–105. [Google Scholar] [CrossRef]
- Litvinov, R.I.; Faizullin, D.A.; Zuev, Y.F.; Weisel, J.W. The α-helix to β-sheet transition in stretched and compressed hydrated fibrin clots. Biophys. J. 2012, 103, 1020–1027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Secundo, F.; Guerrieri, N. ATR-FT/IR Study on the Interactions between Gliadins and Dextrin and Their Effects on Protein Secondary Structure. J. Agric. Food Chem. 2005, 53, 1757–1764. [Google Scholar] [CrossRef]
- Feroz, S.; Muhammad, N.; Ranayake, J.; Dias, G. Keratin—Based materials for biomedical applications. Bioact. Mater. 2020, 5, 496–509. [Google Scholar] [CrossRef]
- Cardamone, J.M. Keratin transamidation. Int. J. Biol. Macromol. 2008, 42, 413–419. [Google Scholar] [CrossRef]
- Jackson, M.; Mantsch, H.H. The Use and Misuse of FTIR Spectroscopy in the Determination of Protein Structure. Crit. Rev. Biochem. Mol. Biol. 2008, 30, 95–120. [Google Scholar] [CrossRef]
- Erra, P.; Gómez, N.; Dolcet, L.M.; Juliá, M.R.; Lewis, D.M.; Willoughby, J.H. FTIR Analysis to Study Chemical Changes in Wool Following a Sulfitolysis Treatment. Text. Res. J. 1997, 67, 397–401. [Google Scholar] [CrossRef]
- Pielichowski, K.; Flejtuch, K. Non-oxidative thermal degradation of poly(ethylene oxide): Kinetic and thermoanalytical study. J. Anal. Appl. Pyrolysis 2005, 73, 131–138. [Google Scholar] [CrossRef]
- Vrandečić, N.S.; Erceg, M.; Jakić, M.; Klarić, I. Kinetic analysis of thermal degradation of poly(ethylene glycol) and poly(ethylene oxide)s of different molecular weight. Thermochim. Acta 2010, 498, 71–80. [Google Scholar] [CrossRef]
- Yoshihara, T.; Tadokoro, H.; Murahashi, S. Normal Vibrations of the Polymer Molecules of Helical Conformation. IV. Polyethylene Oxide and Polyethylene-d4 Oxide. J. Chem. Phys. 2004, 41, 2902. [Google Scholar] [CrossRef]
- Park, C.; Carlson, M.J.; Goddard, W.A. Solvent Effects on the Secondary Structures of Proteins. J. Phys. Chem. A 1999, 104, 2498–2503. [Google Scholar] [CrossRef] [Green Version]
- Christensen, L.F.B.; Nowak, J.S.; Sønderby, T.V.; Frank, S.A.; Otzen, D.E. Quantitating denaturation by formic acid: Imperfect repeats are essential to the stability of the functional amyloid protein FapC. J. Biol. Chem. 2020, 295, 13036–13041. [Google Scholar] [CrossRef]
- Colomer, I.; Batchelor-McAuley, C.; Odell, B.; Donohoe, T.J.; Compton, R.G. Hydrogen Bonding to Hexafluoroisopropanol Controls the Oxidative Strength of Hypervalent Iodine Reagents. J. Am. Chem. Soc. 2016, 138, 8855–8861. [Google Scholar] [CrossRef] [PubMed]
- Guarino, V.; Cirillo, V.; Taddei, P.; Alvarez-Perez, M.A.; Ambrosio, L. Tuning size scale and crystallinity of PCL electrospun fibres via solvent permittivity to address hMSC response. Macromol. Biosci. 2011, 11, 1694–1705. [Google Scholar] [CrossRef]
- Zhang, F.; Zuo, B.Q.; Bai, L. Study on the structure of SF fiber mats electrospun with HFIP and FA and cells behavior. J. Mater. Sci. 2009, 44, 5682–5687. [Google Scholar] [CrossRef]
- Hellwig, P.; Rost, B.; Kaiser, U.; Ostermeier, C.; Michel, H.; Mäntele, W. Carboxyl group protonation upon reduction of the Paracoccus denitrificans cytochrome c oxidase: Direct evidence by FTIR spectroscopy. FEBS Lett. 1996, 385, 53–57. [Google Scholar] [CrossRef] [Green Version]
- Barth, A.; Zscherp, C. What vibrations tell about proteins. Q. Rev. Biophys. 2002, 35, 369–430. [Google Scholar] [CrossRef]
- Zheng, S.; Doucette, A.A. Preventing N- and O-formylation of proteins when incubated in concentrated formic acid. Proteomics 2016, 16, 1059–1068. [Google Scholar] [CrossRef]
- Grewal, M.K.; Chandrapala, J.; Donkor, O.; Apostolopoulos, V.; Stojanovska, L.; Vasiljevic, T. Fourier transform infrared spectroscopy analysis of physicochemical changes in UHT milk during accelerated storage. Int. Dairy J. 2017, 66, 99–107. [Google Scholar] [CrossRef] [Green Version]
- Nandiyanto, A.B.D.; Oktiani, R.; Ragadhita, R. How to read and interpret ftir spectroscope of organic material. Indones. J. Sci. Technol. 2019, 4, 97–118. [Google Scholar] [CrossRef]
- Smith, B.C. The C=O Bond, Part VI: Esters and the Rule of Three. Available online: https://www.spectroscopyonline.com/view/co-bond-part-vi-esters-and-rule-three (accessed on 29 October 2021).
- John Wiley & Sons, Inc. 1,1,1,3,3,3-Hexafluoro-2-Propanol—ATR-IR—Spectrum—SpectraBase. Available online: https://spectrabase.com/spectrum/DMX1NVlNdXc (accessed on 29 October 2021).
- Colomer, I.; Chamberlain, A.E.R.; Haughey, M.B.; Donohoe, T.J. Hexafluoroisopropanol as a highly versatile solvent. Nat. Rev. Chem. 2017, 1, 1–12. [Google Scholar] [CrossRef]
- Bergeron, C.; Perrier, E.; Potier, A.; Delmas, G. A Study of the Deformation, Network, and Aging of Polyethylene Oxide Films by Infrared Spectroscopy and Calorimetric Measurements. Int. J. Spectrosc. 2012, 2012, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Tiiman, A.; Krishtal, J.; Palumaa, P.; Tõugu, V. In vitro fibrillization of Alzheimer’s amyloid-β peptide (1-42). AIP Adv. 2015, 5, 092401. [Google Scholar] [CrossRef] [Green Version]
- Wei, G.; Shea, J.E. Effects of solvent on the structure of the alzheimer amyloid-β(25-35) peptide. Biophys. J. 2006, 91, 1638–1647. [Google Scholar] [CrossRef] [Green Version]
- Sanchez Ramirez, D.O.; Carletto, R.A.; Tonetti, C.; Giachet, F.T.; Varesano, A.; Vineis, C. Wool keratin film plasticized by citric acid for food packaging. Food Packag. Shelf Life 2017, 12, 100–106. [Google Scholar] [CrossRef]
- Hamed, E.; Xu, T.; Keten, S. Poly(ethylene glycol) Conjugation Stabilizes the Secondary Structure of α-Helices by Reducing Peptide Solvent Accessible Surface Area. Biomacromolecules 2013, 14, 4053–4060. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
| Sample | Solvent | Total Polymer Conc. (%wt.) | Polymer Blend (%wt.) | Flow Rate (mL min−1) | Tip-Collector Distance (cm) | Voltage (kV) | Needle i.d. (mm) |
|---|---|---|---|---|---|---|---|
| KSFA | FA | 15 | 100 KS | 0.002 | 15 | 25 | 0.2 |
| KSHFIP | HFIP | 6 | 100 KS | 0.002 | 15 | 25 | 0.9 |
| KSPEO 1 | Water | 7 | 70/30 KS/PEO | 0.010 | 20 | 25 | 0.2 |
| Sample | Intermolecular β–Sheet | Intramolecular β–Sheet | β–Sheet II | β–Turn | Random Coil | α–Helix |
|---|---|---|---|---|---|---|
| KS | 23.9 | 6.5 | 24.2 | 11.3 | 6.7 | 27.4 |
| KSFA-As spun | 21.0 | 4.7 | 21.1 | 20.5 | 25.0 | 7.7 |
| KSFA-180 °C | 20.8 | 6.1 | 17.8 | 20.6 | 27.7 | 7.0 |
| KSHFIP-As spun | 18.3 | 6.3 | 17.5 | 21.2 | 4.0 | 32.6 |
| KSHFIP-180 °C | 19.0 | 7.1 | 18.9 | 21.0 | 4.1 | 29.9 |
| KSPEO-As spun | 20.5 | 0.2 | 25.2 | 26.8 | 19.5 | 7.7 |
| KSPEO-180 °C | 16.7 | 13.9 | 21.9 | 17.5 | 16.4 | 13.6 |
| KSPEO-180 °C, 24 h in water | 23.0 | 4.9 | 19.0 | 22.9 | 18.2 | 12.0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sanchez Ramirez, D.O.; Cruz-Maya, I.; Vineis, C.; Guarino, V.; Tonetti, C.; Varesano, A. Wool Keratin-Based Nanofibres—In Vitro Validation. Bioengineering 2021, 8, 224. https://doi.org/10.3390/bioengineering8120224
Sanchez Ramirez DO, Cruz-Maya I, Vineis C, Guarino V, Tonetti C, Varesano A. Wool Keratin-Based Nanofibres—In Vitro Validation. Bioengineering. 2021; 8(12):224. https://doi.org/10.3390/bioengineering8120224
Chicago/Turabian StyleSanchez Ramirez, Diego Omar, Iriczalli Cruz-Maya, Claudia Vineis, Vincenzo Guarino, Cinzia Tonetti, and Alessio Varesano. 2021. "Wool Keratin-Based Nanofibres—In Vitro Validation" Bioengineering 8, no. 12: 224. https://doi.org/10.3390/bioengineering8120224
APA StyleSanchez Ramirez, D. O., Cruz-Maya, I., Vineis, C., Guarino, V., Tonetti, C., & Varesano, A. (2021). Wool Keratin-Based Nanofibres—In Vitro Validation. Bioengineering, 8(12), 224. https://doi.org/10.3390/bioengineering8120224