
Homogeneous Space Construction and Projection for Single-Cell Expression Prediction Based on Deep Learning


Abstract
:1. Introduction
2. Materials and Methods
2.1. Problem and Theory Explanation
2.2. VAE & -TCVAE
2.3. Assumptions
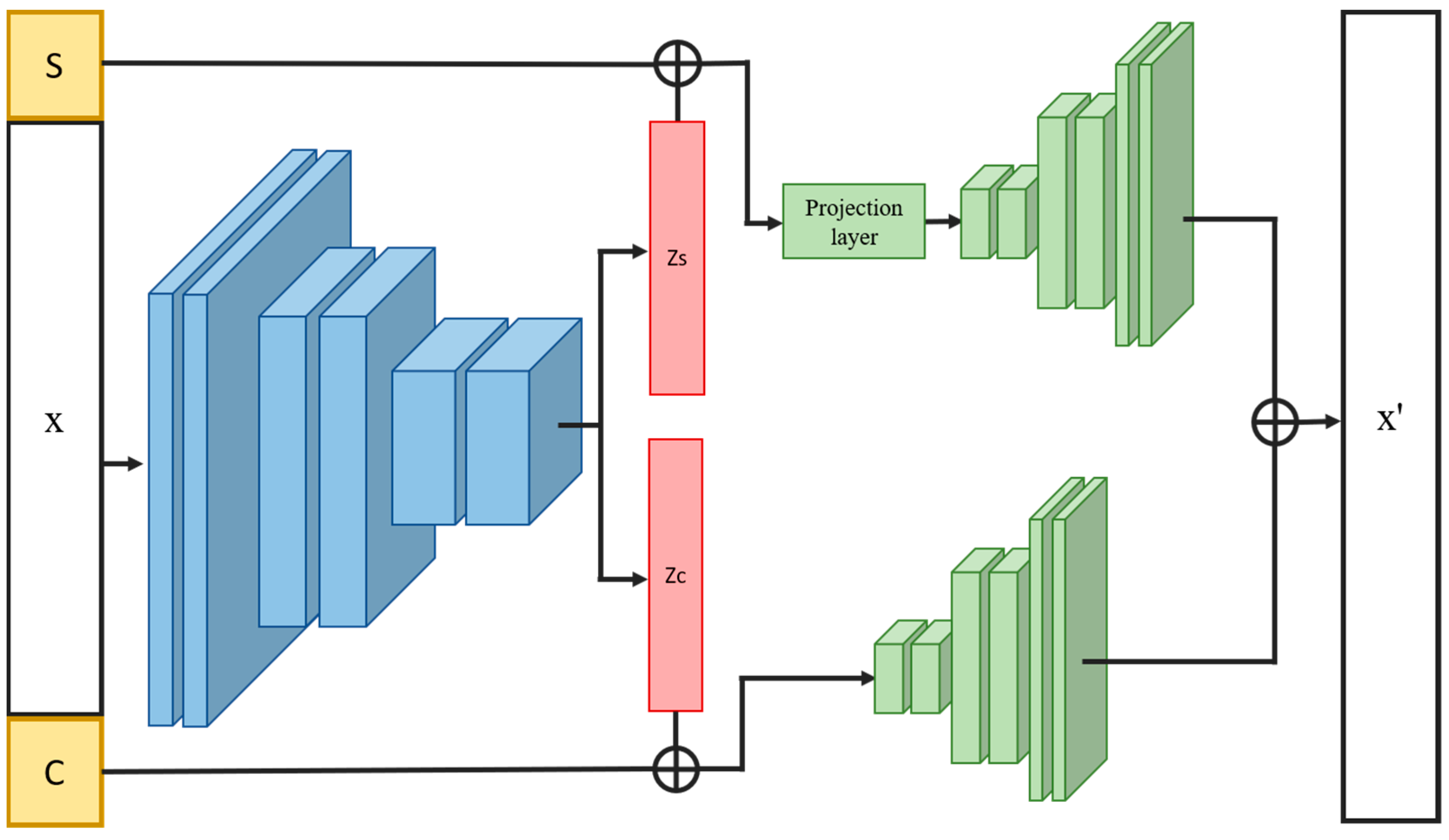
2.4. Overview of INVAE
2.5. Loss Functions
2.5.1. -TCVAE Loss
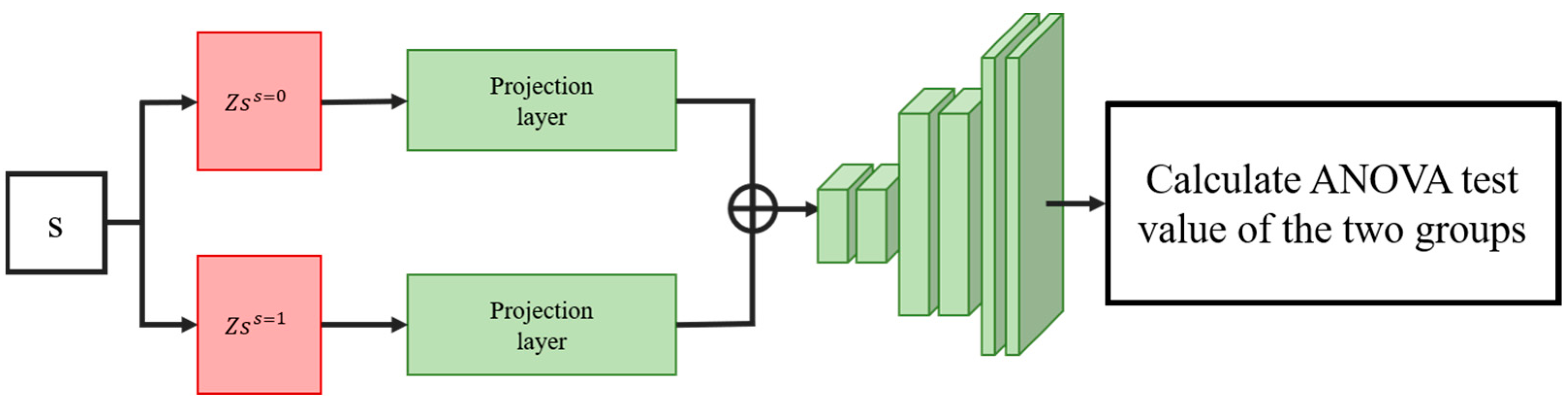
2.5.2. ANOVA Information Navigation Loss
2.5.3. Total Loss Function
2.6. Training Processes
| Algorithm 1: INVAE training process |
| Input: data, X; annotation of cell-type and condition, C, S; encoder, F; decoder1 and 2, ; projection layer, 1. count = 0 2. While FinishTraining! = True: 3. For b in numBatches: 4. Get training batch , , , from X, C, S 5. Z = F(concat(, , )) 6. Split latent variables Z into first and second parts: , 7. = ) 8. = 9. For i in {i: i c}: 10. s {0, 1} 11. Get from which annotation is (c = i) & (s = 0) 12. Get from which annotation is (c = i) & (s = 1) 13. Create set = {, } 14. Create set = {, } 15. End 16. Calculate (5), (6) with Z, , , , , , , 17. If count < : 18. Update model using (5) 19. elif count == : 20. Update model using (6) 21. count = 0 22. End 23. count += 1 24. End 25. End |
3. Datasets and Benchmarks
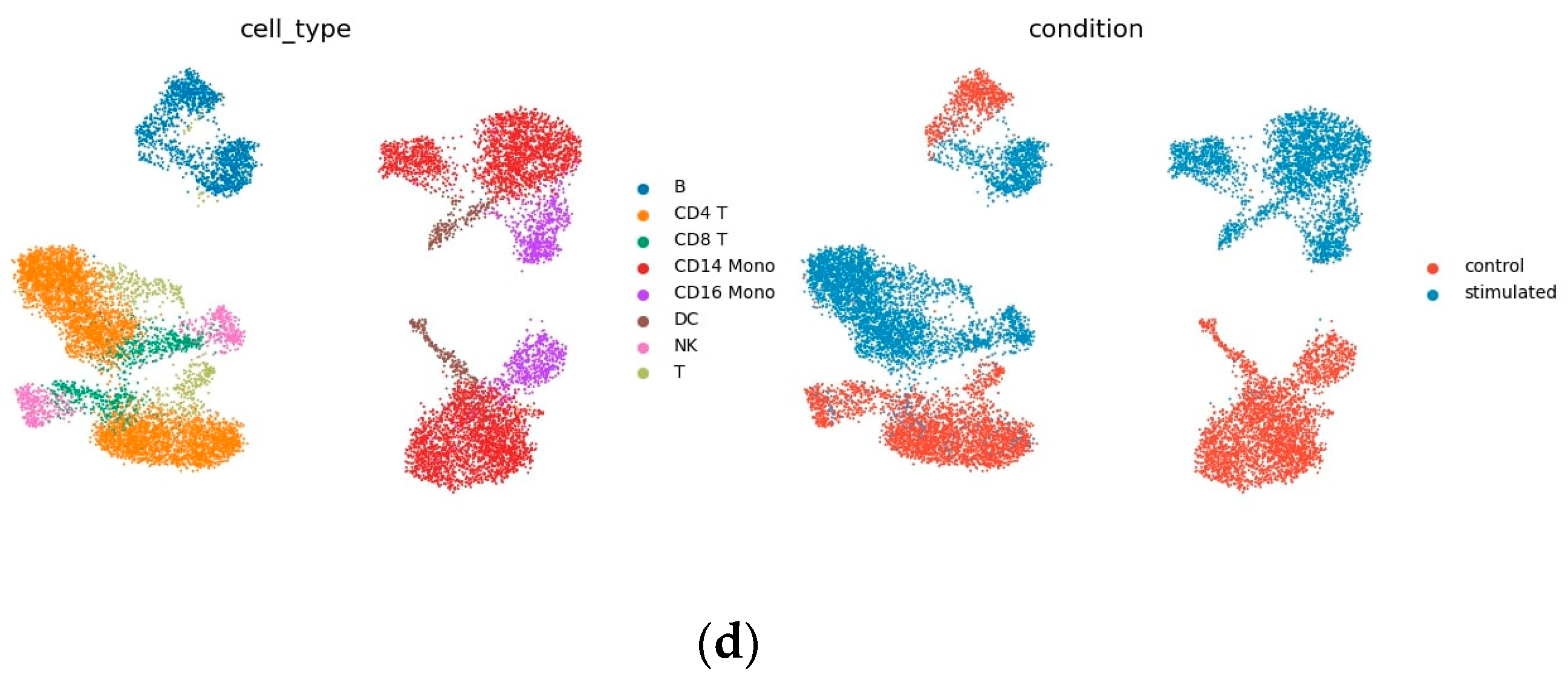
3.1. Haber Dataset
3.2. Kang Dataset
3.3. LPS Dataset
3.4. trVAE
3.5. stVAE
3.6. scPreGAN
3.7. Hyperparameter
4. Results
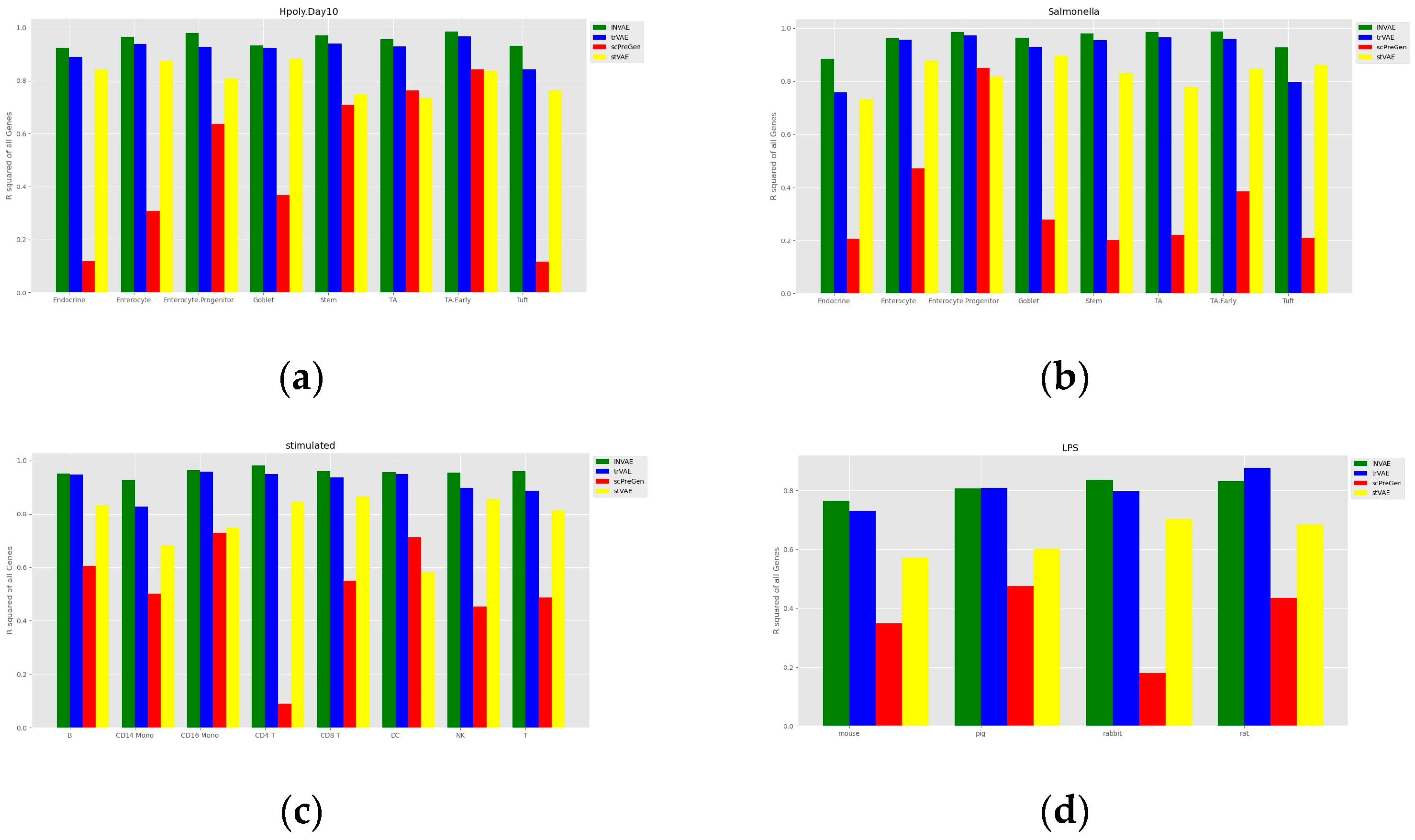
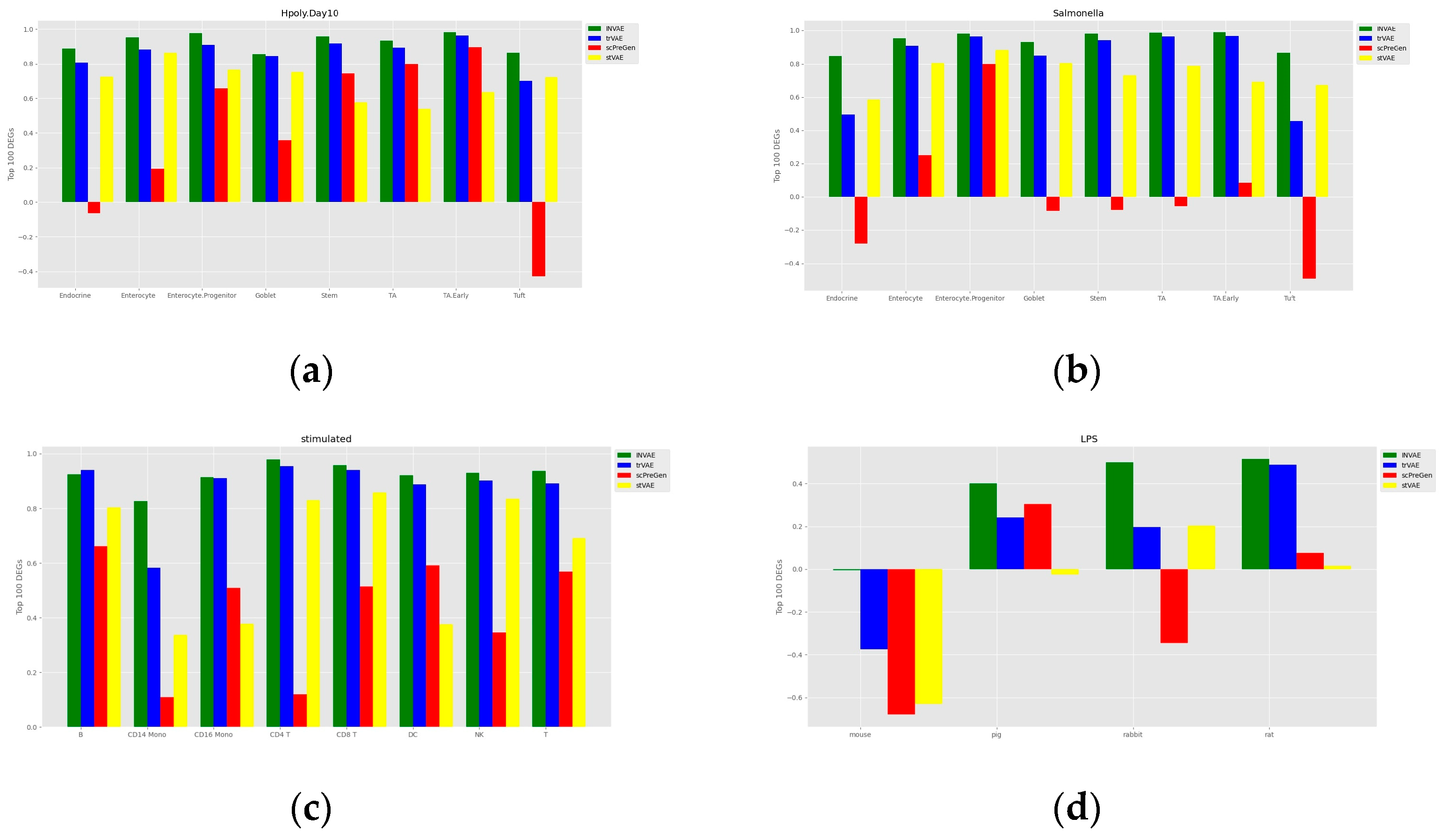
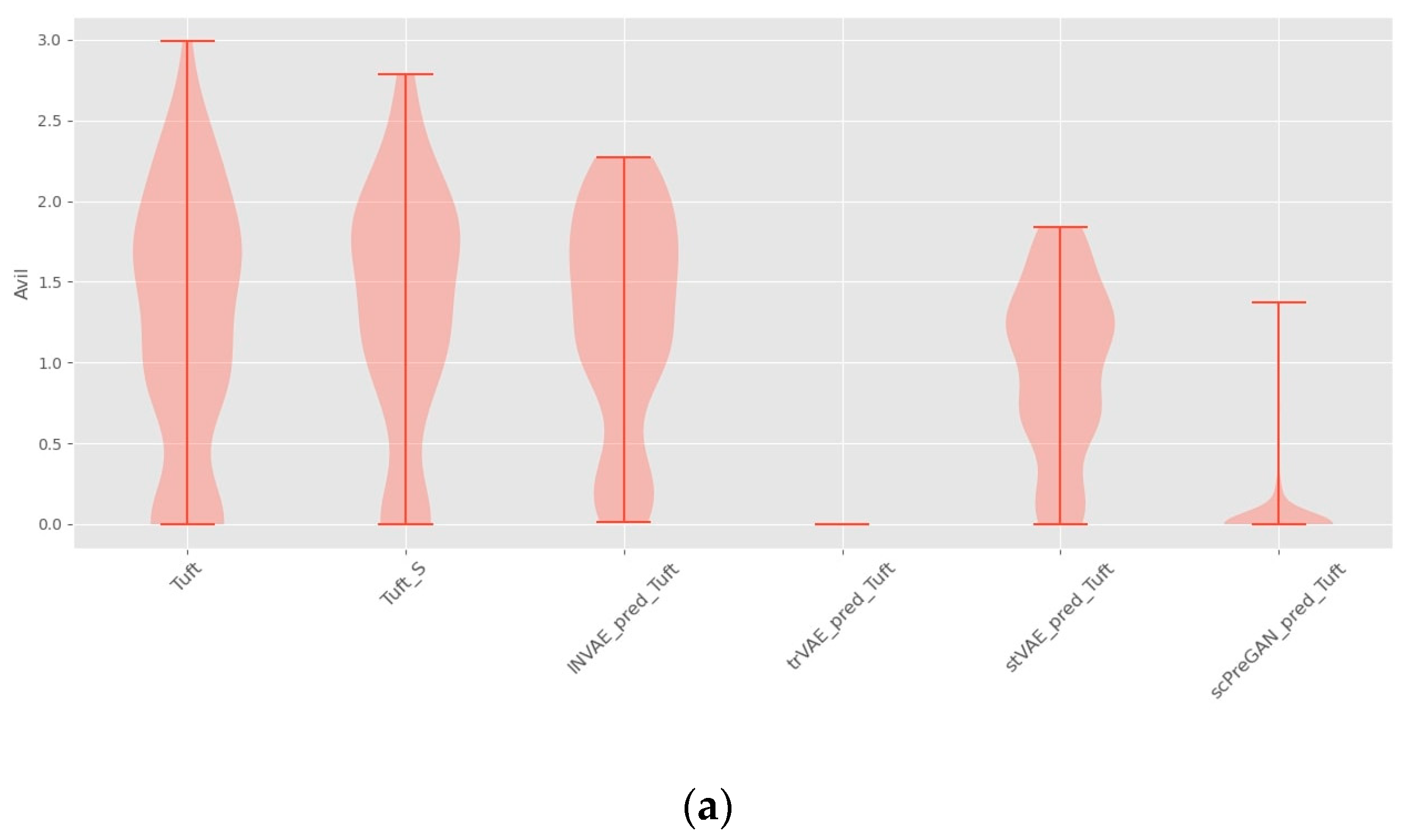
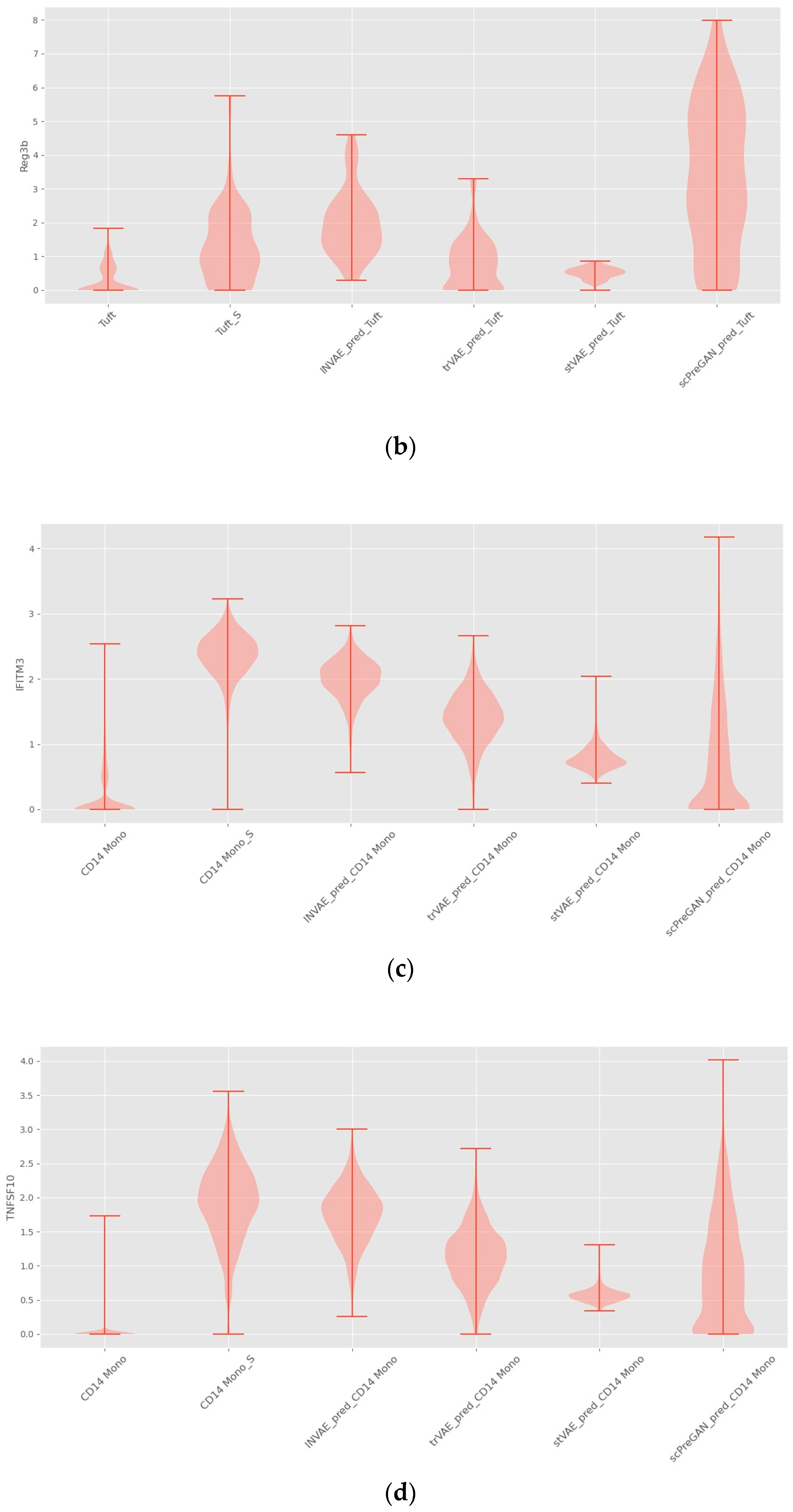
4.1. Prediction Accuracy Comparison
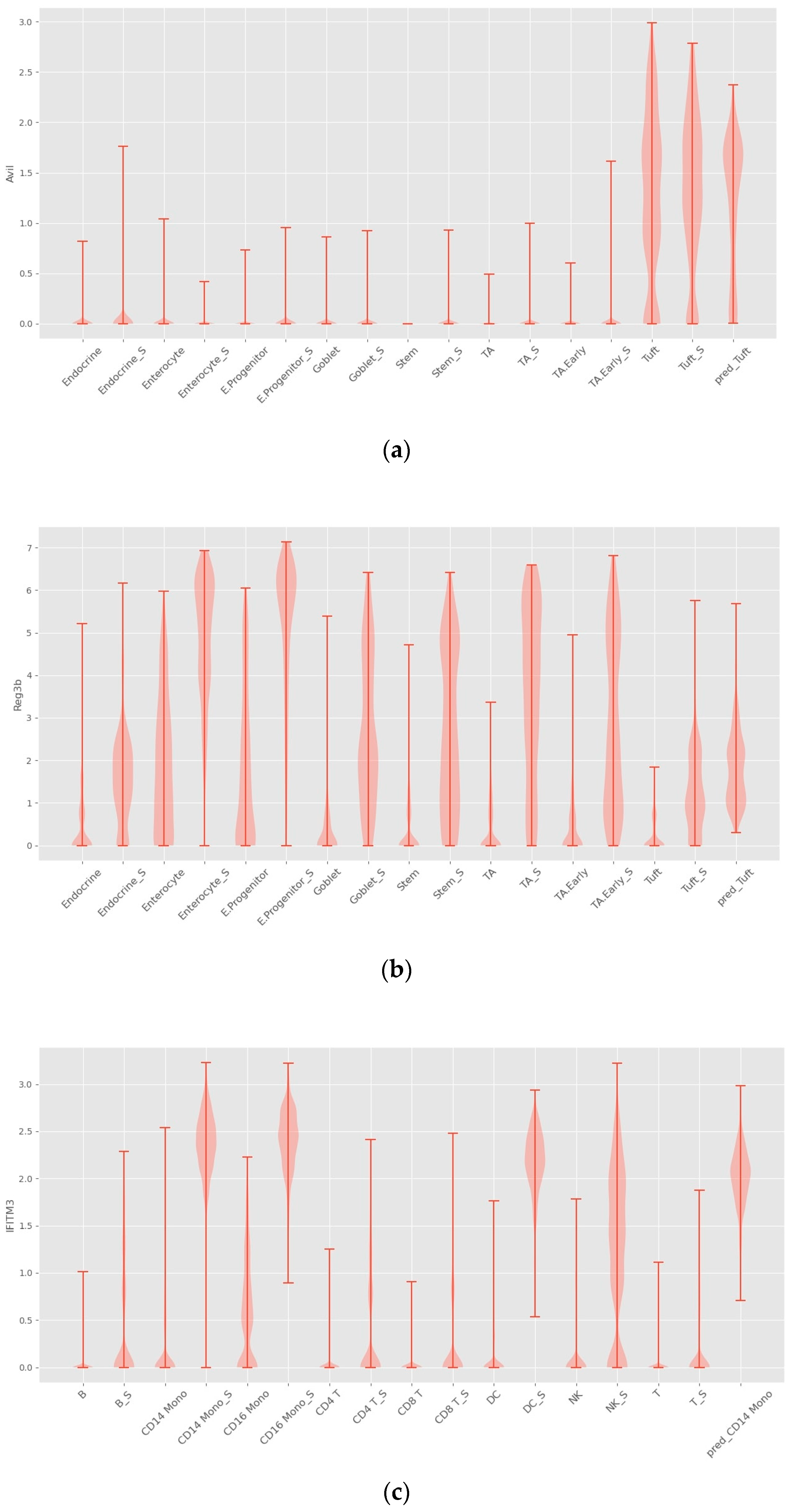
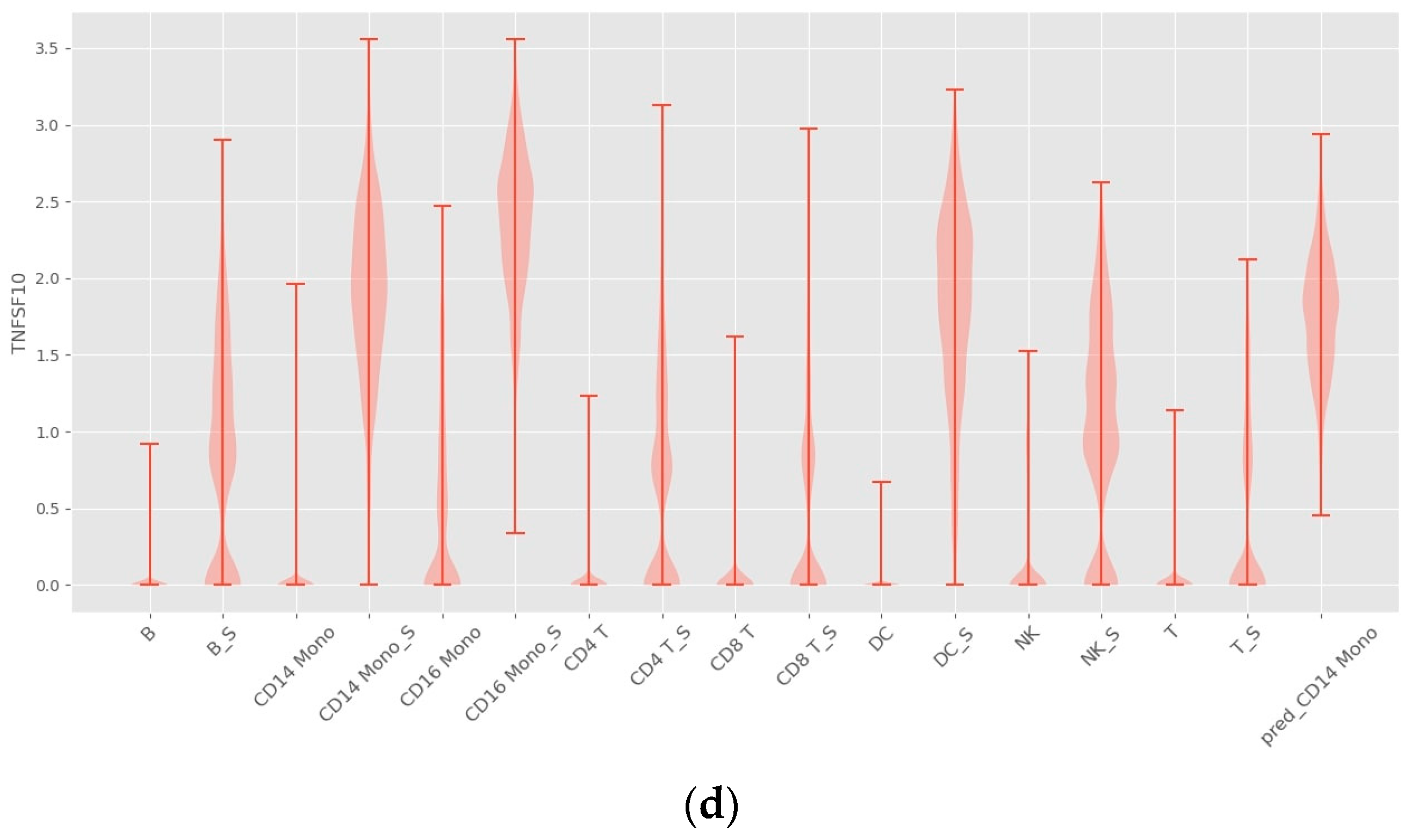
4.2. Interpretability
4.3. INVAE Captures Non-Linearly Gene–Gene Interaction Features
4.4. Ablation Study
4.5. Model Convergence Time
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Efremova, M.; Teichmann, S.A. Computational methods for single-cell omics across modalities. Nat. Methods 2020, 17, 14–17. [Google Scholar] [CrossRef]
- Saliba, A.E.; Westermann, A.J.; Gorski, S.A.; Vogel, J. Single-cell RNA-seq: Advances and future challenges. Nucleic Acids Res. 2014, 42, 8845–8860. [Google Scholar] [CrossRef]
- Stark, R.; Grzelak, M.; Hadfield, J. RNA sequencing: The teenage years. Nat. Rev. Genet. 2019, 20, 631–656. [Google Scholar] [CrossRef] [PubMed]
- Gaublomme, J.T.; Yosef, N.; Lee, Y.; Gertner, R.S.; Yang, L.V.; Wu, C.; Pandolfi, P.P.; Mak, T.; Satija, R.; Shalek, A.K.; et al. Single-cell genomics unveils critical regulators of Th17 cell pathogenicity. Cell 2015, 163, 1400–1412. [Google Scholar] [CrossRef] [PubMed]
- Yofe, I.; Dahan, R.; Amit, I. Single-cell genomic approaches for developing the next generation of immunotherapies. Nat. Med. 2020, 26, 171–177. [Google Scholar] [CrossRef] [PubMed]
- Srivatsan, S.R.; McFaline-Figueroa, J.L.; Ramani, V.; Saunders, L.; Cao, J.; Packer, J.; Pliner, H.A.; Jackson, D.L.; Daza, R.M.; Christiansen, L.; et al. Massively multiplex chemical transcriptomics at single-cell resolution. Science 2020, 367, 45–51. [Google Scholar] [CrossRef]
- Rampášek, L.; Hidru, D.; Smirnov, P.; Haibe-Kains, B.; Goldenberg, A. Dr. VAE: Improving drug response prediction via modeling of drug perturbation effects. Bioinformatics 2019, 35, 3743–3751. [Google Scholar] [CrossRef]
- Alipanahi, B.; Delong, A.; Weirauch, M.T.; Frey, B.J. Predicting the sequence specificities of DNA-and RNA-binding proteins by deep learning. Nat. Biotechnol. 2015, 33, 831–838. [Google Scholar] [CrossRef]
- Chaudhary, K.; Poirion, O.B.; Lu, L.; Garmire, L.X. Deep learning–based multi-omics integration robustly predicts survival in liver cancer using deep learning to predict liver cancer prognosis. Clin. Cancer Res. 2018, 24, 1248–1259. [Google Scholar] [CrossRef]
- Wei, X.; Dong, J.; Wang, F. scPreGAN, a deep generative model for predicting the response of single cell expression to perturbation. Bioinformatics 2022, 38, 3377–3384. [Google Scholar] [CrossRef]
- Targonski, C.; Bender, M.R.; Shealy, B.T.; Husain, B.; Paseman, B.; Smith, M.C.; Feltus, F.A. Cellular state transformations using deep learning for precision medicine applications. Patterns 2020, 1, 100087. [Google Scholar] [CrossRef] [PubMed]
- Goodfellow, I.J.; Pouget-Abadie, J.; Mirza, M.; Xu, B.; Warde-Farley, D.; Ozair, S.; Courville, A.; Bengio, Y. Generative adversarial nets. Adv. Neural Inf. Process. Syst. 2014, 27, 2672–2680. [Google Scholar]
- Russkikh, N.; Antonets, D.; Shtokalo, D.; Makarov, A.; Vyatkin, Y.; Zakharov, A.; Terentyev, E. Style transfer with variational autoencoders is a promising approach to RNA-Seq data harmonization and analysis. Bioinformatics 2020, 36, 5076–5085. [Google Scholar] [CrossRef]
- Lotfollahi, M.; Naghipourfar, M.; Theis, F.J.; Wolf, F.A. Conditional out-of-distribution generation for unpaired data using transfer VAE. Bioinformatics 2020, 36, i610–i617. [Google Scholar] [CrossRef]
- Lotfollahi, M.; Susmelj, A.K.; De Donno, C.; Ji, Y.; Ibarra, I.L.; Wolf, F.A.; Yakubova, N. Learning Interpretable Cellular Responses to Complex Perturbations in High-Throughput Screens. BioRxiv 2021. Available online: https://www.biorxiv.org/content/10.1101/2021.04.14.439903v2.abstract (accessed on 1 January 2021).
- Sohn, K.; Yan, X.; Lee, H. Learning structured output representation using deep conditional generative models. Adv. Neural Inf. Process. Syst. 2015, 28, 3483–3491. [Google Scholar]
- Gretton, A.; Fukumizu, K.; Harchaoui, Z.; Sriperumbudur, B.K. A fast, consistent kernel two-sample test. Adv. Neural Inf. Process. Syst. 2009, 22, 673–681. [Google Scholar]
- Yu, H.; Welch, J.D. MichiGAN: Sampling from disentangled representations of single-cell data using generative adversarial networks. Genome Biol. 2021, 22, 158. [Google Scholar] [CrossRef]
- Kingma, D.P.; Welling, M. Auto-encoding variational bayes. arXiv 2013, arXiv:1312.6114. [Google Scholar]
- Chen, R.T.Q.; Li, X.; Grosse, R.; Duvenaud, D. Isolating sources of disentanglement in variational autoencoders. Adv. Neural Inf. Process. Syst. 2018, 31, 2615–2625. [Google Scholar]
- Hinton, G.E.; Zemel, R. Autoencoders, minimum description length and Helmholtz free energy. Adv. Neural Inf. Process. Syst. 1993, 6, 3–10. [Google Scholar]
- Weiss, K.; Khoshgoftaar, T.M.; Wang, D. A survey of transfer learning. J. Big Data 2016, 3, 9. [Google Scholar] [CrossRef]
- Haber, A.L.; Biton, M.; Rogel, N.; Herbst, R.H.; Shekhar, K.; Smillie, C.; Burgin, G.; Delorey, T.M.; Howitt, M.R.; Katz, Y.; et al. A single-cell survey of the small intestinal epithelium. Nature 2017, 551, 333–339. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.M.; Subramaniam, M.; Targ, S.; Nguyen, M.; Maliskova, L.; McCarthy, E.; Wan, E.; Wong, S.; Byrnes, L.; Lanata, C.M.; et al. Multiplexed droplet single-cell RNA-sequencing using natural genetic variation. Nat. Biotechnol. 2018, 36, 89–94. [Google Scholar] [CrossRef] [PubMed]
- Hagai, T.; Chen, X.; Miragaia, R.J.; Rostom, R.; Gomes, T.; Kunowska, N.; Henriksson, J.; Park, J.-E.; Proserpio, V.; Donati, G.; et al. Gene expression variability across cells and species shapes innate immunity. Nature 2018, 563, 197–202. [Google Scholar] [CrossRef]
- Ahuja, K.; Caballero, E.; Zhang, D.; Gagnon-Audet, J.C.; Bengio, Y.; Mitliagkas, I.; Rish, I. Invariance principle meets information bottleneck for out-of-distribution generalization. Adv. Neural Inf. Process. Syst. 2021, 34, 3438–3450. [Google Scholar]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Operation | Kernel Dim. | Dropout | Activation | Input |
|---|---|---|---|---|---|
| Input | - | input_dim | - | - | - |
| Conditions | - | 1 | - | - | - |
| Cell types | - | 1 | - | - | - |
| FC-1 | FC | 800 | √ | ReLU | [input, conditions, cell types] |
| FC-2 | FC | 800 | √ | ReLU | FC-1 |
| FC-3 | FC | 128 | √ | ReLU | FC-2 |
| Output s&c | FC | 30 + 30 | - | - | FC-3 |
| Name | Operation | Kernel Dim. | Dropout | Activation | Input |
|---|---|---|---|---|---|
| FC-1 | FC | 128 | √ | ReLU | [Encoder output c, Cell types] |
| FC-2 | FC | 800 | √ | ReLU | FC-1 |
| FC-3 | FC | 800 | √ | ReLU | FC-2 |
| Output | FC | input_dim | - | - | FC-3 |
| Name | Operation | Kernel Dim. | Dropout | Activation | Input |
|---|---|---|---|---|---|
| Output | FC | 128 | √ | ReLU | [Encoder output s, conditions] |
| Name | Operation | Kernel Dim. | Dropout | Activation | Input |
|---|---|---|---|---|---|
| FC-1 | FC | 800 | √ | ReLU | Projection layer output |
| FC-2 | FC | 800 | √ | ReLU | FC-1 |
| Output | FC | input_dim | - | - | FC-2 |
| Optimizer | Adam | ||||
| Learning rate | 0.001 | ||||
| Dropout rate | 0.2 |
| INVAE | w/o Training Method | w/o Noise Filer | |
|---|---|---|---|
| All | 0.920 | 0.902 | 0.770 |
| DEGs 100 | 0.786 | 0.734 | 0.588 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yeh, C.-H.; Chen, Z.-G.; Liou, C.-Y.; Chen, M.-J. Homogeneous Space Construction and Projection for Single-Cell Expression Prediction Based on Deep Learning. Bioengineering 2023, 10, 996. https://doi.org/10.3390/bioengineering10090996
Yeh C-H, Chen Z-G, Liou C-Y, Chen M-J. Homogeneous Space Construction and Projection for Single-Cell Expression Prediction Based on Deep Learning. Bioengineering. 2023; 10(9):996. https://doi.org/10.3390/bioengineering10090996
Chicago/Turabian StyleYeh, Chia-Hung, Ze-Guang Chen, Cheng-Yue Liou, and Mei-Juan Chen. 2023. "Homogeneous Space Construction and Projection for Single-Cell Expression Prediction Based on Deep Learning" Bioengineering 10, no. 9: 996. https://doi.org/10.3390/bioengineering10090996
APA StyleYeh, C.-H., Chen, Z.-G., Liou, C.-Y., & Chen, M.-J. (2023). Homogeneous Space Construction and Projection for Single-Cell Expression Prediction Based on Deep Learning. Bioengineering, 10(9), 996. https://doi.org/10.3390/bioengineering10090996