1. Introduction
Type one diabetes mellitus (T1DM) is a complex autoimmune disorder characterized by the immune-mediated destruction of insulin-producing beta cells. This condition leads not only to increased blood glucose levels but also to alterations in the micro and macro blood circulation, which implies an increased risk to develop nephropathy, retinopathy, and neuropathy [
1]. Currently, T1DM therapy consists chiefly of daily administration of exogenous insulin via multiple subcutaneous injections or through insulin infusion. Unfortunately, insulin mono-therapy presents a considerable risk of hypoglycemia and strict control of glucose levels is mandatory. For these reasons, new frontiers of therapies for T1DM are being studied in recent years to treat the disease but also hopefully to reverse it in the future. These therapies include immunotherapies that target T cells as well as cell therapy [
2]. The differentiation of various stem and progenitor cell populations into functional islet cells has provided new hope for the field [
3]. Indeed, the use of stem cells could be a valid approach to making progress in the understanding of biological systems and, in the future, to the development of new cellular therapy. Among the various sources of cells used in cell therapy, particular interest has been given in the past 20 years to perinatal cells and their potential application in regenerative medicine. Perinatal cells are isolated from term placentas and fetal annexes. Since perinatal tissues are discarded immediately after birth the resulting cells are free of ethical implications and present several advantages, including their easy collection and the high yield in terms of cell number obtained after isolation [
4,
5,
6].
In our study, we focus on two different perinatal cells: amniotic epithelial cells (AECs), isolated from the inner part of the amniotic membrane, and Wharton’s jelly mesenchymal stromal cells (WJ-MSCs), isolated from the umbilical cord. Due to their early development, perinatal cells retain characteristics found in pluripotent stem cell populations and have been successfully induced to differentiate towards all three germ layers under specific culture conditions [
7]. Importantly, AECs and WJ-MSCs have been successfully differentiated into the pancreatic–endodermic lineage [
4,
8,
9,
10]. Moreover, cell populations deriving from the placenta are physiologically involved in the achievement of feto-maternal tolerance, whose role is to avoid the immune-mediated rejection of the embryo during pregnancy. The immunomodulatory properties of perinatal cells are a fundamental aspect for regenerative medicine applications and for the treatment of auto-immune diseases, including T1DM [
11].
Thanks to the advancements in matrices and scaffold engineering, studies on three-dimensional (3D) cultures have increased in recent years. Spheroid formation is usually achieved through the use of these techniques: (a) forced floating, (b) hanging drop, (c) spinning flask, (d) rotating vessel, (e) electrical-force assisted, (f) magnetic-force assisted, and (g) matrix-based [
12]. Three-dimensional models mimic the in vivo cell environment better than a classical two-dimension (2D) cell culture, improving cell viability, morphology, proliferation, response to stimuli, and differentiation. Moreover, 3D cultures have greater stability and longer lifespans than cell cultures in 2D.
Among 3D models, spheroids are dense cellular structures that are promising to increase the therapeutic potential of cell-based therapies. Specifically, spheroids retain their extracellular matrix (ECM), which is fundamental to increasing cell survival in harsh conditions and up-regulating trophic factor secretion. Thanks to the presence of ECM and cell–cell interactions, spheroids better mimic the micro-environment found in native tissue [
3,
12]. Moreover, spheroids can be constituted of different types of cells giving rise to more physiological cell–cell interactions. The aim of our work is to create a 3D model consisting of a co-culture of AECs and WJ-MSCs. In this way, it is possible to combine the ability of AECs to differentiate into insulin-producing cells, already demonstrated by our laboratory [
4], with the scaffolding support and ECM secretion exerted by WJ-MSCs, which are crucial for the stability of a 3D model [
13]. A co-culture of epithelial cells and mesenchymal cells could be a valid model to mimic the pancreatic micro-environment in which a tight correlation between glandular cells and stromal cells is present. One more important aspect is that both AECs and WJ-MSCs have immunomodulatory activity, specifically by stimulating the tolerogenic cell component. In the circumstance of T1DM, the immunomodulatory effect could be a valid approach to preserve beta cells from the hyperactivation of the immune system [
14].
2. Materials and Methods
2.1. Ethics Statement
This study was approved by the Local Ethical Committee (IRCCS St. Orsola-Malpighi University Hospital Ethical Committee, protocol n° 2481/2017, ref n° 68/2017/U/Tess). Placentas were obtained from healthy donor mothers undergoing elective caesarean section at term (37–40 weeks) after written informed consent. Perinatal tissues were maintained under sterile conditions until specific cell isolation was performed.
2.2. Isolation of Human Amniotic Epithelial Cells (AECs)
Fetal membranes were washed with ice-cold phosphate-buffered saline (PBS, Corning, NY, USA) with 1% penicillin–streptomycin solution (10,000 U/mL penicillin, 10,000 U/mL streptomycin, Corning, Steuben County, NY, USA). The amniotic membrane was mechanically peeled off the underlying chorion layer to remove any blood clots. Then the tissues were incubated for 10 min at room temperature with PBS/ethylenediaminetetraacetic acid (EDTA) 0.5 mM. The amniotic membrane was then minced into small pieces (4 cm2 approximately) and digested twice for 30 min at 37 °C using trypsin-EDTA 0.25% (Corning, Steuben County, NY, USA) with gentle shaking. For both digestion steps, trypsin was inactivated with fetal bovine serum (FBS, Gibco, Life Technologies, Carlsbad, CA, USA) and the cell suspension was centrifuged for 10 min at 390× g. The cell pellet was resuspended in basal culture medium, Dulbecco’s Modified Eagle’s Medium–high glucose (DMEM H., Corning, Steuben County, NY, USA) containing 10% FBS, 1% penicillin-streptomycin solution, and 10 ng/mL of epithelial growth factor (EGF, Sigma-Aldrich, St. Louis, MO, USA). The single-cell suspension was counted and tested for viability using erythrosin B (Sigma-Aldrich, St. Louis, MO, USA). Only samples with >90% viability were used for further assays.
2.3. Isolation of Wharton’s Jelly Mesenchymal Stromal Cells (WJ-MSCs)
First, the umbilical cord (UC) was removed from the placenta. UC was minced into pieces of approximately 2–3 cm lengths and rinsed 3–4 times with phosphate-buffered saline (PBS, Corning, NY, USA) containing 1% penicillin–streptomycin (10,000 U/mL penicillin, 10,000 U/mL streptomycin, Corning, Steuben County, NY, USA). After the removal of two arteries and a vein, Wharton’s jelly (WJ) was chopped into pieces of 3–5 mm with a scalpel. These small pieces were transferred onto the culture dishes and covered with a drop of Dulbecco’s Modified Eagle’s Medium (DMEM) with 10% FBS (fetal bovine serum) to prevent the pieces of WJ from drying out. The dishes were placed in the incubator with 5% CO2 for at least two hours and then 10 mL of medium was added to the dishes. The culture dishes were maintained and left untouched for 5 days in the incubator. On day 5 of culture, 3 mL of media was added. After approximately 7–14 days, WJ-MSCs were isolated. The explanted tissues were gently removed and the isolated cells were detached by the surface using Accutase® (P10-21500, Pan Biotech, Bayern, Germany) and centrifuged at 300× g for 5 min. After centrifugation, cells were recovered and the single-cell suspension was counted and tested for viability using erythrosin B (Sigma-Aldrich, St. Louis, MO, USA).
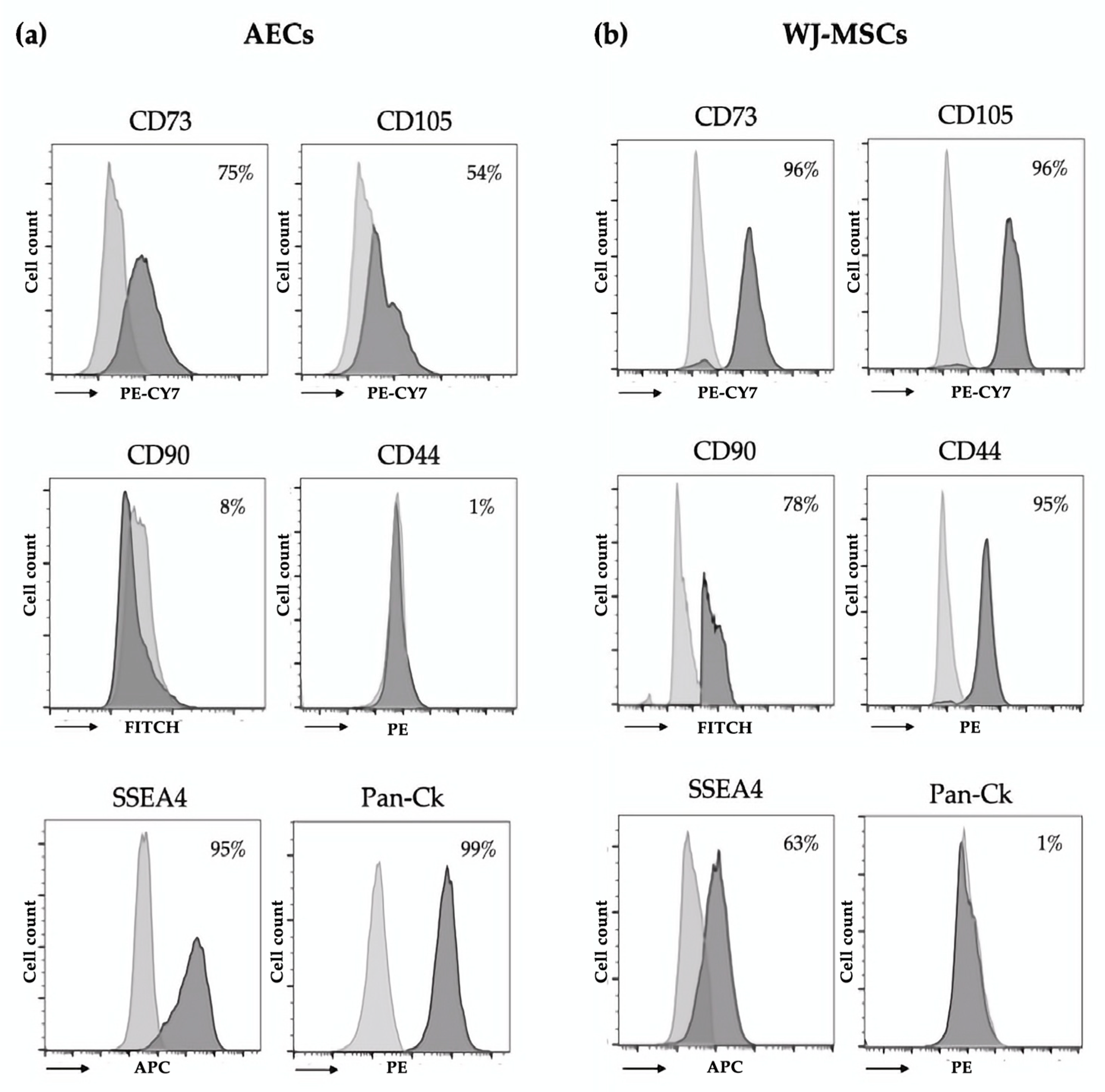
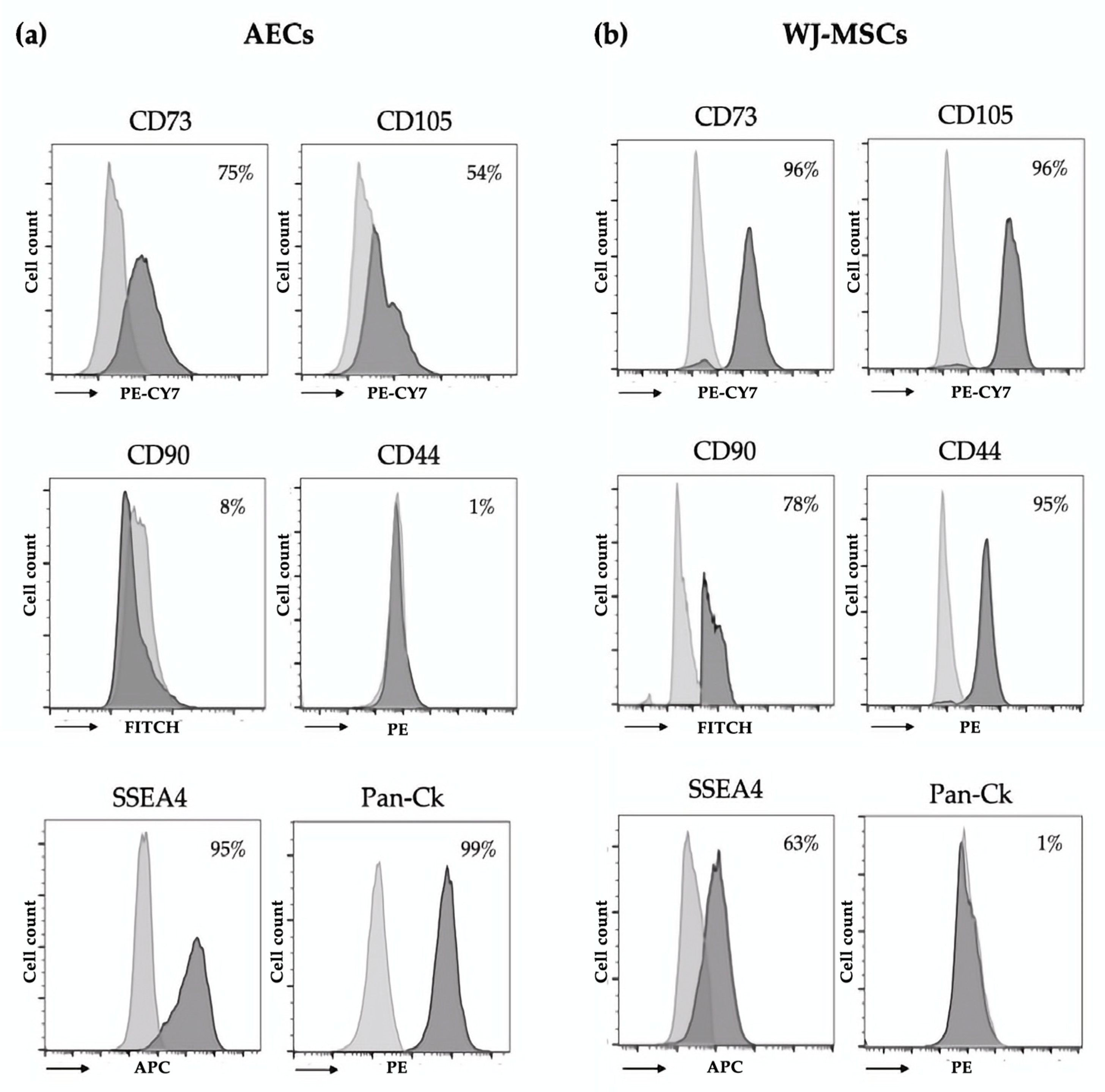
2.4. Immunophenotype of AECs and WJ-MSCs
Immunophenotypic characterization of isolated AECs and WJ-MSCs was performed using flow cytometry. Cells were fixed for 10 min at room temperature using Intraprep Kit (Beckman–Coulter Inc., Brea, CA, USA) and washed twice with PBS. Cells were incubated for 30 min at 4 °C with conjugated primary antibodies (1 μg/mL) specific for epithelial (anti-pan Cytokeratin (Pan-Ck)-PE, Santa Cruz Biotechnology, Santa Cruz, CA, USA), mesenchymal (anti-CD44-PE, anti-CD73-PECY7, anti-CD90-FITC, anti-CD105-PECY7, Beckman-Coulter Inc., Brea, CA, USA), hematopoietic (anti-CD14-PE, anti-CD34-PerCP, anti-CD45-FITC, Beckman-Coulter Inc., Brea, CA, USA), and stem cell (anti-state-specific embryonic antigen-4 (SSEA4) APC, Miltenyi Biotech, Germany) markers. For the analysis of Pan-Ck, we used the Pan-Ck Type I/II Antibody Cocktail (MA5-13156, Thermo Scientific, Waltham, MA, USA) and anti-mouse–Alexa Fluor 488 (A11001, Thermo Scientific, Waltham, MA, USA) as a secondary antibody. After incubation, cells were washed with PBS and analyzed using the FACS Navio FC (Beckman-Coulter Inc., Brea, CA, USA) cytometer and the Kaluza FC C 1.2 Analysis software (Beckman-Coulter Inc., Brea, CA, USA).
2.5. 3D Culture, Perinatal Spheroid Generation
Spheroids were generated starting from isolated WJ-MSCs seeded as mono-culture or by combining WJ-MSCs with AECs using 96-well round-bottom cell-repellant plates (Greiner Bio-One, Kremsmünster, Austria). Briefly, 200 μL of DMEM with 10% FBS containing a total of 5000 cells/well were seeded in each well in both mono- and co-culture spheroids. In the co-culture setting, 2500 cells/well of each cellular subtype and EGF (10 ng/mL) were added. Immediately after seeding, the plates were centrifuged at 50× g for 3 min and placed in an incubator at 37 °C with 5% CO2. The culture media was changed every day thereafter.
2.6. Spheroid Formation
Co-culture spheroids were generated starting from isolated WJ-MSCs stained using Vybrant™ DiD Cell-Labeling Solutions (Molecular Probes, Eugene, OR, USA) and AECs stained using Vybrant™ DiO Cell-Labeling Solutions (Molecular Probes, Eugene, OR, USA). Biefly, 1 × 106 cells/mL in serum-free DMEM were incubated for 20 min at 37 °C with the corresponding labelling solution (5 μL/mL). Cells were washed twice in PBS and seeded in 96-well round-bottom cell-repellant plates. Immediately after seeding, the plates were centrifuged at 50× g for 3 min and placed in the IncuCyte® S3 live imaging system (Sartorius) for 96 h at 37 °C in 5% CO2. Bright-field and immunofluorescence images were acquired using 10× magnification every 3 h from the seeding to the complete spheroid formation after 96 h.
2.7. Prestoblue® Assay for Spheroid Viability
To determine the presence of metabolically active cells in our spheroids the resazurin reduction assay was performed [
15]. The assay is based on the principle that resorufin, which is the reduced form of resazurin, can be measured using a fluorescent method, thus indirectly determining the viability rate of incubated spheroids. Following 4 and 14 days of 3D culture, the spheroids were transferred to a non-ULA 96 well plate and incubated for 24 h to allow the spheroids to adhere. After 24 h 100 μL of 10% Prestoblue
® stock solution was added and incubated for a maximum of 24 h at 37 °C under 5% CO
2 humidified conditions. The reduction of the reagent was measured at 595 nm at 2, 4, 6, 8, and 24 h using a multiplate reader (Victor II—Perkin Elmer, Waltham, MA, USA).
2.8. Live and Dead Staining
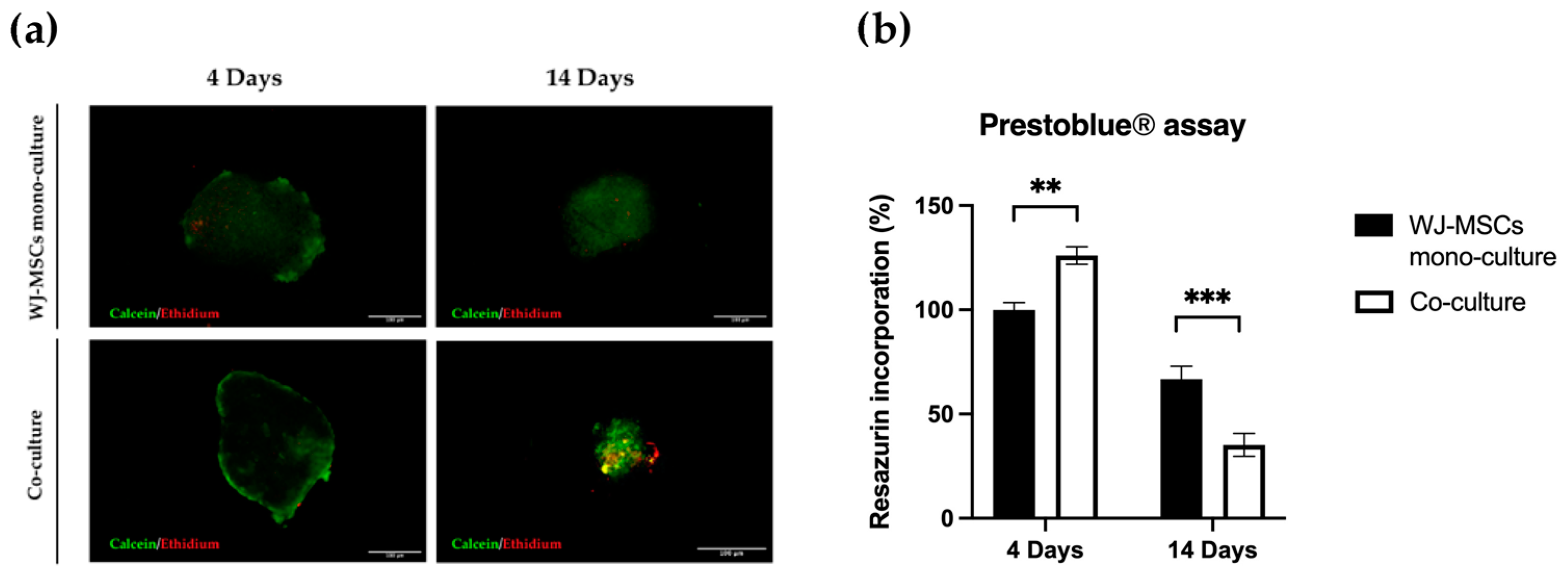
Cell viability within the spheroids was tested using the Live/Dead Cell Assay Kit (Life Technologies). This assay was used to visually determine the group of cells within spheroids maintaining viability after clusterization indispensable for spheroid formation. Spheroids of mono- and co-culture at 4 and 14 days were incubated with a solution containing 1 μM calcein AM and 2 μM ethidium homodimer-1 at 37 °C for 45 min. After washing with PBS, spheroids were imaged with laser excitation of the sample at 488 nm and 561 nm using a Nikon Inverted Microscope (Nikon Instruments, Tokyo, Japan), and images were acquired with a Digital Sight camera DS-03 using the imaging software NIS-Elements 4.1 (Nikon Corporation, Tokyo, Japan).
2.9. Slide Preparation and Histology
The spheroids were washed in PBS and transferred to 1.5 mL Eppendorf tubes to encapsulate them with Epredia™ HistoGel™ Specimen Processing Gel (Fisher-Scientific, Loughborough, UK). Each gel specimen was moved to a cassette and loaded inside a tissue processor (Histo-line laboratories, Pantigliate, Italy). The samples were embedded in paraffin blocks using an automated inclusor (Medite Medical, Burgdorf, Germany). Microtome (Microme, Bio-Optica, Milano, Italy) sections of 10 µm were placed on Super Frost glass slides (DiaPath, Menzel, Martinengo, Italy) and allowed to dry. The sections were deparaffinized with 2 changes of xylene for 5 min and rehydrated with 2 changes of 100% ethanol, followed by washes in 90% ethanol and 70% ethanol for 3 min. Finally, the slides were rinsed in distilled water.
2.10. Hematoxylin and Eosin Staining
The slides were deparaffinized, rehydrated, and stained with hematoxylin and eosin. The sections were stained with Gill 2 hematoxylin (Bio-Optica) for 5 min, washed in tap water for 3 min, rinsed with distilled water, and subsequently stained with 1% eosin G (Bio-Optica) for 10 min, dehydrated in ethanol of ascending concentration (70, 90, and 100%), clarified in pure xylene, and mounted with Bio Mount balm (Bio-Optica). Stained sections were observed using a Leica DM 750 equipped with a Leica ICC50 HD digital camera.
2.11. Biophysical Properties Analysis of Spheroids (W8 Analysis)
The W8 instrument (CellDynamics SRL) is able to accurately measure the size, weight, and density of the spheroids. Single spheroids free-fall into a vertical flow channel dedicated to analyzing their terminal velocity. The physical approach of this analysis and the mathematical equations used for the calculation is extensively described by Cristaldi et al. [
16]. Before the measurement, the spheroids were fixed with paraformaldehyde (PFA) 4% for 72 h at 4 °C, resuspended in 4 mL of phosphate-buffered saline (PBS, Corning, NY, USA), transferred in a centrifuge conical tube, and then analyzed. A minimum of 10 spheroids was analyzed for both mono- and co-culture at different time points.
2.12. Immunofluorescence Analysis
Antigen retrieval was performed by incubating slides of sectioned spheroids in sodium citrate pH 6 at 95 °C for 30 min. Sections were washed with PBS and permeabilized by adding PBS 0.3% Triton (Triton X-100, Sigma-Aldrich, Co., St. Louis, MO, USA) for 10 min. Slides were incubated for 30 min with a blocking solution containing PBS 4% bovine serum albumin (BSA, Sigma-Aldrich, St. Louis, MO, USA) and 0.3% Triton, then incubated overnight at 4 °C with the primary antibodies anti-vimentin (1:200, #MA5-14564, Thermo Fisher Scientific, Waltham, MA, USA), anti-pan cytokeratin (1:200, #MA5-13156, Thermo Fisher Scientific, Waltham, MA, USA), anti-laminin (1:100, #L9393, Sigma-Aldrich, Co., St. Louis, MO, USA), anti-fibronectin (1:200, #F3648, Sigma-Aldrich, Co., St. Louis, MO, USA), anti-cadherin (1:200, #71-7100, Thermo Fisher Scientific, Waltham, MA, USA), and anti-collagen I (1:200, #BK72026S, Cell Signaling Technology, Danvers MA, USA) diluted in blocking solution. Secondary anti-mouse Alexa Fluor 488 (1:500, #A11001, Thermo Scientific, Waltham, MA, USA) and Alexa Fluor 594 (1:500, #A32754, Thermo Fisher Scientific, Waltham, MA, USA) were used for 1 h incubation at room temperature. After three washes with PBS, coverslips were mounted using the Prolong Gold Antifade Mountant with DAPI (Thermo Fisher Scientific, Monza, Italy). Stained cells were observed using a Nikon Inverted Microscope (Nikon Instruments, Tokyo, Japan), and images were acquired with a Digital Sight camera DS-03 using the imaging software NIS-Elements 4.1 (Nikon Corporation, Tokyo, Japan).
2.13. Hypoxia Probe
The hypoxia level of whole-mounted spheroids was evaluated using the hypoxia LOX-1 probe (Organogenix- MBL) according to the manufacturer’s instructions. The probe was added to the culture medium at a final concentration of 2 μM 24 h before detection. Images visualizing the hypoxic area in mono- and co-culture spheroids were obtained using a Nikon Inverted Microscope (Nikon Instruments, Tokyo, Japan), and images were acquired with a Digital Sight camera DS-03 using the imaging software NIS-Elements 4.1(Nikon Corporation, Tokyo, Japan). The percentage of the hypoxic area within a total spheroid area was determined.
2.14. Statistical Analysis
All the experiments, except the one for which details were already described, were performed on at least three human samples in technical triplicate. Data are presented as mean ± standard deviation (SD) and were analyzed with two-way ANOVA or t-test using Graph Pad Prism 9.0 software (San Diego, CA, USA). The significance threshold was set at p < 0.05.
4. Discussion
Currently, T1DM therapy is based almost exclusively on the control of insulin levels through the administration of exogenous insulin. Patients with diabetes have a high risk of incurring other pathologies, which can considerably shorten life expectancy [
23,
24]. Alternative solutions are highly demanded to find new treatments for diabetes. An ideal therapeutic approach may act on the reduction of both blood glucose levels and auto-immune response. In this context, cell therapy can be considered a suitable option.
Perinatal cells offer a wise solution as a candidate cell source for cell therapy against T1DM [
2]. In particular, AECs and WJ-MSCs have immunomodulatory activity, inhibiting the activation of the immune system and stimulating the tolerogenic cell component. In the context of T1DM, the immunomodulatory properties exerted by perinatal cells could be a valid tool to preserve beta cells from the hyper-activation of the immune system [
10]. Thus, our work aims to develop a 3D cellular model that is suitable for endo-pancreatic differentiation protocols as a possible step in the development of a cell therapy against T1DM. Three-dimensional models of WJ-MSC, such as spheroids, show an increased physiological relevance for future clinical applications based on their greater immunomodulatory and differentiating properties [
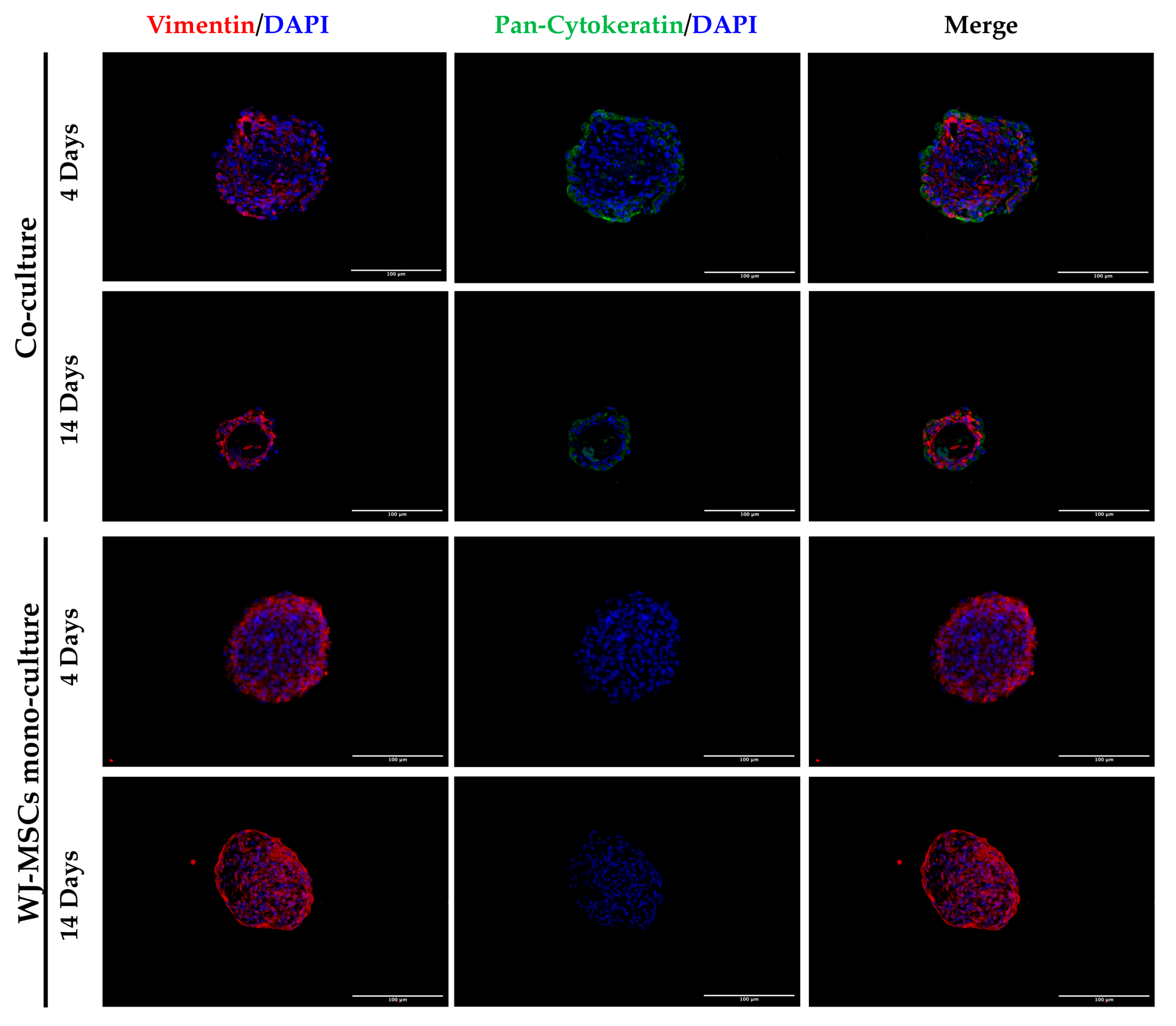
25]. While the assembly of WJ-MSC spheroid mono-cultures has been reported and largely characterized, less information is available for AEC spheroids. Our group has already developed 3D cultures of AECs, which were shown to be able to secrete insulin and glucagon [
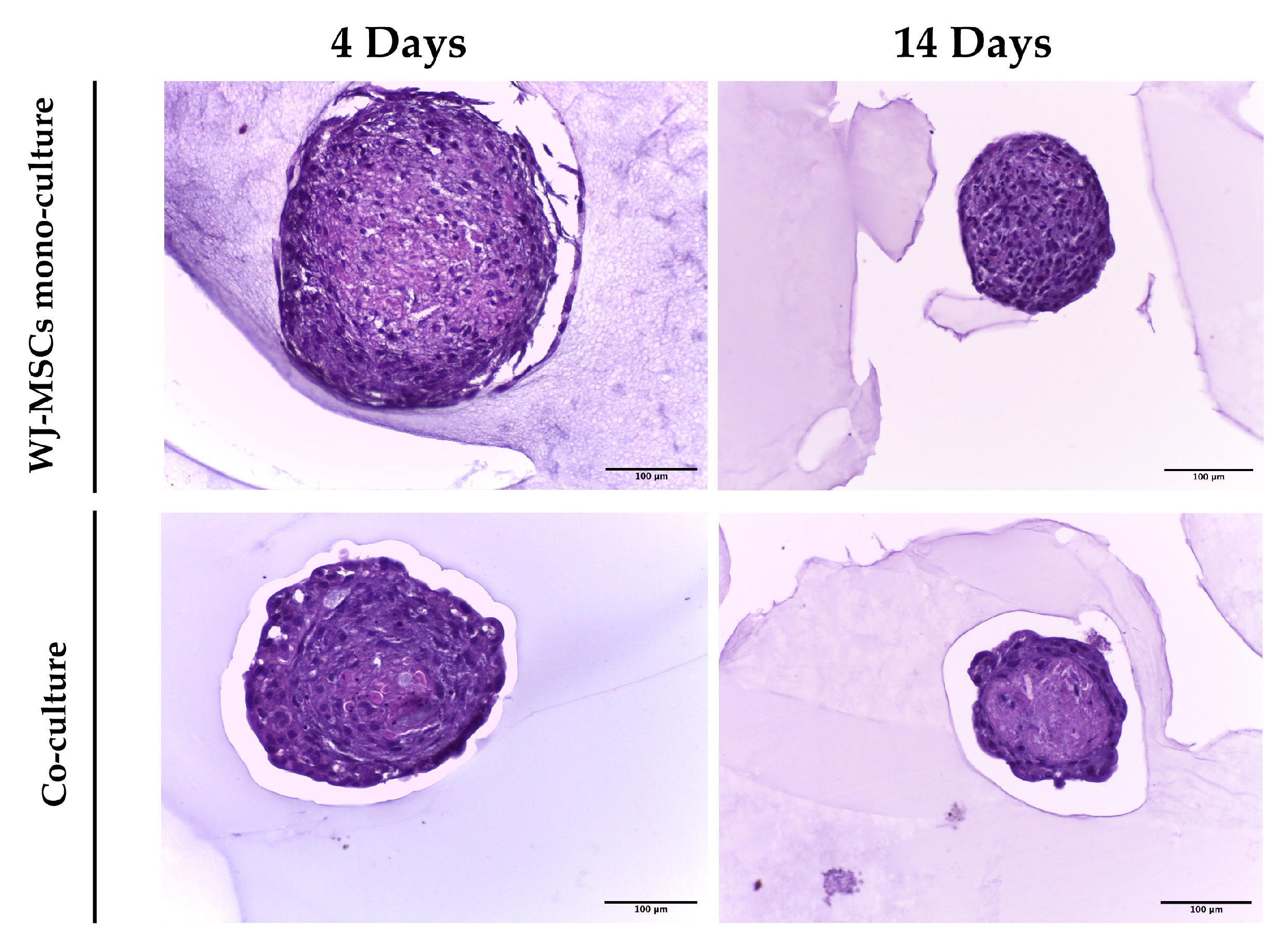
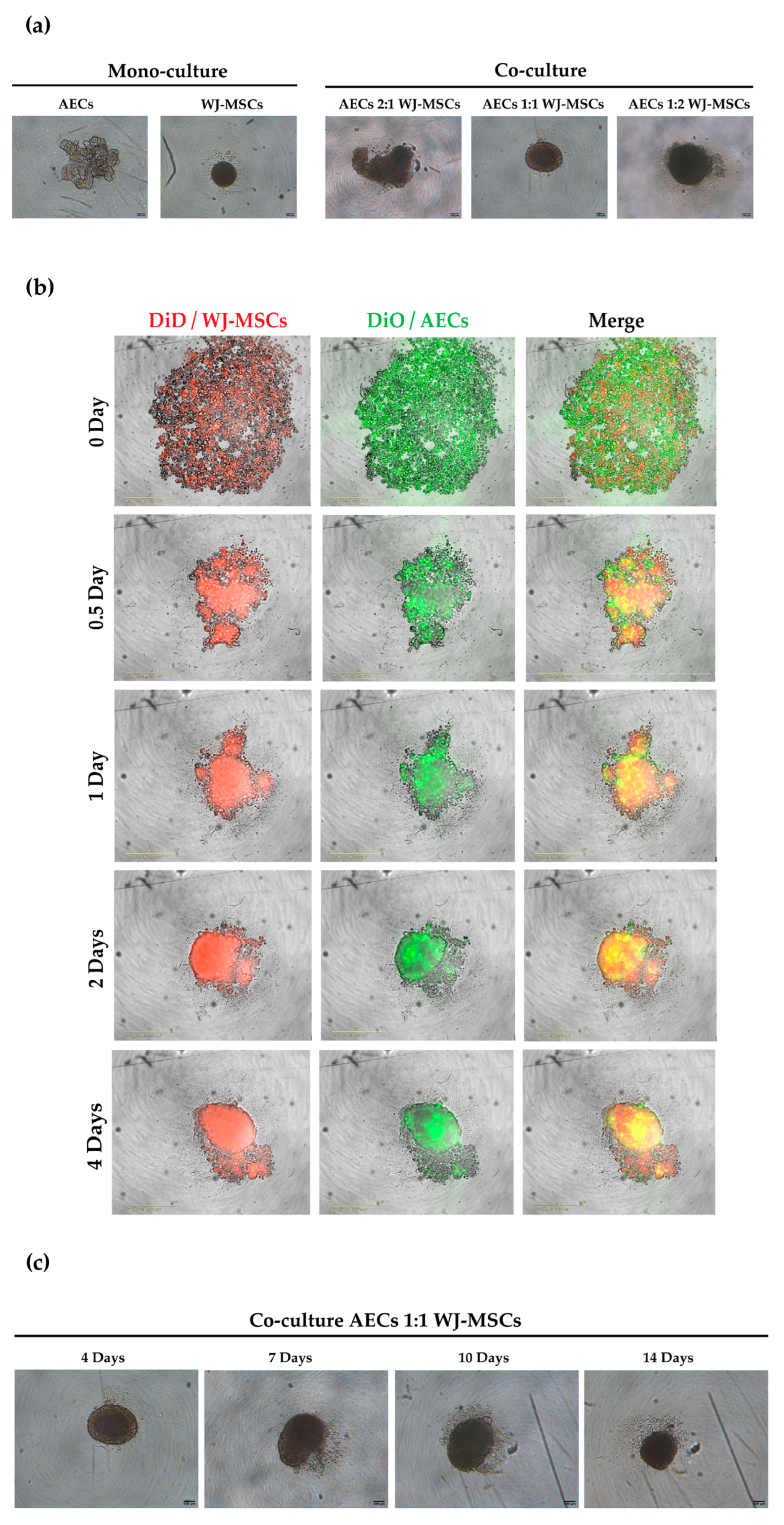
4], thus representing a good cell source for endocrine pancreatic differentiation. However, the levels of insulin secreted were not high enough to be considered physiologically relevant and, as shown in our experiment, AECs alone have difficulty forming spheroids with regular edges and roundness (
Figure 2). For this reason, establishing a new optimized 3D model is fundamental. To address this, we develop a 3D spheroid co-culture of WJ-MSCs and AECs. WJ-MSCs act as structural support for the development of more consistent spheroids. Moreover, the addition of a mesenchymal population can better mimic the structural and functional characteristics of native tissue micro-environments, such as pancreatic islets, where epithelial and stromal cells are closely connected. Furthermore, WJ-MSCs have several advantages in spheroid formation, including the secretion of factors that promote tight junctions, cell–cell adhesion, and extracellular matrix formation. Additionally, WJ-MSCs have been found to enhance the production of growth factors and cytokines, and have increased differentiation and stemness potential, which could increase insulin secretion [
26,
27].
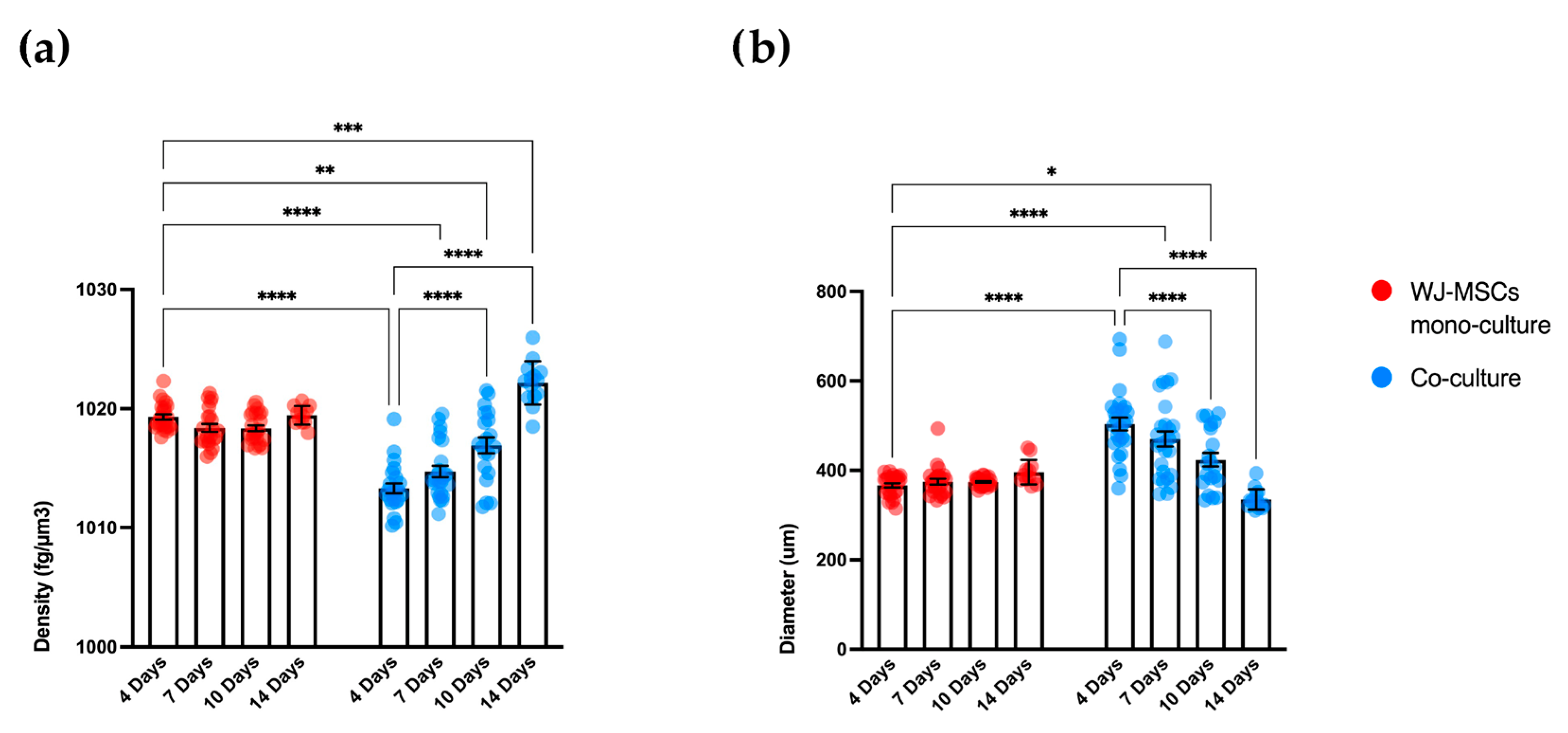
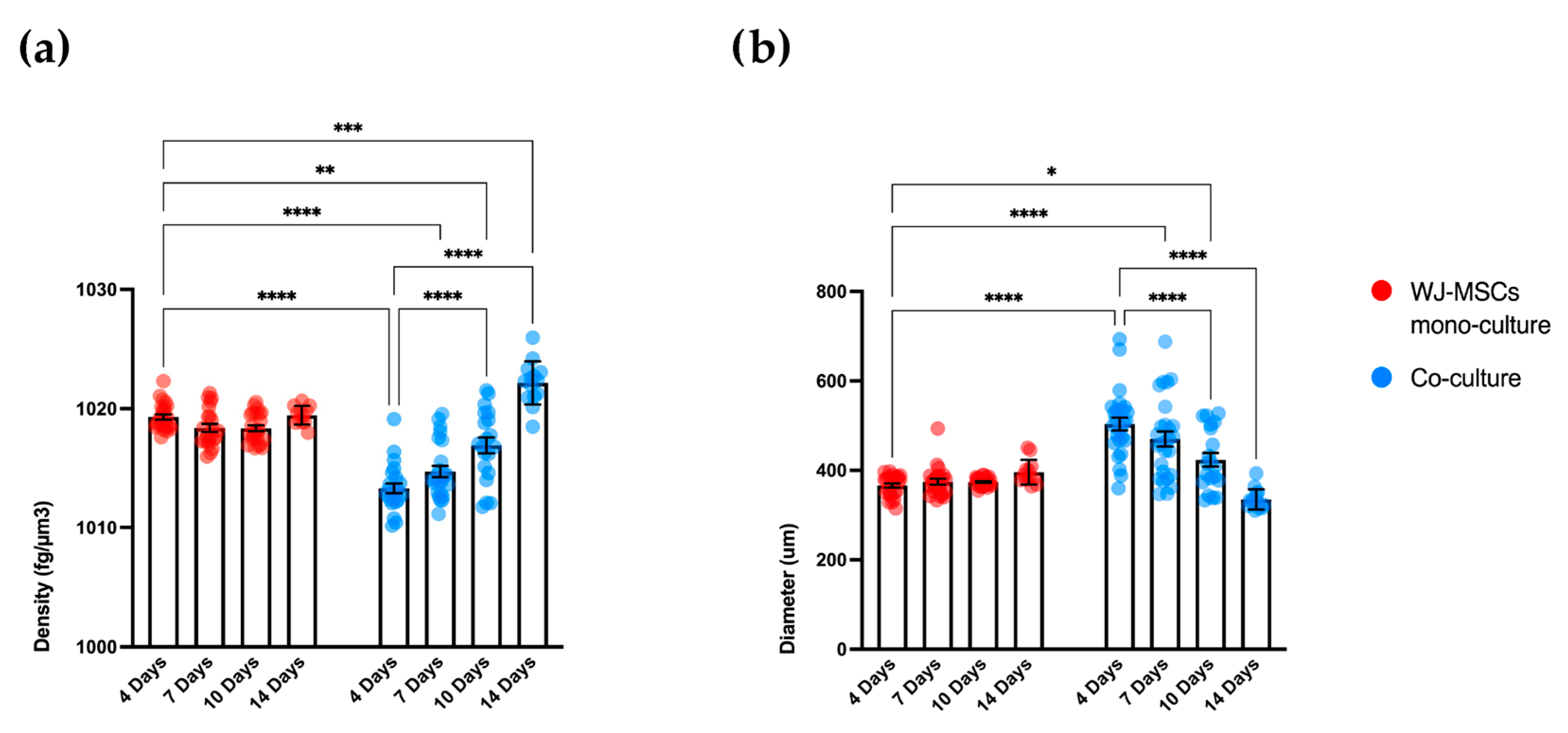
As a first step in creating co-cultures, we combined AEC and WJ-MSC cultures in different ratios. By using a ratio enabling a double quantity of AECs in comparison to the WJ-MSCs, co-cultures did not efficiently form spheroid-like shapes. Among the tested ratios, the most suitable and consistent cell aggregation was found in the one-to-one ratio. Co-culture in a one-to-one ratio appeared to store all cell types and have a consistent and reproducible shape and dimensions among donors and repeated experiments. Moreover, the diameter of co-culture spheroids, which is about 400 µm at the end of its formation (4 days) (
Figure 4 and
Figure 5b), does not exceed the maximum diameter of human pancreatic islets, which is 500 µm [
27,
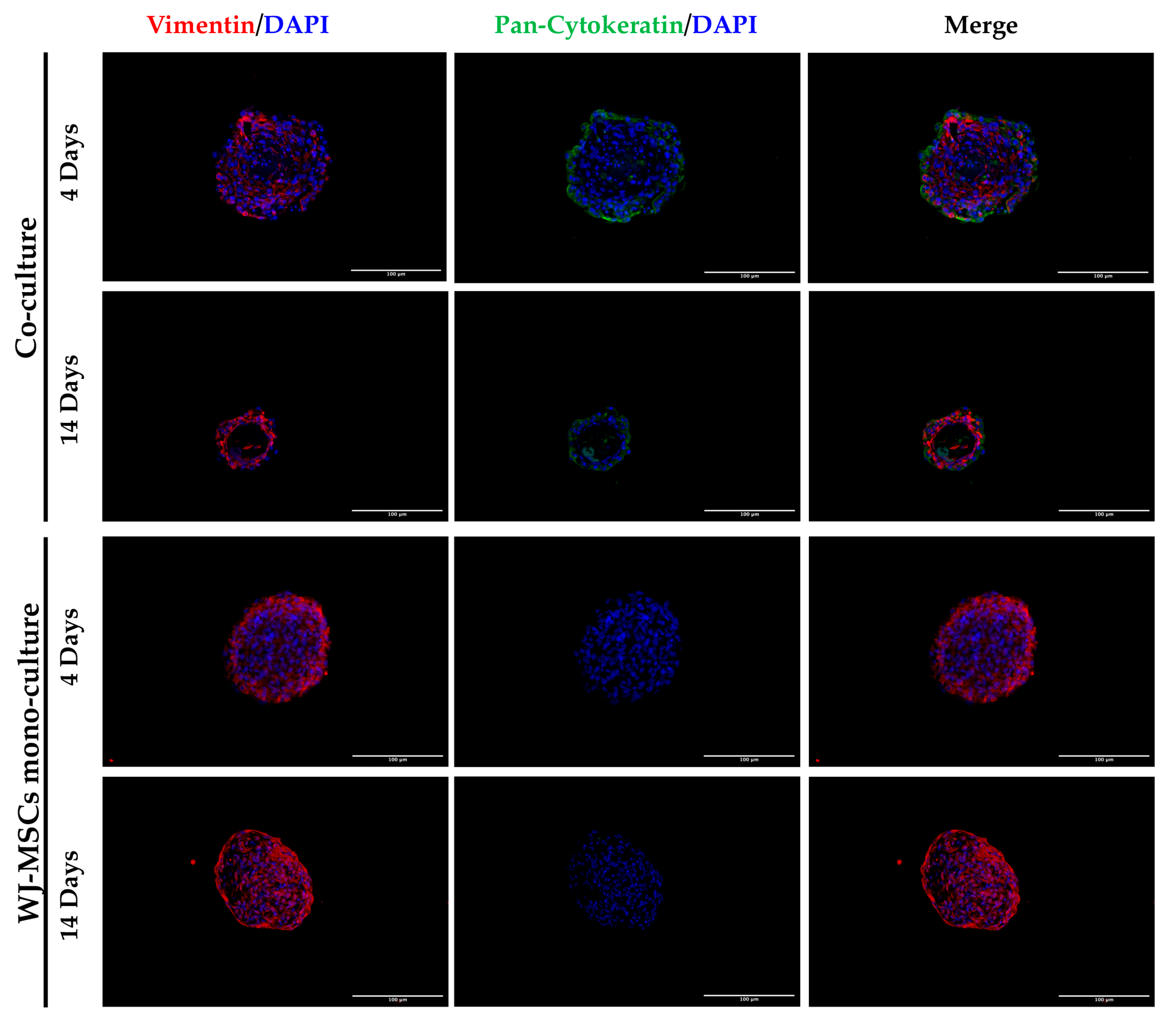
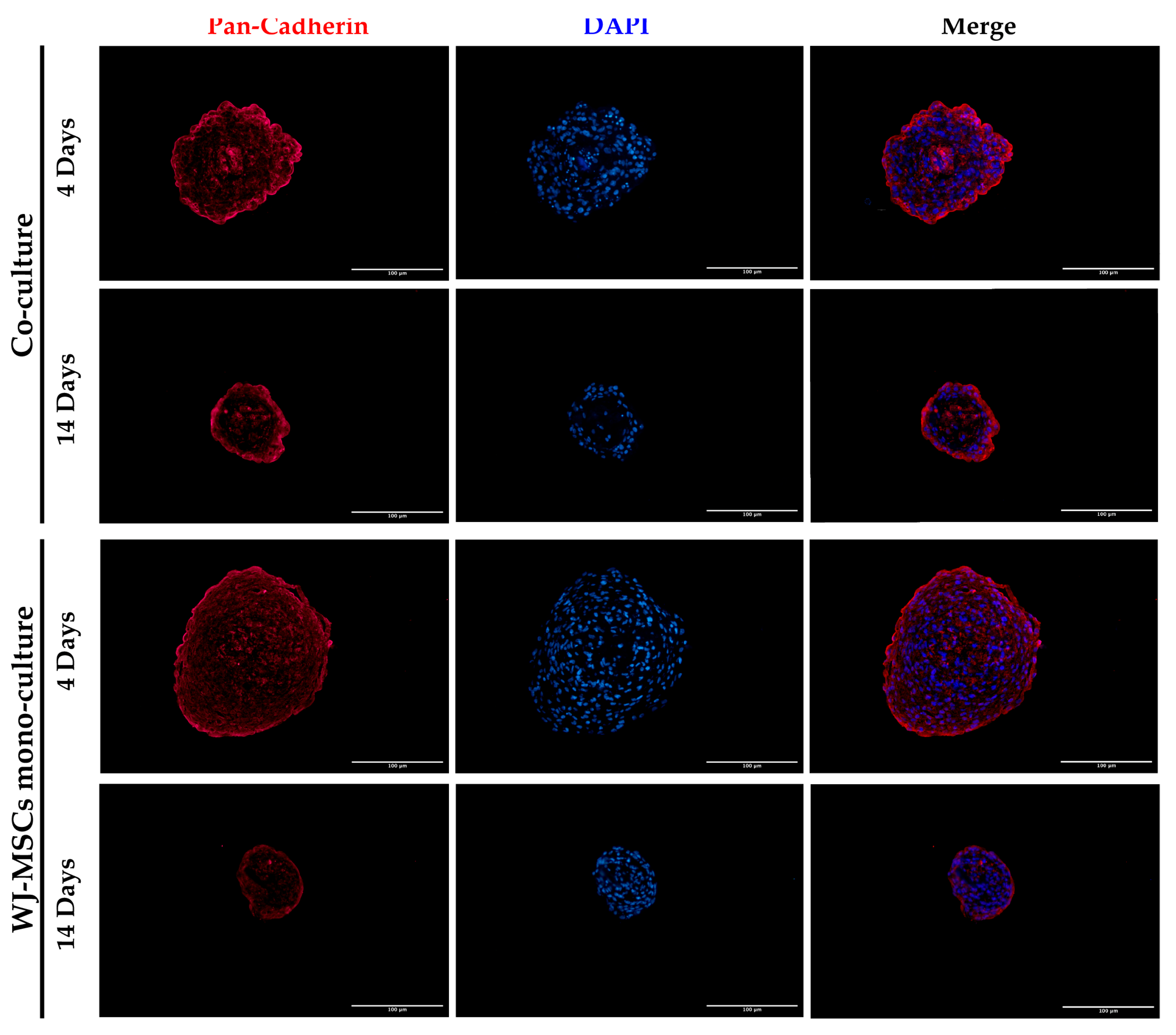
28]. Spheroids with this dimension have a great advantage to be easily manipulated during media change, since they are visible to the naked eye, and to be transferred to downstream analysis without high-throughput equipment. In addition, since AECs are potentially involved in beta cell differentiation, it is fundamental to set up a model that could include a higher number of AECs in comparison to our previously developed AEC-only spheroids. This consideration was impossible at an AEC:WJ-MSC ratio of 1:2, confirming the selection of the one-to-one ratio. Moreover, in our co-culture model, AECs (Pan-Ck positive cells) localized spontaneously at the periphery of the spheroid (
Figure 7), as following with increasing spheroid diameter would contain a higher number of AECs than smaller spheroids, still resembling the minimum size of human pancreatic islets [
28,
29] (e.g., spheroid with 50 µm diameter). We hypothesize that a greater number of AECs on the surface of the spheroid may better enable the release of insulin derived from an eventual successful differentiation into beta cells. The assembled spheroid co-cultures are in the range of measurement of an innovative instrument, the W8 Physical Cytometer, able to analyze the mass density of the spheroids with a label-free approach [
30]. To further investigate the bio-physical characteristics of the assembled spheroid co-cultures, W8 analysis was performed (
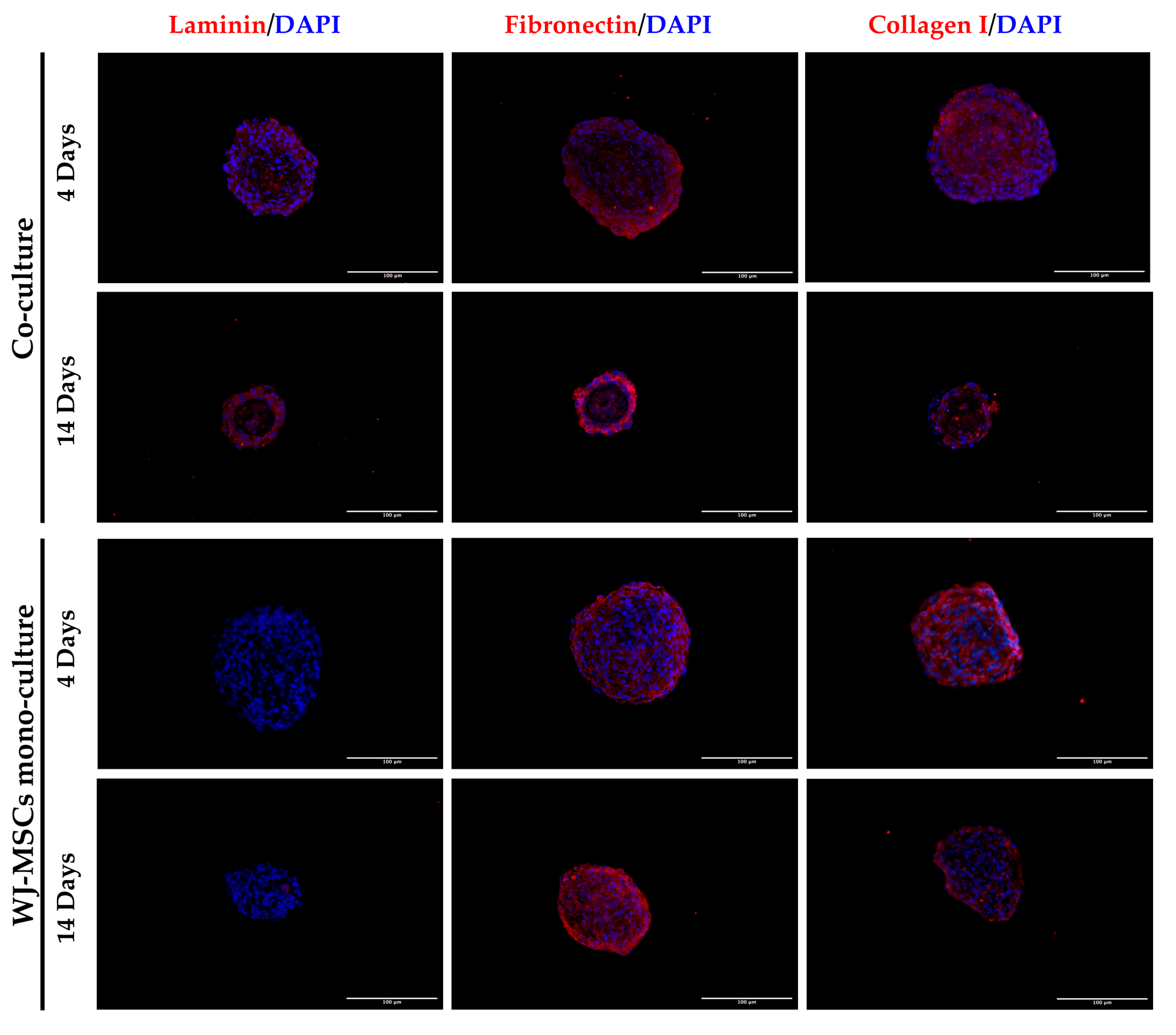
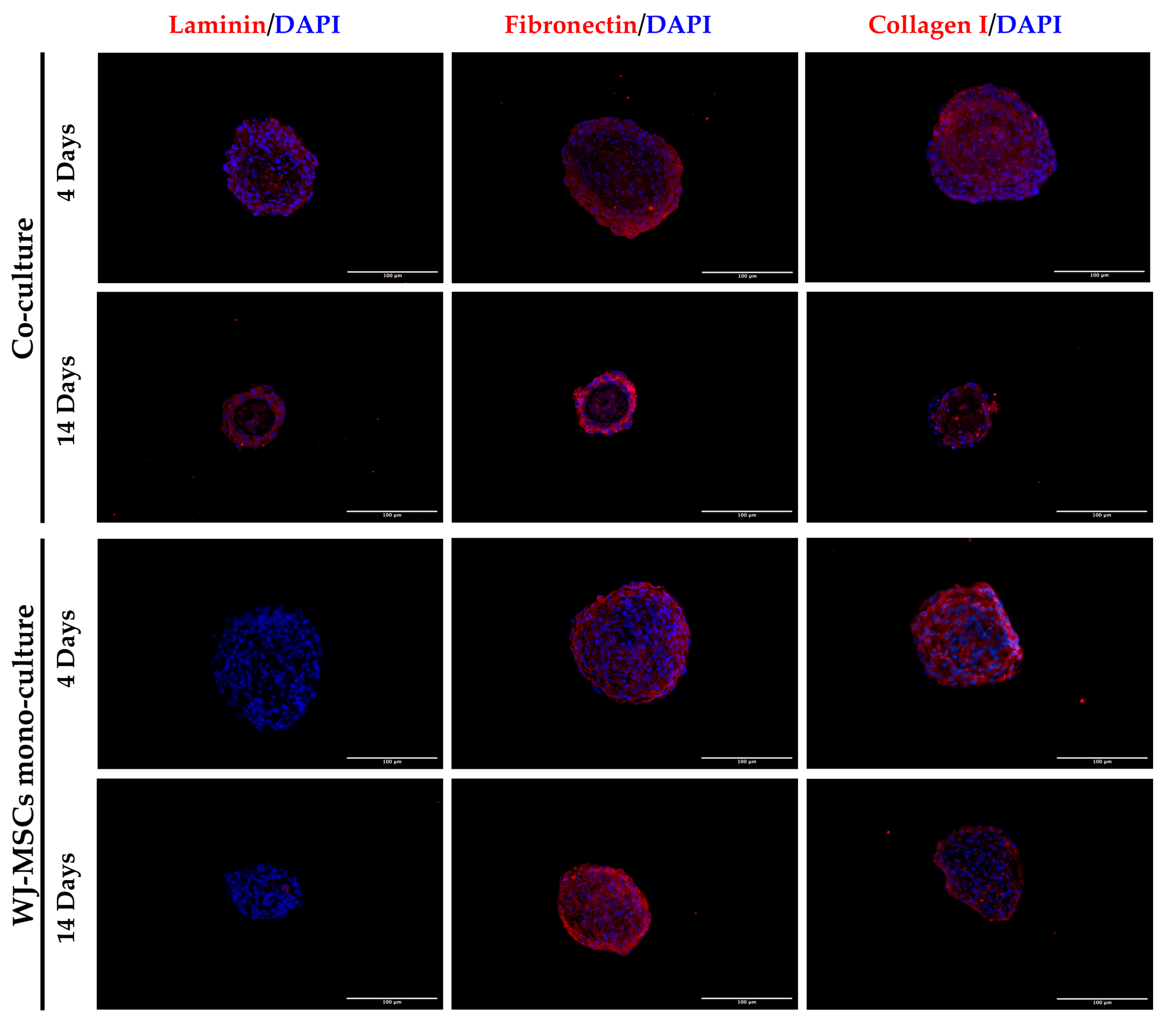
Figure 5). The results showed that the mono-culture control of only WJ-MSCs was quite stable after formation, but the co-culture spheroids decreased their diameter and increased their mass density over time. These results find a correlation with the immunofluorescence analysis of different ECM proteins secreted within the spheroid formation (
Figure 8). Type I collagen and fibronectin belong to the interstitial matrix network that is interspersed around the cells. The deposition of fibronectin and collagen type I, usually secreted by MSCs and stromal cells [
13], was visible in the spheroids at 4 days after cell seeding, the time required for consistent spheroid formation in both mono-cultures and co-cultures. The formation of the ECM network is fundamental not only for spheroid formation, but also for mirroring the micro-environment present in pancreatic islets. Indeed, laminin and type I collagen are naturally present in the pancreatic islets [
23] and fibronectin is fundamental for their physiologic development, and it is thought to play a role in endocrine cell motility during islet formation [
22]. Moreover, ECM can enhance secreted factor storage and differentiating agent retention; thus, the appearance of a 3D mesh of fibronectin and collagen I may support the induction protocol of differentiation [
31,
32].
Laminin is a protein occurring in the basal membrane, which is a lining structure of the ECM at the interface between the epithelium and the stromal compartment of several organs [
20]. Laminin staining was found, only in co-culture spheroids, close to the region occupied by AECs, likely suggesting that they are the main secreting cell type, as already demonstrated in previously developed AEC-only spheroids [
4]. In addition, cell–cell interaction is demonstrated with cadherin staining, a suggested marker of compaction of the spheroids [
15]. By increasing inter-cellular contacts, the diameter is likely to decreased over time. However, despite the ECM still being present at 14 days of culture, the nuclei of a few cells in the core of the spheroids were lost (
Figure 4 and
Figure 8), where more WJ-MSCs were present in comparison to AECs edging the spheroid co-culture structure. As a consequence of this, WJ-MSCs could work better as ECM producers more than cell players during beta cell induction. Thus, the increase in cell density during long-term cultures could be a result of an augmented ratio of ECM:MSCs in the spheroid.
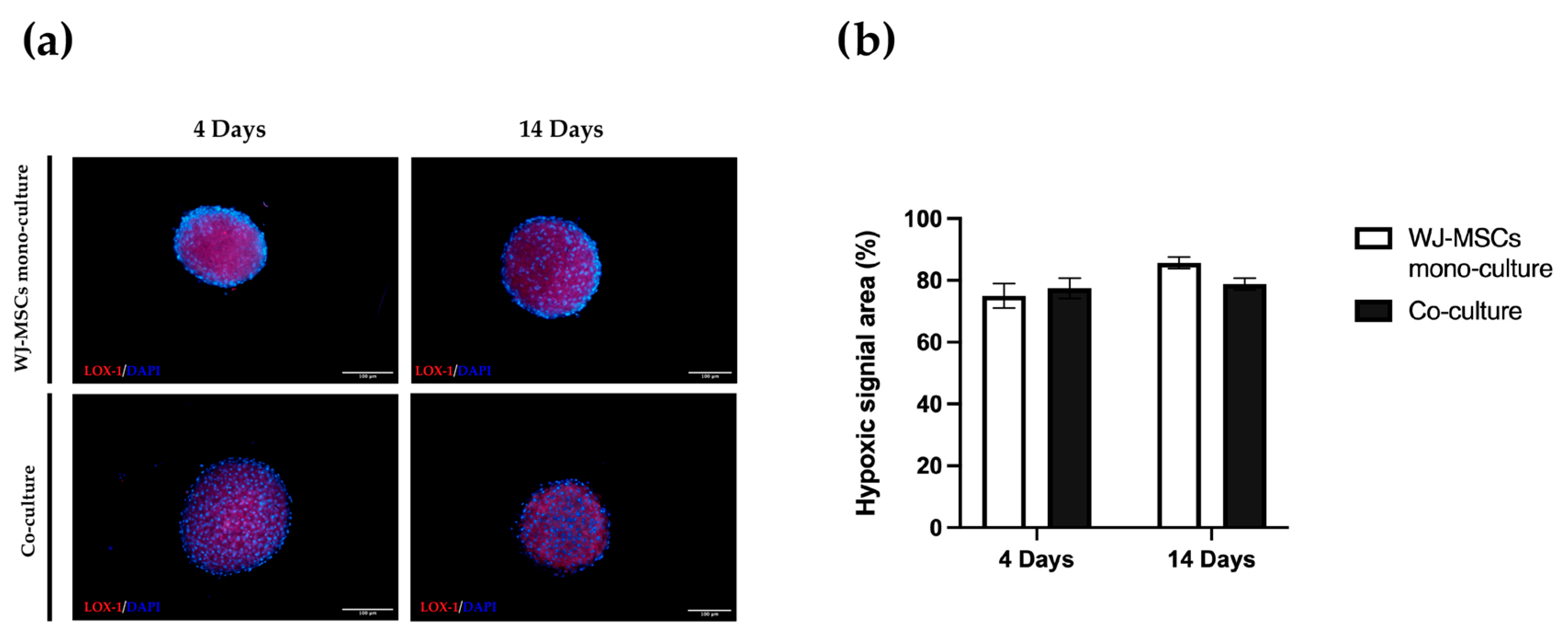
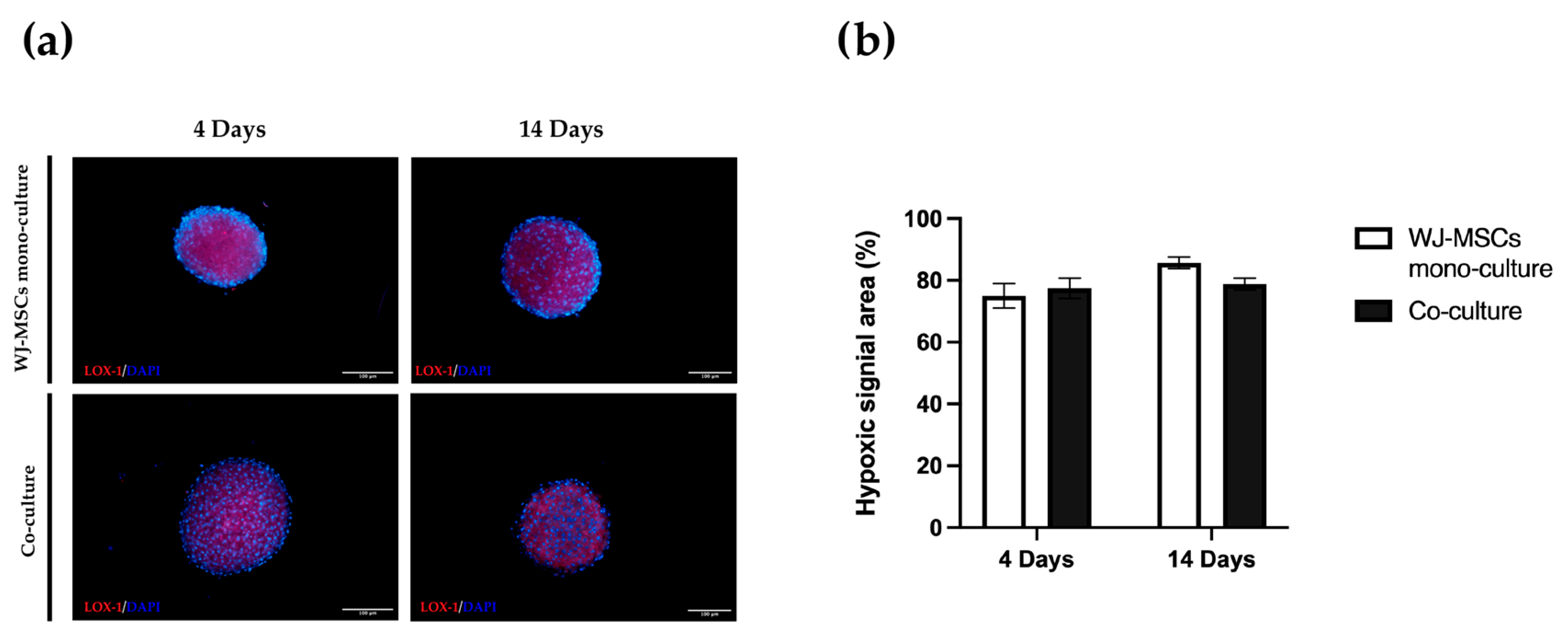
An important consideration is that spheroids derived from perinatal isolated cells, unlike organoids derived from insulin-producing cells or tissues, do not possess self-renewal capacity. A prolonged time in culture can be detrimental to cell viability. For this reason, is fundamental to also consider culture duration ahead of starting the differentiation protocol. Long-term cultured spheroids suffer metabolites and gas exchange, similar to tumorspheres; the hypoxic level can increase with negative consequences and apoptotic/necrotic cores can form in the inner part of the cell aggregates. Overall, our co-culture characterization indicates short-term cultured spheroids including AECs and WJ-MSCs are superior to long-term cultured spheroids, at least for our static culture conditions. Hyperoxia, often characterizing in vitro manipulation conditions, may cause oxidative stress by the generation of reactive oxygen species that will affect the functionality of cultured stem cells. On the contrary, a controlled level of hypoxia is a physiological stimulus characterizing many stem cell niches, including the MSC one [
33]. Hyperoxia can sustain stemness maintenance in vitro [
34,
35] and control the properties of in vitro cultured WJ-MSCs [
36,
37,
38]. The hypoxia level did not change between 4 days and 14 days (
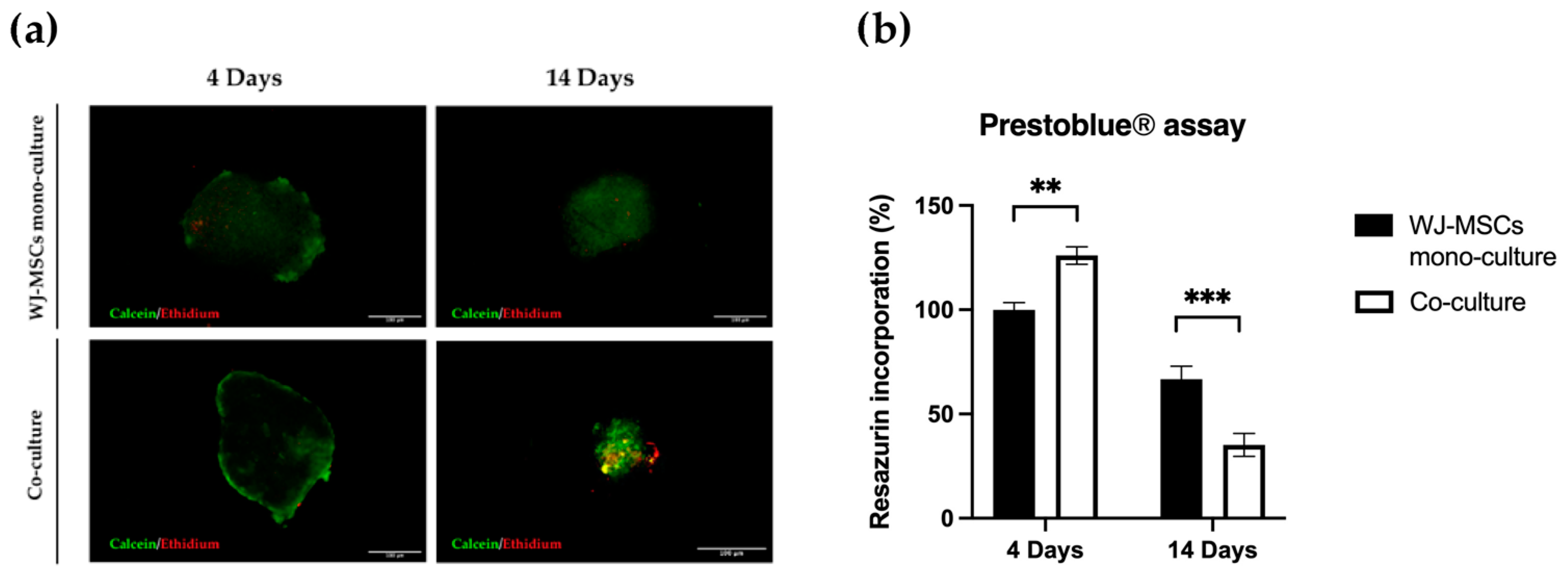
Figure 9), but cell viability evaluated by both a metabolism indicator (
Figure 3a) and a cell-membrane-integrity signal (
Figure 3b) decreased. Based on our results, spheroids taken soon after their formation could be more suitable to proceed to any induction of differentiation, owing to a better viability rate.
In the future, our designed co-cultures can be optimized for dynamic culturing with bioreactors [
39], in an effort to maintain the properties of the spheroid seen at its formation.
Another limitation of a co-culture is the difficulty to dissociate the spheroids to perform subsequent analyses of the single-cell type. Preliminary experiments of dissociation through enzymatic and mechanical separation were unsuccessful. A very low yield in terms of cell number and low viability levels was recovered, in particular, after the onset of intercellular junctions at both homo- and hetero-typic levels, and following the formation of strong attachment to the ECM. Moreover, any subsequent analyses of the single-cell type should require additional protocols to isolate it, creating an issue, for example, in gene expression analysis.
Regarding the viability of our model, based on our results (
Figure 3 and
Figure 4), we would likely lose WJ-MSCs if a long-term culture were to be applied. An aspect to take strongly into consideration is that such long-term cultures mirror an undifferentiated state by employing a basal cell medium. The differentiation medium may sustain stromal MSC viability or even stimulate proliferation. However, we demonstrated that AECs, which are the best candidate for becoming beta cell replacements, are still detected 14 days after seeding, suggesting that AECs can entirely survive before their induction towards endo-pancreatic differentiation. A proper differentiation into a pancreatic lineage seems to require at least more than the time used to generate our spheroid co-cultures. This is not relevant if freshly generated spheroids were to be induced. Since AEC populations present a well-known inter- and intra-donor variability, future protocols could take advantage of this and involve the use of instruments for high-throughput, gentle, label-free cell selection [
30,
40].



 ,
, 





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}