Predictive Effect of Helicobacter pylori in Gastric Carcinoma Development: Systematic Review and Quantitative Evidence Synthesis

 , , ,
, , ,
Abstract
:1. Introduction
1.1. H. pylori Infection and Gastric Cancer
1.2. Subpopulation Variance of H. pylori and Gastric Cancer
2. Materials and Methods
2.1. Study Design
2.2. Design Rationale
2.3. QES Significance and Relevance
2.4. QES Dynamism and Intervention Mapping
2.5. Search Engine and Strategies
2.6. Study Eligibility
2.7. Data Extraction
2.8. Study Variables
2.9. Data Analysis
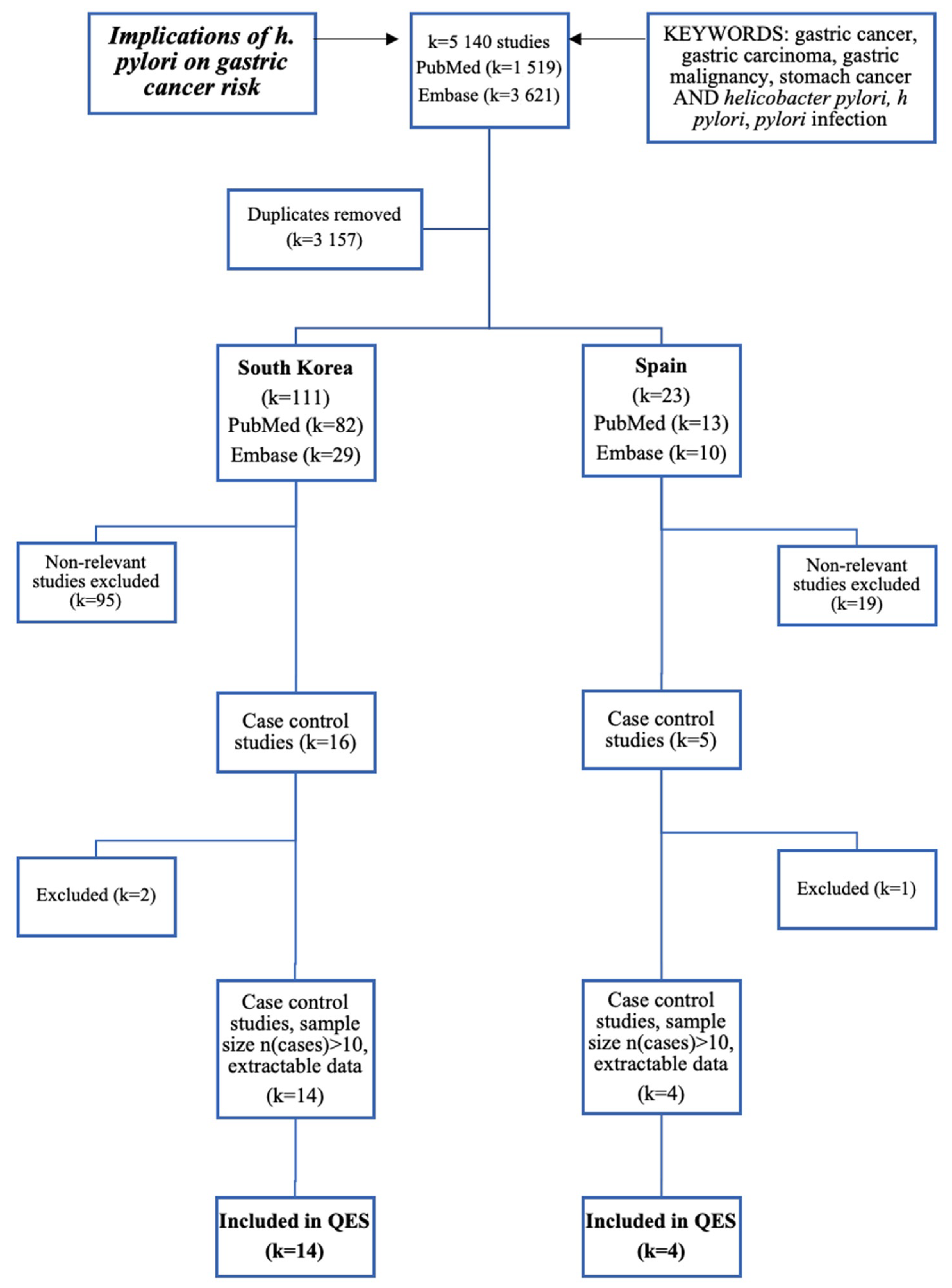
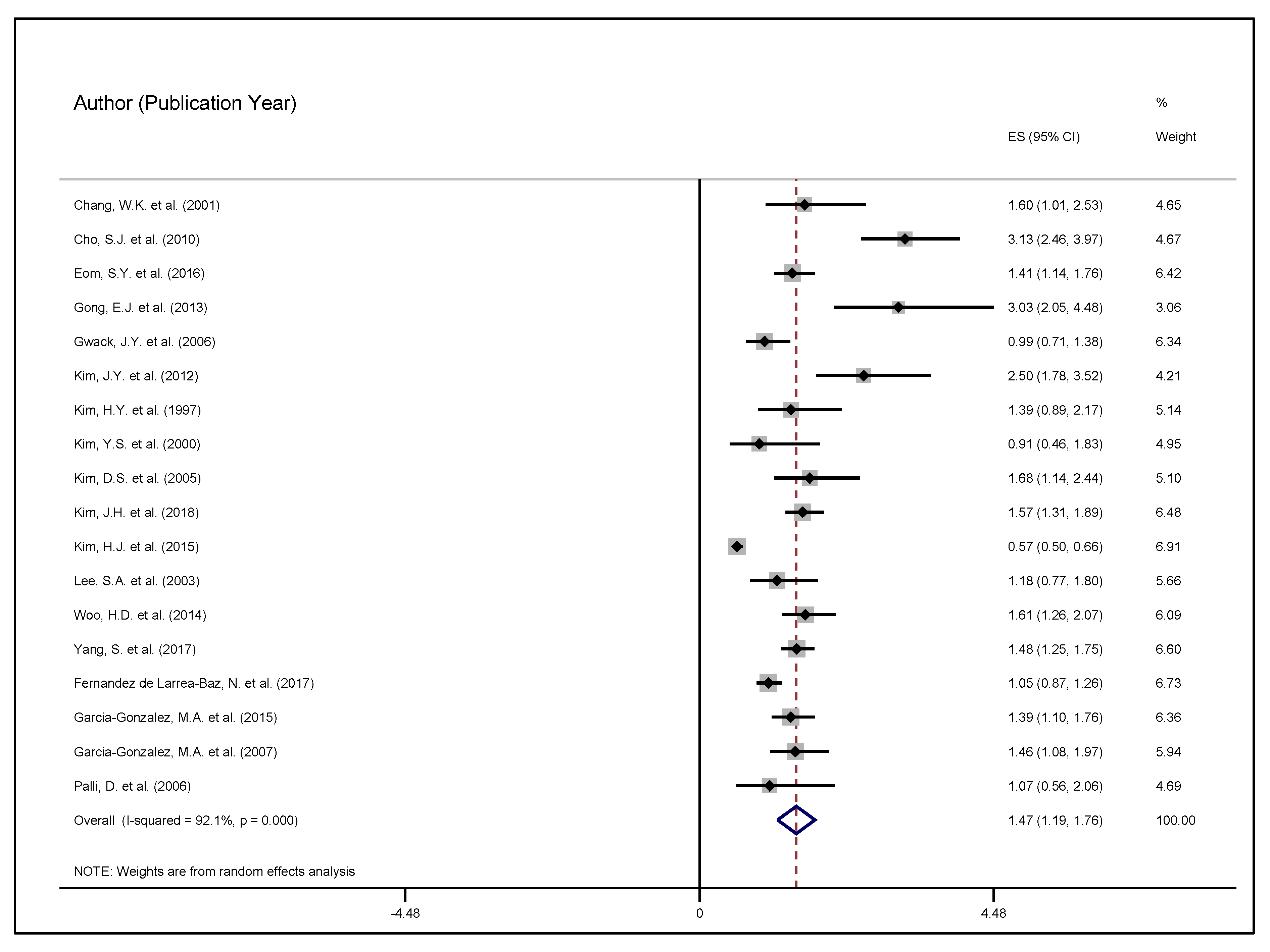
3. Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Burucoa, C.; Axon, A. Epidemiology of Helicobacter pylori infection. Helicobacter 2017, 22, e12403. [Google Scholar] [CrossRef] [PubMed]
- Rosario, D.F. Helicobacter Pylori. Available online: https://kidshealth.org/en/parents/h-pylori.htm (accessed on 25 June 2020).
- Wroblewski, L.E.; Peek, R.M.; Wilson, K.T. Helicobacter pylori and gastric cancer: Factors that modulate disease risk. Clin. Microbiol. Rev. 2010, 23, 713–739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Polk, D.B.; Peek, R.M. Helicobacter pylori: Gastric cancer and beyond. Nat. Rev. Cancer 2010, 10, 403–414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haley, K.P.; Gaddy, J.A. Helicobacter pylori: Genomic Insight into the Host-Pathogen Interaction. Int. J. Genom. 2015, 2015, 386905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conteduca, V.; Sansonno, D.; Lauletta, G.; Russi, S.; Ingravallo, G.; Dammacco, F.H. pylori infection and gastric cancer: State of the art (review). Int. J. Oncol. 2013, 42, 5–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bourke, B.; Ceponis, P.; Chiba, N.; Czinn, S.; Ferraro, R.; Fischbach, L.; Gold, B.; Hyunh, H.; Jacobson, K.; Jones, N.L.; et al. Canadian Helicobacter Study Group Consensus Conference: Update on the approach to Helicobacter pylori infection in children and adolescents—An evidence-based evaluation. Can. J. Gastroenterol. 2005, 19, 399–408. [Google Scholar]
- Karkhah, A.; Ebrahimpour, S.; Rostamtabar, M.; Koppolu, V.; Darvish, S.; Vasigala, V.K.R.; Validi, M.; Nouri, H.R. Helicobacter pylori evasion strategies of the host innate and adaptive immune responses to survive and develop gastrointestinal diseases. Microbiol. Res. 2019, 218, 49–57. [Google Scholar] [CrossRef]
- Parsonnet, J. Bacterial infection as a cause of cancer. Environ. Health Perspect. 1995, 103 (Suppl. 8), 263–268. [Google Scholar] [CrossRef] [Green Version]
- Sallas, M.L.; Dos Santos, M.P.; Orcini, W.A.; David, É.; Peruquetti, R.L.; Payão, S.L.M.; Rasmussen, L.T. Status (on/off) of oipA gene: Their associations with gastritis and gastric cancer and geographic origins. Arch. Microbiol. 2019, 201, 93–97. [Google Scholar] [CrossRef]
- Rassow, J. Helicobacter pylori vacuolating toxin A and apoptosis. Cell Commun. Signal. 2011, 9, 26. [Google Scholar] [CrossRef] [Green Version]
- Montecucco, C.; Rappuoli, R. Living dangerously: How Helicobacter pylori survives in the human stomach. Nat. Rev. Mol. Cell Biol. 2001, 2, 457–466. [Google Scholar] [CrossRef] [PubMed]
- Ricci, V.; Romano, M.; Boquet, P. Molecular cross-talk between Helicobacter pylori and human gastric mucosa. World J. Gastroenterol. 2011, 17, 1383–1399. [Google Scholar] [CrossRef] [PubMed]
- Ebule, I.A.; Longdoh, A.N.; Paloheimo, I.L. Helicobacter pylori infection and atrophic gastritis. Afr. Health Sci. 2013, 13, 112–117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lahner, E.; Carabotti, M.; Annibale, B. Treatment of. World J. Gastroenterol. 2018, 24, 2373–2380. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.S.; Wong, I.O.; Leung, W.K. Helicobacter pylori associated gastric intestinal metaplasia: Treatment and surveillance. World J. Gastroenterol. 2016, 22, 1311–1320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olmez, S.; Aslan, M.; Erten, R.; Sayar, S.; Bayram, I. The Prevalence of Gastric Intestinal Metaplasia and Distribution of Helicobacter pylori Infection, Atrophy, Dysplasia, and Cancer in Its Subtypes. Gastroenterol. Res. Pract. 2015, 2015, 434039. [Google Scholar] [CrossRef] [Green Version]
- Clyne, M.; Labigne, A.; Drumm, B. Helicobacter pylori requires an acidic environment to survive in the presence of urea. Infect. Immun. 1995, 63, 1669–1673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ansari, S.; Yamaoka, Y. Survival of Helicobacter pylori in gastric acidic territory. Helicobacter 2017, 22. [Google Scholar] [CrossRef]
- Violeta Filip, P.; Cuciureanu, D.; Sorina Diaconu, L.; Maria Vladareanu, A.; Silvia Pop, C. MALT lymphoma: Epidemiology, clinical diagnosis and treatment. J. Med. Life 2018, 11, 187–193. [Google Scholar] [CrossRef]
- Liu, H.; Chen, Y.T.; Wang, R.; Chen, X.Z. Helicobacter pylori infection, atrophic gastritis, and pancreatic cancer risk: A meta-analysis of prospective epidemiologic studies. Medicine 2017, 96, e7811. [Google Scholar] [CrossRef]
- Al-Mulhim, A.S.; Mohammad, H.A. Hypergastrinemia and H. pylori infection as a risk factor for colorectal cancer. Ann. Saudi Med. 2002, 22, 252–255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- GonzÁlez, I.; Araya, P.; Rojas, A. Helicobacter Pylori Infection and Lung Cancer: New Insights and Future Challenges. Zhongguo Fei Ai Za Zhi 2018, 21, 658–662. [Google Scholar] [CrossRef] [PubMed]
- George, S.; Lucero, Y.; Torres, J.P.; Lagomarcino, A.J.; O’Ryan, M. Gastric Damage and Cancer-Associated Biomarkers in. Front. Microbiol. 2020, 11, 90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orellana-Manzano, A.; O’Ryan, M.G.; Lagomarcino, A.J.; George, S.; Muñoz, M.S.; Mamani, N.; Serrano, C.A.; Harris, P.R.; Ramilo, O.; Mejías, A.; et al. Infection Is Associated with Decreased Expression of SLC5A8, a Cancer Suppressor Gene, in Young Children. Front. Cell. Infect. Microbiol. 2016, 6, 121. [Google Scholar] [CrossRef] [PubMed]
- Rothenbacher, D.; Brenner, H. Burden of Helicobacter pylori and H. pylori-related diseases in developed countries: Recent developments and future implications. Microbes. Infect. 2003, 5, 693–703. [Google Scholar] [CrossRef]
- Jafri, W.; Yakoob, J.; Abid, S.; Siddiqui, S.; Awan, S.; Nizami, S.Q. Helicobacter pylori infection in children: Population-based age-specific prevalence and risk factors in a developing country. Acta Paediatr. 2010, 99, 279–282. [Google Scholar] [CrossRef]
- Ghoshal, U.C.; Chaturvedi, R.; Correa, P. The enigma of Helicobacter pylori infection and gastric cancer. Indian J. Gastroenterol. 2010, 29, 95–100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Misra, V.; Pandey, R.; Misra, S.P.; Dwivedi, M. Helicobacter pylori and gastric cancer: Indian enigma. World J. Gastroenterol. 2014, 20, 1503–1509. [Google Scholar] [CrossRef]
- Moon, H.S.; Yun, G.Y.; Kim, J.S.; Eun, H.S.; Kang, S.H.; Sung, J.K.; Jeong, H.Y.; Song, K.S. Risk factors for metachronous gastric carcinoma development after endoscopic resection of gastric dysplasia: Retrospective, single-center study. World J. Gastroenterol. 2017, 23, 4407–4415. [Google Scholar] [CrossRef]
- Epplein, M.; Signorello, L.B.; Zheng, W.; Peek, R.M.; Michel, A.; Williams, S.M.; Pawlita, M.; Correa, P.; Cai, Q.; Blot, W.J. Race, African ancestry, and Helicobacter pylori infection in a low-income United States population. Cancer Epidemiol. Biomarkers Prev. 2011, 20, 826–834. [Google Scholar] [CrossRef] [Green Version]
- Khalifa, M.M.; Sharaf, R.R.; Aziz, R.K. Helicobacter pylori: A poor man’s gut pathogen? Gut Pathog. 2010, 2, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, S.; Metz, D.C.; Ellenberg, S.; Kaplan, D.E.; Goldberg, D.S. Risk Factors and Incidence of Gastric Cancer After Detection of Helicobacter pylori Infection: A Large Cohort Study. Gastroenterology 2020, 158, 527–536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huerta-Franco, M.R.; Banderas, J.W.; Allsworth, J.E. Ethnic/racial differences in gastrointestinal symptoms and diagnosis associated with the risk of. Clin. Exp. Gastroenterol. 2018, 11, 39–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goh, K.L.; Cheah, P.L.; Md, N.; Quek, K.F.; Parasakthi, N. Ethnicity and H. pylori as risk factors for gastric cancer in Malaysia: A prospective case control study. Am. J. Gastroenterol. 2007, 102, 40–45. [Google Scholar] [CrossRef]
- Gutiérrez-Escobar, A.J.; Méndez-Callejas, G.; Acevedo, O.; Bravo, M.M. Rapid evolution of the. PeerJ 2018, 6, e4846. [Google Scholar] [CrossRef]
- Kadhim, M.; Thacker, M.; Kadhim, A.; Holmes, L. Treatment of unicameral bone cyst: Systematic review and meta analysis. J. Child Orthop. 2014, 8, 171–191. [Google Scholar] [CrossRef] [Green Version]
- Holmes, L.; Lim, A.; Comeaux, C.R.; Dabney, K.W.; Okundaye, O. DNA Methylation of Candidate Genes (ACE II, IFN-γ, AGTR 1, CKG, ADD1, SCNN1B and TLR2) in Essential Hypertension: A Systematic Review and Quantitative Evidence Synthesis. Int. J. Environ. Res. Public Health 2019, 16, 4829. [Google Scholar] [CrossRef] [Green Version]
- Holmes, L.; Shutman, E.; Chinaka, C.; Deepika, K.; Pelaez, L.; Dabney, K.W. Aberrant Epigenomic Modulation of Glucocorticoid Receptor Gene (NR3C1) in Early Life Stress and Major Depressive Disorder Correlation: Systematic Review and Quantitative Evidence Synthesis. Int. J. Environ. Res. Public Health 2019, 16, 4280. [Google Scholar] [CrossRef] [Green Version]
- DerSimonian, R. Meta-analysis in the design and monitoring of clinical trials. Stat. Med. 1996, 15, 1237–1248. [Google Scholar] [CrossRef]
- DerSimonian, R.; Laird, N. Meta-analysis in clinical trials. Control. Clin. Trials 1986, 7, 177–188. [Google Scholar] [CrossRef]
- Chang, W.K.; Kim, H.Y.; Kim, D.J.; Lee, J.; Park, C.K.; Yoo, J.Y.; Kim, H.J.; Kim, M.K.; Choi, B.Y.; Choi, H.S.; et al. Association between Helicobacter pylori infection and the risk of gastric cancer in the Korean population: Prospective case-controlled study. J. Gastroenterol. 2001, 36, 816–822. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.J.; Choi, I.J.; Kim, C.G.; Lee, J.Y.; Kook, M.C.; Seong, M.W.; Park, S.R.; Lee, J.S.; Kim, Y.W.; Ryu, K.W.; et al. Helicobacter pylori Seropositivity Is Associated with Gastric Cancer Regardless of Tumor Subtype in Korea. Gut Liver 2010, 4, 466–474. [Google Scholar] [CrossRef] [Green Version]
- Eom, S.Y.; Hong, S.M.; Yim, D.H.; Kwon, H.J.; Kim, D.H.; Yun, H.Y.; Song, Y.J.; Youn, S.J.; Hyun, T.; Park, J.S.; et al. Additive interactions between PRKAA1 polymorphisms and Helicobacter pylori CagA infection associated with gastric cancer risk in Koreans. Cancer Med. 2016, 5, 3236–3335. [Google Scholar] [CrossRef] [PubMed]
- Gong, E.J.; Ahn, J.Y.; Jung, H.Y.; Lim, H.; Choi, K.S.; Lee, J.H.; Kim, D.H.; Choi, K.D.; Song, H.J.; Lee, G.H.; et al. Risk factors and clinical outcomes of gastric cancer identified by screening endoscopy: A case-control study. J. Gastroenterol. Hepatol. 2014, 29, 301–309. [Google Scholar] [CrossRef]
- Gwack, J.; Shin, A.; Kim, C.S.; Ko, K.P.; Kim, Y.; Jun, J.K.; Bae, J.; Park, S.K.; Hong, Y.C.; Kang, D.; et al. CagA-producing Helicobacter pylori and increased risk of gastric cancer: A nested case-control study in Korea. Br. J. Cancer 2006, 95, 639–641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.Y.; Lee, H.S.; Kim, N.; Shin, C.M.; Lee, S.H.; Park, Y.S.; Hwang, J.H.; Kim, J.W.; Jeong, S.H.; Lee, D.H.; et al. Prevalence and clinicopathologic characteristics of gastric cardia cancer in South Korea. Helicobacter 2012, 17, 358–368. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.Y.; Cho, B.D.; Chang, W.K.; Kim, D.J.; Kim, Y.B.; Park, C.K.; Shin, H.S.; Yoo, J.Y. Helicobacter pylori infection and the risk of gastric cancer among the Korean population. J. Gastroenterol. Hepatol. 1997, 12, 100–103. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.S.; Park, H.A.; Kim, B.S.; Yook, J.H.; Lee, M.S. Efficacy of screening for gastric cancer in a Korean adult population: A case-control study. J. Korean Med. Sci. 2000, 15, 510–515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, D.S.; Lee, M.S.; Kim, Y.S.; Kim, D.H.; Bae, J.M.; Shin, M.H.; Ahn, Y.O. Effect modification by vitamin C on the relation between gastric cancer and Helicobacter pylori. Eur. J. Epidemiol. 2005, 20, 67–71. [Google Scholar] [CrossRef]
- Kim, J.H.; Lee, J.; Choi, I.J.; Kim, Y.I.; Kwon, O.; Kim, H.; Kim, J. Dietary Carotenoids Intake and the Risk of Gastric Cancer: A Case-Control Study in Korea. Nutrients 2018, 10, 1031. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.J.; Kim, N.; Kim, H.Y.; Lee, H.S.; Yoon, H.; Shin, C.M.; Park, Y.S.; Park, D.J.; Kim, H.H.; Lee, K.H.; et al. Relationship between body mass index and the risk of early gastric cancer and dysplasia regardless of Helicobacter pylori infection. Gastric Cancer 2015, 18, 762–773. [Google Scholar] [CrossRef] [Green Version]
- Woo, H.D.; Lee, J.; Choi, I.J.; Kim, C.G.; Lee, J.Y.; Kwon, O.; Kim, J. Dietary flavonoids and gastric cancer risk in a Korean population. Nutrients 2014, 6, 4961–4973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, S.; Lee, J.; Choi, I.J.; Kim, Y.W.; Ryu, K.W.; Sung, J.; Kim, J. Effects of alcohol consumption, ALDH2 rs671 polymorphism, and Helicobacter pylori infection on the gastric cancer risk in a Korean population. Oncotarget 2017, 8, 6630–6641. [Google Scholar] [CrossRef] [PubMed]
- Fernández de Larrea-Baz, N.; Pérez-Gómez, B.; Michel, A.; Romero, B.; Lope, V.; Pawlita, M.; Fernández-Villa, T.; Moreno, V.; Martín, V.; Willhauck-Fleckenstein, M.; et al. Helicobacter pylori serological biomarkers of gastric cancer risk in the MCC-Spain case-control Study. Cancer Epidemiol. 2017, 50, 76–84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- García-González, M.A.; Bujanda, L.; Quintero, E.; Santolaria, S.; Benito, R.; Strunk, M.; Sopeña, F.; Thomson, C.; Pérez-Aisa, A.; Nicolás-Pérez, D.; et al. Association of PSCA rs2294008 gene variants with poor prognosis and increased susceptibility to gastric cancer and decreased risk of duodenal ulcer disease. Int. J. Cancer 2015, 137, 1362–1373. [Google Scholar] [CrossRef] [PubMed]
- García-González, M.A.; Lanas, A.; Quintero, E.; Nicolás, D.; Parra-Blanco, A.; Strunk, M.; Benito, R.; Angel Simón, M.; Santolaria, S.; Sopeña, F.; et al. Gastric cancer susceptibility is not linked to pro-and anti-inflammatory cytokine gene polymorphisms in whites: A Nationwide Multicenter Study in Spain. Am. J. Gastroenterol. 2007, 102, 1878–1892. [Google Scholar] [CrossRef] [PubMed]
- Uemura, N.; Okamoto, S.; Yamamoto, S.H. pylori infection and the development of gastric cancer. Keio J. Med. 2002, 51 (Suppl. 2), 63–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herrera, V.; Parsonnet, J. Helicobacter pylori and gastric adenocarcinoma. Clin. Microbiol. Infect. 2009, 15, 971–976. [Google Scholar] [CrossRef] [Green Version]
- Takahashi-Kanemitsu, A.; Knight, C.T.; Hatakeyama, M. Molecular anatomy and pathogenic actions of Helicobacter pylori CagA that underpin gastric carcinogenesis. Cell. Mol. Immunol. 2020, 17, 50–63. [Google Scholar] [CrossRef] [Green Version]
- Lan, J.; Xiong, Y.Y.; Lin, Y.X.; Wang, B.C.; Gong, L.L.; Xu, H.S.; Guo, G.S. Helicobacter pylori infection generated gastric cancer through p53-Rb tumor-suppressor system mutation and telomerase reactivation. World J. Gastroenterol. 2003, 9, 54–58. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Author & Date | Sample Population | Sample Size (Exposure) | Variables Studied | Measure of Effect | Results |
|---|---|---|---|---|---|
| Chang et al., 2001 [42] | 136 GC, 13 controls | 126 exposed, 146 unexposed | Age, sex, marital status, occupation, education, salary, H. pylori serology | OR 1.82, (1.10–3.00) | + effect |
| Cho et al., 2010 [43] | 2819 GC, 564 controls | 2763 exposed, 618 unexposed | Age, sex, smoking, drinking, family history, living conditions, education, SES, family structure | OR 3.13, (2.46–3.97) | + effect |
| Eom et al., 2016 [44] | 846 GC, 846 controls | 1252 exposed, 440 unexposed | Age, sex, smoking, drinking, education, BMI, H. pylori infection, cagA | OR 1.43, (1.12–1.81) | + effect |
| Gong et al., 2013 [45] | 327 GC, 327 controls | 448 exposed, 169 unexposed | Age, sex, BMI, H. pylori serology, family history, smoking, drinking | OR 2.93, (1.88–4.59) | + effect |
| Gwack et al., 2006 [46] | 100 GC, 400 controls | 449 exposed, 51 unexposed | Age, sex, smoking, drinking, education, diagnosis, H. pylori serology, cagA and vacA | OR 0.96, (0.68–1.36) | N/A |
| Kim et al., 2012 [47] | 829 GC, 270 controls | 917 exposed, 180 unexposed | Age, sex, smoking, drinking, family history, BMI, H. pylori serology, eradication efforts, past diagnoses | GCC, OR 2.50, (1.78–3.52) GNCC, OR 2.10, (0.98–4.48) | + effect |
| Kim et al., 1997 [48] | 160 GC, 160 controls | 179 exposed, 141 unexposed | Age, sex, H. pylori serology | OR 1.39, (0.89–2.17) | + effect |
| Kim et al., 2000 [49] | 287 GC, 33 controls | 142 exposed, 178 unexposed | Age, sex, urbanity, SES, education, BMI, H. pylori serology, smoking, drinking | 43.9% GC group, 48.5% non-GC group | N/A |
| Kim et al., 2005 [50] | 295 GC, 295 controls | N/A | Age, sex, SES, education, BMI, H. pylori serology, smoking, drinking, family history | OR 1.68, (1.14–2.44) | + effect |
| Kim et al., 2018 [51] | 415 GC, 830 controls | 868 exposed, 353 unexposed | Age, sex, BMI, H. pylori serology, family history, smoking, drinking, exercise, education, marital status, occupation, salary, diagnoses | 92.1% GC 58.6% non-GC | + effect |
| Kim et al., 2015 [52] | 998 GC, 1288 controls | 1817 exposed, 469 unexposed | Age, sex, BMI, smoking, drinking, family history, H. pylori serology, diagnosis | M: 86.2% GC M: 77.1% non-GC W: 87.2% GC W: 71.1% non-GC | + effect |
| Woo et al., 2014 [53] | 334 GC, 334 controls | 457 exposed, 211 unexposed | Age, sex, BMI, H. pylori serology, family history, smoking, drinking, exercise, education, marital status, occupation | 84.4% GC 52.4% non-GC | + effect |
| Yang et al., 2017 [54] | 450 GC, 1050 controls | 1051 exposed, 449 unexposed | Age, ALDH2 genotype, drinking, smoking, education, salary, diet, H. pylori serology, family history | OR 7.07, (4.60–10.86) | + effect |
| Author & Date | Sample Population | Sample Size (Expose) | Variables Studied | Measure of Effect | Results |
|---|---|---|---|---|---|
| Fernandez de Larrea-Baz et al., 2017 [55] | 264 GC, 2071 controls | 2017 exposed, 259 unexposed | Age, sex, race, education, SES, BMI, smoking, family history, diagnosis | OR 1.31, (0.82–2.11) | + effect |
| Garcia-Gonzalez et al., 2015 [56] | 603 GC, 675 controls | 794 exposed, 484 unexposed | Age, sex, smoking, family history, cagA vagA | OR 1.39, (1.10–1.76) | + effect |
| Garcia-Gonzalez et al., 2007 [57] | 404 GC, 404 controls | 506 exposed, 302 unexposed | Age, sex, smoking, family history, cagA vagA | OR 1.46, (1.08–1.97) | + effect |
| Author and Publishing Year | ES (OR) | 95% CI | Weight | |
|---|---|---|---|---|
| South Korea (Asia) | ||||
| Chang, W.K. et al. (2001) | 1.60 | 1.01 | 2.53 | 4.65 |
| Cho, S.J. et al. (2010) | 3.13 | 2.46 | 3.97 | 4.67 |
| Eom, S.Y. et al. (2016) | 1.41 | 1.14 | 1.76 | 6.42 |
| Gong, E.J. et al. (2013) | 3.03 | 2.05 | 4.48 | 3.06 |
| Gwack, J.Y. et al. (2006) | 0.99 | 0.71 | 1.38 | 6.34 |
| Kim, J.Y. et al. (2012) | 2.50 | 1.78 | 3.52 | 4.21 |
| Kim, H.Y. et al. (1997) | 1.39 | 0.89 | 2.17 | 5.14 |
| Kim, Y.S. et al. (2000) | 0.91 | 0.46 | 1.83 | 4.95 |
| Kim, D.S. et al. (2005) | 1.68 | 1.14 | 2.44 | 5.10 |
| Kim, J.H. et al. (2018) | 1.57 | 1.31 | 1.89 | 6.48 |
| Kim, H.J. et al. (2015) | 0.57 | 0.50 | 0.66 | 6.91 |
| Lee, S.A. et al. (2003) | 1.18 | 0.77 | 1.80 | 5.66 |
| Woo, H.D. et al. (2014) | 1.61 | 1.26 | 2.07 | 6.09 |
| Yang, S. et al. (2017) | 1.48 | 1.25 | 1.75 | 6.60 |
| Sub-Total D + L Pooled ES | 1.56 | 1.19 | 1.94 | 76.29 |
| Spain (Western Europe) | ||||
| Fernandez de Larrea-Baz, N. et al. (2017) | 1.05 | 0.87 | 1.26 | 6.73 |
| Garcia-Gonzalez, M.A. et al. (2015) | 1.39 | 1.10 | 1.76 | 6.36 |
| Garcia-Gonzalez, M.A. et al. (2007) | 1.46 | 1.08 | 1.97 | 5.94 |
| Palli, D. et al. (2006) | 1.07 | 0.56 | 2.06 | 4.69 |
| Sub-Total D + L Pooled ES | 1.23 | 1.00 | 1.45 | 23.71 |
| Overall D + L Pooled ES | 1.47 | 1.19 | 1.76 | 100.00 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Holmes, L., Jr.; Rios, J.; Berice, B.; Benson, J.; Bafford, N.; Parson, K.; Halloran, D. Predictive Effect of Helicobacter pylori in Gastric Carcinoma Development: Systematic Review and Quantitative Evidence Synthesis. Medicines 2021, 8, 1. https://doi.org/10.3390/medicines8010001
Holmes L Jr., Rios J, Berice B, Benson J, Bafford N, Parson K, Halloran D. Predictive Effect of Helicobacter pylori in Gastric Carcinoma Development: Systematic Review and Quantitative Evidence Synthesis. Medicines. 2021; 8(1):1. https://doi.org/10.3390/medicines8010001
Chicago/Turabian StyleHolmes, Laurens, Jr., Jasmine Rios, Betyna Berice, Jacqueline Benson, Nastocia Bafford, Kadedrah Parson, and Daniel Halloran. 2021. "Predictive Effect of Helicobacter pylori in Gastric Carcinoma Development: Systematic Review and Quantitative Evidence Synthesis" Medicines 8, no. 1: 1. https://doi.org/10.3390/medicines8010001
APA StyleHolmes, L., Jr., Rios, J., Berice, B., Benson, J., Bafford, N., Parson, K., & Halloran, D. (2021). Predictive Effect of Helicobacter pylori in Gastric Carcinoma Development: Systematic Review and Quantitative Evidence Synthesis. Medicines, 8(1), 1. https://doi.org/10.3390/medicines8010001