
Sulforaphane (SFN): An Isothiocyanate in a Cancer Chemoprevention Paradigm

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
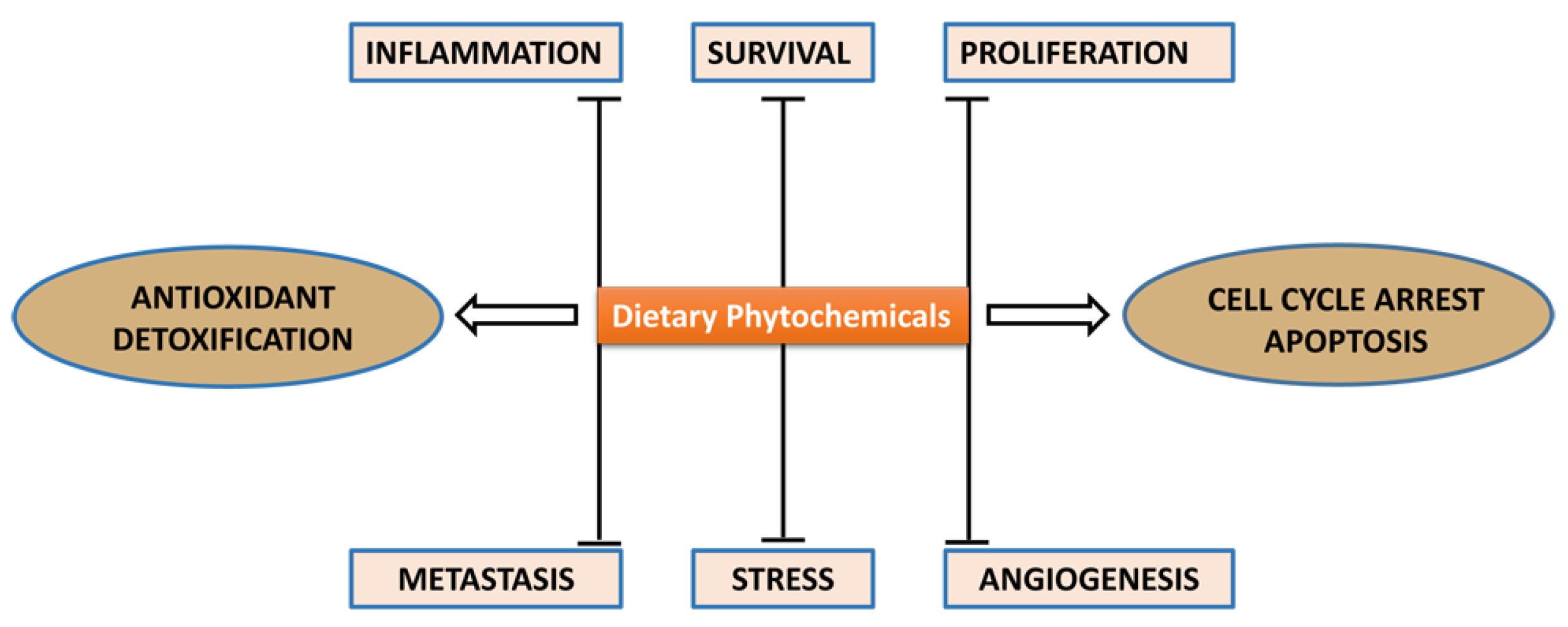
:1. Introduction


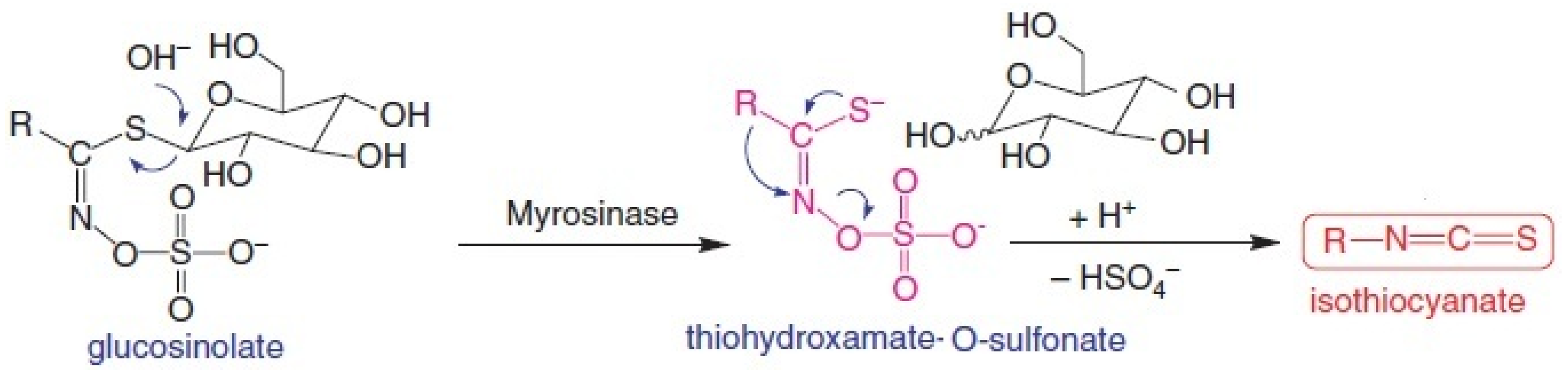
2. Anticancer Action Mechanisms of Sulforaphane
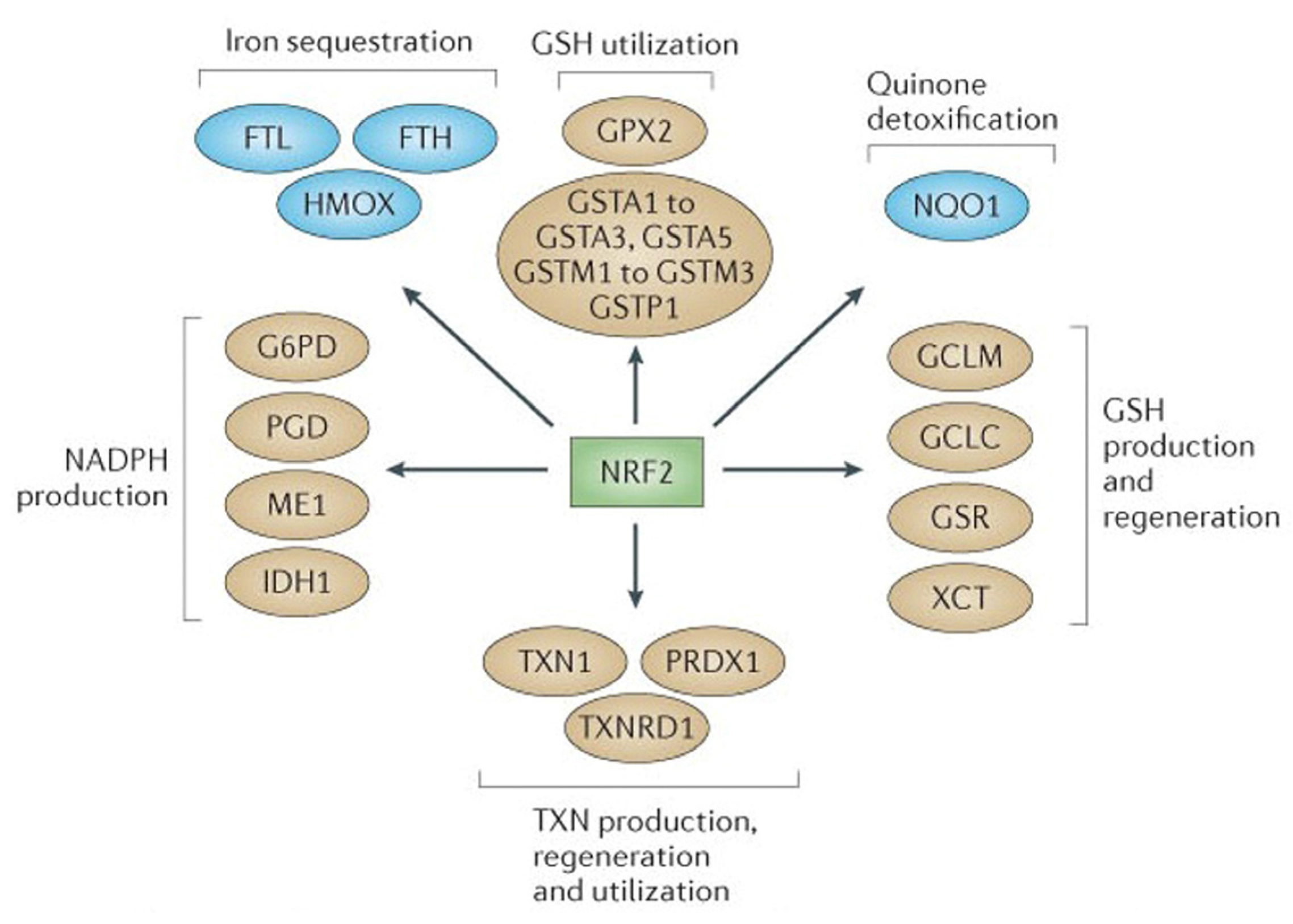
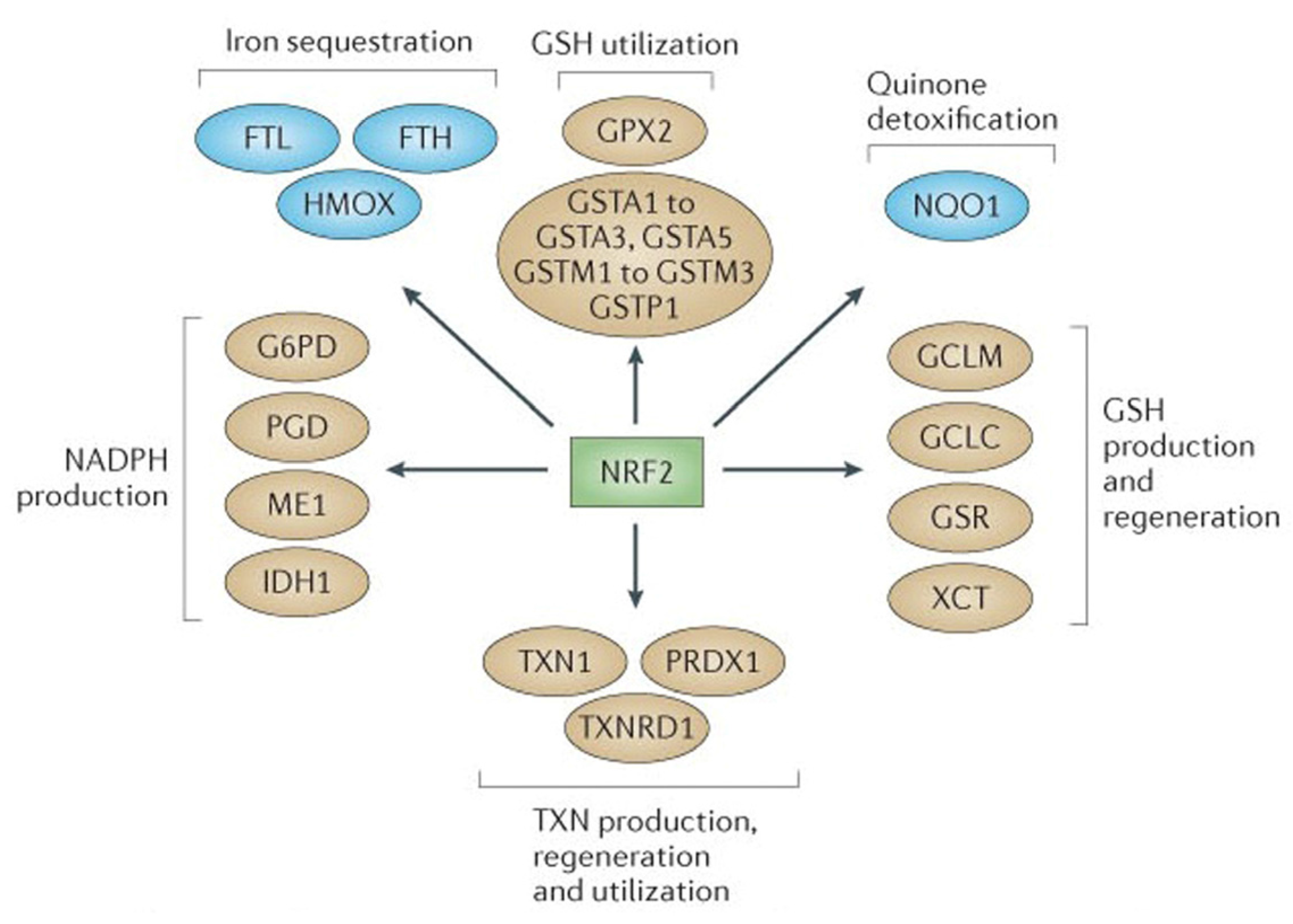
2.1. NRF2-Mediated Elevation of Antioxidant Defense by Sulforaphane Reduces the Incidence of ROS-Induced Genomic Insult
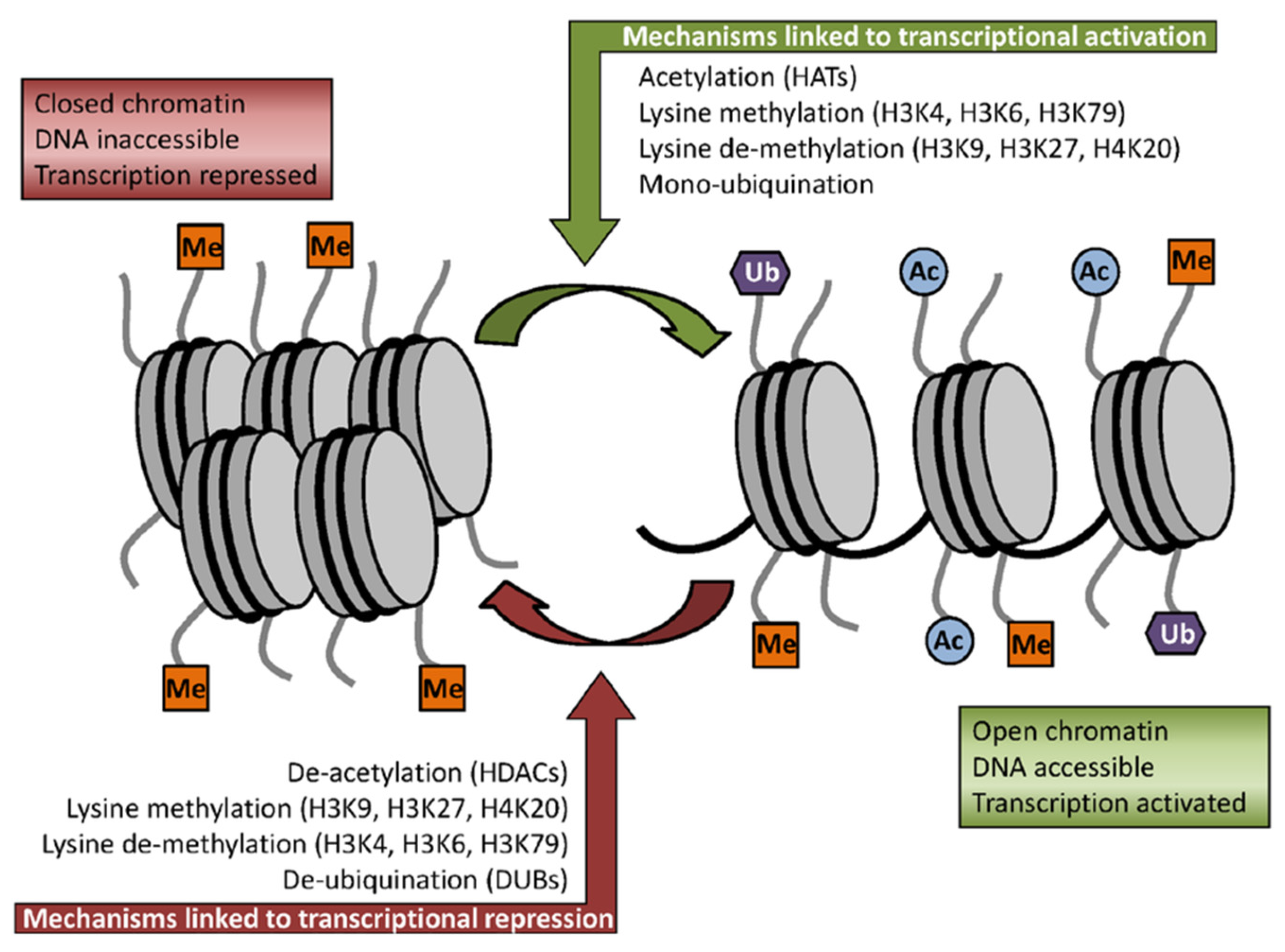
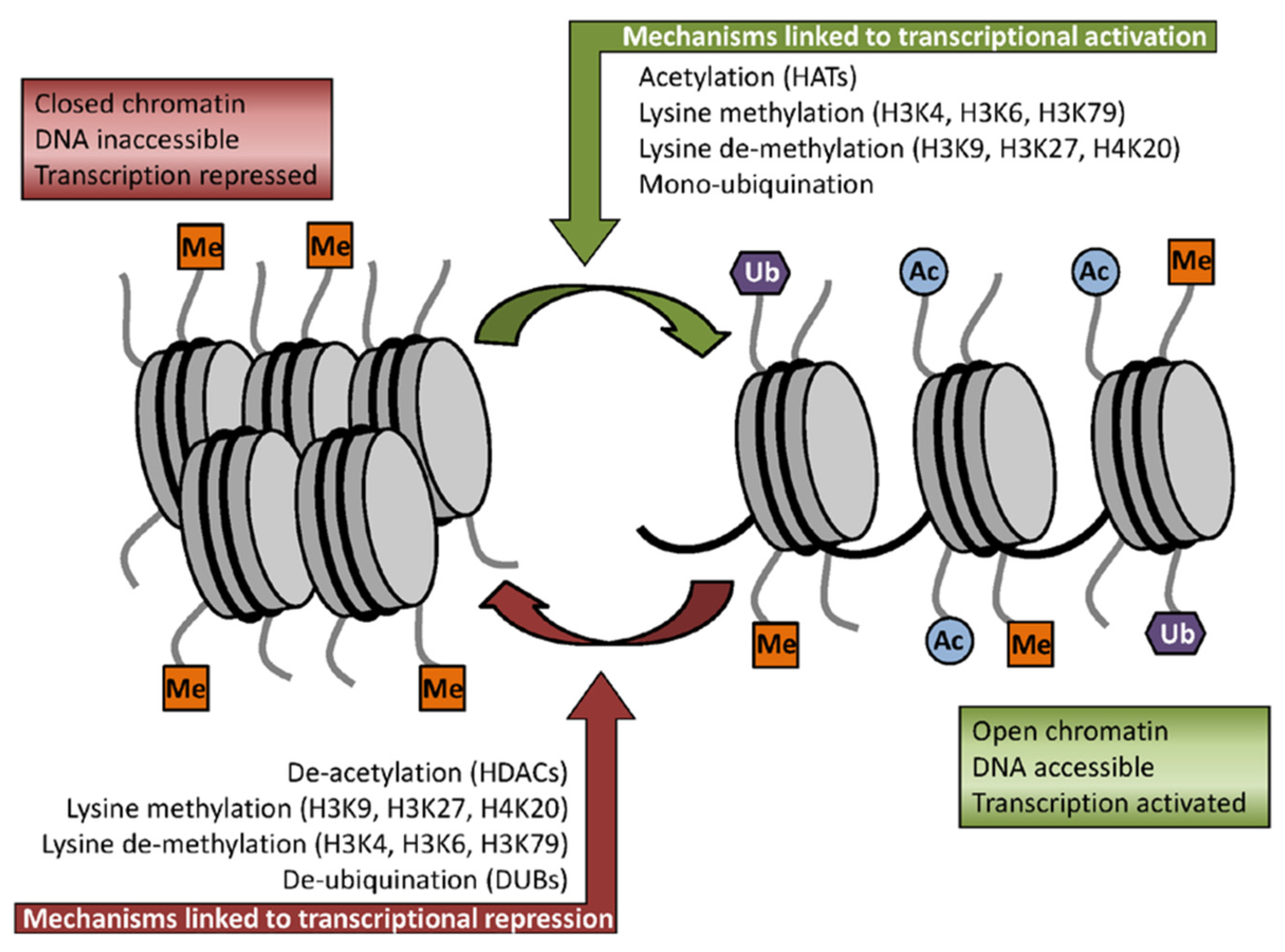
2.2. Sulforaphane as Inhibitor of HDACs Challenges the Pro-Oncogenic Epigenetic Pattern in Cancer Cells
2.3. Induction of Cell Cycle Arrest and Apoptosis in Cancer Cells by Sulforaphane

2.4. Sulforaphane Influences Transcriptional Factors and Cellular Signaling in Cancer Cells
3. Bioavailability and Safety Issues
4. Conclusions
Acknowledgments
Conflicts of Interest
References
- World Health Organization. Cancer Fact Sheet. February 2011. Available online: http://www.who.int/mediacentre/factsheets/fs297/en/index.html (accessed on 30 March 2012).
- Ashwell, M. Functional foods: A simple scheme for establishing the scientific basis for all claims. Public Health Nutr. 2001, 4, 859–862. [Google Scholar] [PubMed]
- Zohary, M. Plants of the Bible: A Complete Handbook to All the Plants with 200 Full-Color Plates Taken in the Natural Habitat; Cambridge University Press: Cambridge, UK, 1982. [Google Scholar]
- Ullah, M.F.; Bhat, S.H.; Hussain, E.; Abu-Duhier, F.; Hadi, S.M.; Sarkar, F.H.; Ahmad, A. Cancer chemopreventive pharmacology of phytochemicals derived from plants of dietary and non-dietary origin:implication for alternative and complementary approaches. Phytochem. Rev. 2014, 13, 811–833. [Google Scholar] [CrossRef]
- Block, G.; Patterson, B.; Subar, A. Fruit, vegetables, and cancer prevention: A review of the epidemiological evidence. Nutr. Cancer 1992, 18, 1–29. [Google Scholar] [CrossRef] [PubMed]
- Abuajah, C.I.; Ogbonna, A.C.; Osuji, C.M. Functional components and medicinal properties of food: A review. J. Food Sci. Technol. 2015, 52, 2522–2529. [Google Scholar] [CrossRef] [PubMed]
- Anand, P.; Kunnumakara, A.B.; Sundaram, C.; Harikumar, K.B.; Tharakan, S.T.; Lai, O.S.; Sung, B.; Aggarwal, B.B. Cancer is a Preventable Disease that Requires Major Lifestyle Changes. Pharm. Res. 2008, 25, 2097–2116. [Google Scholar] [CrossRef] [PubMed]
- Latte, K.P.; Appel, K.E.; Lampen, A. Health benefits and possible risks of broccoli—An overview. Food Chem. Toxicol. 2011, 49, 3287–3309. [Google Scholar] [CrossRef] [PubMed]
- Herr, I.; Buchler, M.W. Dietary constituents of broccoli and other cruciferous vegetables: Implications for prevention and therapy of cancer. Cancer Treat. Rev. 2010, 36, 377–383. [Google Scholar] [CrossRef] [PubMed]
- Moreno, D.A.; Carvajal, M.; López-Berenguer, C.; García-Viguera, C. Chemical and biological characterisation of nutraceutical compounds of broccoli. J. Pharm. Biomed. Anal. 2006, 41, 1508–1522. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.V.; Singh, K. Cancer chemoprevention with dietary isothiocyanates mature for clinical translational research. Carcinogenesis 2012, 33, 1833–1842. [Google Scholar] [CrossRef] [PubMed]
- Wattenberg, L.W. Inhibition of carcinogenic effects of polycyclic hydrocarbons by benzyl isothiocyanate and related compounds. J. Natl. Cancer Inst. 1977, 58, 395–398. [Google Scholar] [PubMed]
- Fahey, J.W.; Zalcmann, A.T.; Talalay, P. The chemical diversity and distribution of glucosinolates and isothiocyanates among plants. Phytochemistry 2001, 56, 5–51. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, T.A.; Fahey, J.W.; Wade, K.L.; Stephenson, K.K.; Talalay, P. Human metabolism and excretion of cancer chemoprotectiveglucosinolates and isothiocyanates of cruciferous vegetables. Cancer Epidemiol. Biomark. Prev. 1998, 7, 1091–1100. [Google Scholar]
- Zhang, Y.; Tang, L. Discovery and development of sulforaphane as a cancer chemopreventive phytochemical. Acta Pharmacol. Sin. 2007, 28, 1343–1354. [Google Scholar] [CrossRef] [PubMed]
- Myzak, M.C.; Karplus, P.A.; Chung, F.L.; Dashwood, R.H. A novel mechanism of chemoprotection by sulphoraphane:inhibition of histone deacetylase. Cancer Res. 2004, 64, 5767–5774. [Google Scholar] [CrossRef] [PubMed]
- Cornblatt, B.S.; Ye, L.; Dinkova-Kostova, A.T.; Erb, M.; Fahey, J.W.; Singh, N.K.; Chen, M.S.; Stierer, T.; Garrett-Mayer, E.; Argani, P.; et al. Preclinical and clinical evaluation of sulphoraphane for chemoprevention in the breast. Carcinogenesis 2007, 28, 1485–1490. [Google Scholar] [CrossRef] [PubMed]
- Myzak, M.C.; Tong, P.; Dashwood, W.M.; Dashwood, R.H.; Ho, E. Sulphoraphane retards the growth of human PC-3 xenografts and inhibits HDAC activity in human subjects. Exp. Biol. Med. 2007, 232, 227–234. [Google Scholar]
- Zeng, H.; Trujillo, O.N.; Moyer, M.P.; Botnen, J.H. Prolonged sulforaphane treatment activates survival signaling in nontumorigenic NCM460 colon cells but apoptotic signaling in tumorigenic HCT116 colon cells. Nutr. Cancer 2011, 63, 248–255. [Google Scholar] [CrossRef] [PubMed]
- Gorrini, C.; Harris, I.S.; Mak, T.W. Modulation of oxidative stress as an anticancer strategy. Nat. Rev. Drug Discov. 2013, 12, 931–947. [Google Scholar] [CrossRef] [PubMed]
- Janssen-Heininger, Y.M.; Mossman, B.T.; Heintz, N.H.; Forman, H.J.; Kalyanaraman, B.; Finkel, T.; Stamler, J.S.; Rhee, S.G.; van der Vliet, A. Redox-based regulation of signal transduction: Principles, pitfalls, and promises. Free Radic. Biol. Med. 2008, 45, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Sporn, M.B.; Liby, K.T. NRF2 and cancer: The good, the bad and the importance of context. Nat. Rev. Cancer 2012, 12, 564–571. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.S.; Surh, Y.J. Nrf2 as a novel molecular target for chemoprevention. Cancer Lett. 2005, 224, 171–184. [Google Scholar] [CrossRef] [PubMed]
- Taguchi, K.; Motohashi, H.; Yamamoto, M. Molecular mechanisms of the Keap1-Nrf2 pathway in stress response and cancer evolution. Genes Cells 2011, 16, 123–140. [Google Scholar] [CrossRef] [PubMed]
- Misiewicz, I.; Skupińska, K.; Kowalska, E.; Lubiński, J.; Kasprzycka-Guttman, T. Sulforaphane-mediated induction of a phase 2 detoxifying enzyme NAD(P)H:quinone reductase and apoptosis in human lymphoblastoid cells. Acta Biochim. Pol. 2004, 51, 711–721. [Google Scholar] [PubMed]
- Dinkova-Kostova, A.T.; Holtzclaw, W.D.; Cole, R.N.; Itoh, K.; Wakabayashi, N.; Katoh, Y.; Yamamoto, M.; Talalay, P. Direct evidence that sulfhydryl groups of Keap1 are the sensors regulating induction of phase 2 enzymes that protect against carcinogens and oxidants. Proc. Natl. Acad. Sci. USA 2002, 99, 11908–11913. [Google Scholar] [CrossRef] [PubMed]
- Hong, F.; Sekhar, K.R.; Freeman, M.L.; Liebler, D.C. Identification of sensor cysteines in human Keap1 modified by the cancer chemopreventive agent sulforaphane. Chem. Res. Toxicol. 2005, 18, 1917–1926. [Google Scholar] [CrossRef] [PubMed]
- Kwak, M.K.; Wakabayashi, N.; Kensler, T.W. Chemoprevention through the Keap1-Nrf2 signaling pathway by phase 2 enzyme inducers. Mutat. Res. 2004, 555, 133–148. [Google Scholar] [CrossRef] [PubMed]
- Thimmulappa, R.K.; Mai, K.H.; Srisuma, S.; Kensler, T.W.; Yamamoto, M.; Biswal, S. Identification of Nrf2-regulated genes induced by the chemopreventive agent sulforaphane by oligonucleotide microarray. Cancer Res. 2002, 62, 5196–5203. [Google Scholar] [PubMed]
- Lee, Y.J.; Lee, S.H. Sulforaphane Induces Antioxidative and Antiproliferative Responses by Generating Reactive Oxygen Species in Human Bronchial Epithelial BEAS-2B Cells. J. Korean Med. Sci. 2011, 26, 1474–1482. [Google Scholar] [CrossRef] [PubMed]
- Juge, N.; Mithen, R.F.; Traka, M. Molecular basis for chemoprevention by sulforaphane: A comprehensive review. Cell. Mol. Life Sci. 2007, 64, 1105–1127. [Google Scholar] [CrossRef] [PubMed]
- Manda, G.; Nechifor, M.T.; Neagu, T.M. Reactive oxygen species, cancer and anticancer therapies. Curr. Chem. Biol. 2009, 3, 342–366. [Google Scholar] [CrossRef]
- Dashwood, R.H.; Ho, E. Dietary histone deacetylase inhibitors: From cells to mice to man. Sem. Cancer Biol. 2007, 17, 363–369. [Google Scholar] [CrossRef] [PubMed]
- Struhl, K. Histone acetylation and transcriptional regulatory mechanisms. Genes Dev. 1998, 12, 599–606. [Google Scholar] [CrossRef] [PubMed]
- Basset, S.A.; Barnett, M.P.G. The Role of Dietary Histone Deacetylases (HDACs) Inhibitors in Health and Disease. Nutrients 2014, 6, 4273–4301. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.H.; Kim, M.; Kwon, H.J. Histone deacetylase in carcinogenesis and its inhibitors as anti-cancer agents. J. Biochem. Mol. Biol. 2003, 36, 110–119. [Google Scholar] [CrossRef] [PubMed]
- Marks, P.A.; Xu, W.S. Histone deacetylase inhibitors: Potential in cancer therapy. J. Cell. Biochem. 2009, 107, 600–608. [Google Scholar] [CrossRef] [PubMed]
- Clarkea, J.D.; Dashwood, R.H.; Emily, H. Multi-targeted prevention of cancer by sulforaphane. Cancer Lett. 2008, 269, 291–304. [Google Scholar] [CrossRef] [PubMed]
- Seligson, D.B.; Horvath, S.; Shi, T.; Yu, H.; Tze, S.; Grunstein, M.; Kurdistani, S.K. Global histone modification patterns predict risk of prostate cancer recurrence. Nature 2005, 435, 1262–1266. [Google Scholar] [CrossRef] [PubMed]
- Fronsdal, K.; Saatcioglu, F. Histone deacetylase inhibitors differentially mediate apoptosis in prostate cancer cells. Prostate 2005, 62, 299–306. [Google Scholar] [CrossRef] [PubMed]
- Butler, L.M.; Agus, D.B.; Scher, H.I.; Higgins, B.; Rose, A.; Cordon-Cardo, C.; Thaler, H.T.; Rifkind, R.A.; Marks, P.A.; Richon, V.M.; et al. Suberoylanilidehydroxamic acid, an inhibitor of histone deacetylase, suppresses the growth of prostate cancer cells in vitro and in vivo. Cancer Res. 2000, 60, 5165–5170. [Google Scholar] [PubMed]
- Clarke, J.D.; Hs, A.; Yu, Z.; Dashwood, R.H.; Ho, E. Differential effects of sulforaphane on histone deacetylases, cell cycle arrest and apoptosis in normal prostate cells versus hyperplastic and cancerous prostate cells. Mol. Nutr. Food Res. 2011, 55, 999–1009. [Google Scholar] [CrossRef] [PubMed]
- Rajendran, P.; Kidane, A.I.; Yu, T.W.; Dashwood, W.M.; Bisson, W.H.; Löhr, C.V.; Ho, E.; Williams, D.E.; Dashwood, R.H. HDAC turnover, CtIP acetylation and dysregulated DNA damage signaling in colon cancer cells treated with sulforaphane and related dietary Isothiocyanates. Epigenetics 2013, 8, 612–623. [Google Scholar] [CrossRef] [PubMed]
- Myzak, M.C.; Dashwood, W.M.; Orner, G.A.; Ho, E.; Dashwood, R.H. Sulforaphane inhibits histone deacetylase in vivo and suppresses tumorigenesis in apc-minus mice. FASEB J. 2006, 20, 506–508. [Google Scholar] [PubMed]
- Li, Y.; Yuan, Y.Y.; Meeran, S.M.; Tollefsbol, T.O. Synergistic epigenetic reactivation of estrogen receptor-alpha (ERalpha) by combined green tea polyphenol and histone deacetylase inhibitor in ERalpha-negative breast cancer cells. Mol. Cancer 2010, 9. [Google Scholar] [CrossRef] [PubMed]
- Meeran, S.M.; Patel, S.N.; Li, Y.; Shukla, S.; Tollefsbol, T.O. Bioactive Dietary Supplements Reactivate ER Expression in ER-Negative Breast Cancer Cells by Active Chromatin Modifications. PLoS ONE 2012, 7, e37748. [Google Scholar] [CrossRef] [PubMed]
- Gamet-Payrastre, L.; Li, P.; Lumeau, S.; Cassar, G.; Dupont, M.A.; Chevolleau, S.; Gasc, N.; Tulliez, J.; Tercé, F. Sulforaphane, a Naturally Occurring Isothiocyanate, Induces Cell Cycle Arrest and Apoptosis in HT29 Human Colon Cancer Cells. Cancer Res. 2000, 60, 1426–1433. [Google Scholar] [PubMed]
- Wang, M.; Chen, S.; Wang, S.; Sun, D.; Chen, J.; Li, Y.; Han, W.; Yang, X.; Gao, H.Q. Effects of Phytochemicals sulforaphane on Uridine Diphosphate-Glucuronosyltransferase Expression as well as Cell-Cycle Arrest and Apoptosis in Human Colon Cancer Caco-2 Cells. Chin. J. Physiol. 2012, 55, 134–144. [Google Scholar] [PubMed]
- Kanematsu, S.; Uehara, N.; Miki, H.; Yoshizawa, K.; Kawanaka, A.; Yuri, T.; Tsubura, A. Autophagy Inhibition Enhances Sulforaphane-induced Apoptosis in Human Breast Cancer Cells. Anticancer Res. 2010, 30, 3381–3390. [Google Scholar] [PubMed]
- Tseng, E.; Scott-Ramsay, E.A.; Morris, M.E. Dietary organic isothiocyanates are cytotoxic in human breast cancer MCF-7 and mammary epithelial MCF-12A cell lines. Exp. Biol. Med. 2004, 229, 835–842. [Google Scholar]
- Suppipat, K.; Park, C.S.; Shen, Y.; Zhu, X.; Lacorazza, H.D. Sulforaphane Induces Cell Cycle Arrest and Apoptosis in Acute Lymphoblastic Leukemia Cells. PLoS ONE 2012, 7, e51251. [Google Scholar] [CrossRef] [PubMed]
- Dang, M.Y.; Huang, G.; Chen, Y.R.; Dang, Z.F.; Chen, C.; Liu, F.L.; Guo, Y.F.; Xie, X.D. Sulforaphane Inhibits the Proliferation of the BIU87 Bladder Cancer Cell Line via IGFBP-3 Elevation. Asian Pac. J. Cancer Prev. 2014, 15, 1517–1520. [Google Scholar] [CrossRef] [PubMed]
- Abbaoui, B.; Riedl, K.M.; Ralston, R.A.; Thomas-Ahner, J.M.; Schwartz, S.J.; Clinton, S.K.; Mortazavi, A. Inhibition of Bladder Cancer by Broccoli IsothiocyanatesSulforaphane and Erucin: Characterization, Metabolism and Interconversion. Mol. Nutr. Food Res. 2012, 56, 1675–1687. [Google Scholar] [CrossRef] [PubMed]
- Chaudhuri, D.; Orsulic, S.; Ashok, B.T. Antiproliferative activity of sulforaphane in Akt-overexpressing ovarian cancer cells. Mol. Cancer Ther. 2007, 6, 334–345. [Google Scholar] [CrossRef] [PubMed]
- Bryant, C.S.; Kumar, S.; Chamala, S.; Shah, J.; Pal, J.; Haider, M.; Seward, S.; Qazi, A.M.; Morris, R.; Semaan, A.; et al. Sulforaphane induces cell cycle arrest by protecting RB-E2F-1 complex in epithelial ovarian cancer cells. Mol. Cancer 2010, 9. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Landen, C.N.; Li, Y.; Alvarez, R.D.; Trygve, O.; Tollefsbol, T.O. Epigallocatechin Gallate and Sulforaphane Combination Treatment Induce Apoptosis in Paclitaxel-Resistant Ovarian Cancer Cells through hTERT and Bcl-2 Down-regulation. Exp. Cell Res. 2013, 319, 697–706. [Google Scholar] [CrossRef] [PubMed]
- Chu, W.F.; Wu, D.M.; Liu, W.; Wu, L.J.; Li, D.Z.; Xu, D.Y.; Wang, X.F. Sulforaphane induces G2-M arrest and apoptosis in high metastasis cell line of salivary gland adenoid cystic carcinoma. Oral Oncol. 2009, 45, 998–1004. [Google Scholar] [CrossRef] [PubMed]
- Matsui, T.A.; Murata, H.; Sakabe, T.; Sowa, Y.; Horie, N.; Nakanishi, R.; Sakai, T.; Kubo, T. Sulforaphane induces cell cycle arrest and apoptosis in murine osteosarcoma cells in vitro and inhibits tumor growth in vivo. Oncol. Rep. 2007, 18, 1263–1268. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.-J.; Tang, W.-Y.; Hsu, C.-W.; Tsai, Y.-T.; Wu, J.-F.; Wu, C.-W.; Cheng, Y.-M.; Hsu, Y.-C. Apoptosis Induction in Primary Human Colorectal Cancer Cell Lines and Retarded Tumor Growth in SCID Mice by Sulforaphane. Evid.-Based Complement. Altern. Med. 2012. [Google Scholar] [CrossRef] [PubMed]
- Huang, T.Y.; Chang, W.C.; Wang, M.Y.; Yang, Y.R.; Hsu, Y.C. Effect of sulforaphane on growth inhibition in human brain malignant glioma GBM 8401 cells by means of mitochondrial- and MEK/ERK-mediated apoptosis pathway. Cell Biochem. Biophys. 2012, 63, 247–259. [Google Scholar] [CrossRef] [PubMed]
- Liang, H.; Lai, B.; Yuan, Q. Sulforaphane induces cell-cycle arrest and apoptosis in cultured human lung adenocarcinoma LTEP-A2 cells and retards growth of LTEP-A2 xenografts in vivo. J. Nat. Prod. 2008, 71, 1911–1914. [Google Scholar] [CrossRef] [PubMed]
- Bhamre, S.; Sahoo, D.; Tibshirani, R.; Dill, D.L.; Brooks, J.D. Temporal changes in gene expression induced by sulforaphane in human prostate cancer cells. Prostate 2009, 69, 181–190. [Google Scholar] [CrossRef] [PubMed]
- Hahm, E.R.; Chandra-Kuntal, K.; Desai, D.; Amin, S.; Singh, S.V. Notch Activation Is Dispensable for D, L-Sulforaphane-Mediated Inhibition of Human Prostate Cancer Cell Migration. PLoS ONE 2012, 7, e44957. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, C.; Shen, G.; Chen, C.; Gélinas, C.; Kong, A.T. Suppression of NF-kB and NF-kB-regulated gene expression by sulforaphane and PEITC through IjBa, IKK pathway in human prostate cancer PC-3 cells. Oncogene 2005, 24, 4486–4495. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.K.; Kim, S.J. Sulforaphane Regulates Differentiation via PI-3K/AKT Pathway in Human Chondrosarcoma Cell Line, HTB-94 Cells. Cancer Prev. Res. 2013, 18, 26–32. [Google Scholar]
- Jakubikova, J.; Cervi, D.; Ooi, M.; Kim, K.; Nahar, S.; Klippel, S.; Cholujova, D.; Leiba, M.; Daley, J.F.; Delmore, J.; et al. Anti-tumor activity and signaling events triggered by the isothiocyanates, sulforaphane and phenethylisothiocyanate, in multiple myeloma. Haematologica 2011, 96, 1170–1179. [Google Scholar] [CrossRef] [PubMed]
- Tarapore, R.S.; Siddiqui, I.A.; Mukhtar, H. Modulation of Wnt/b-catenin signaling pathway by bioactive food components. Carcinogenesis 2012, 33, 483–491. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhang, T.; Korkaya, H.; Liu, S.; Lee, H.F.; Newman, B.; Yu, Y.; Clouthier, S.G.; Schwartz, S.J.; Wicha, M.S.; et al. Sulforaphane, a Dietary Component of Broccoli/Broccoli Sprouts, Inhibits Breast Cancer Stem Cells. Clin. Cancer Res. 2010, 16, 2580–2590. [Google Scholar] [CrossRef] [PubMed]
- Buttner, R.; Mora, L.B.; Jove, R. Activated STAT signaling in human tumors provides novel molecular targets for therapeutic intervention. Clin. Cancer Res. 2002, 8, 945–954. [Google Scholar]
- Bowman, T.; Garcia, R.; Turkson, J.; Jove, R. STATs in oncogenesis. Oncogene 2000, 19, 2474–2488. [Google Scholar] [CrossRef] [PubMed]
- Pinz, S.; Unser, S.; Rascle, A. Natural chemopreventive agent sulforaphane inhibits STAT 5 activity. PLoS ONE 2014, 9, e99391. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, T.A.; Fahey, J.W.; Dinkova-Kostova, A.T.; Holtzclaw, W.D.; Stephenson, K.K.; Wade, K.L.; Ye, L.; Talalay, P. Safety, tolerance, and metabolism of broccoli sprout glucosinolates and isothiocyanates: A clinical phase I study. Nutr. Cancer 2006, 55, 53–62. [Google Scholar] [CrossRef] [PubMed]
- Ye, L.; Dinkova-Kostova, A.T.; Wade, K.L.; Zhang, Y.; Shapiro, T.A.; Talalay, P. Quantitative determination of dithiocarbamates in human plasma, serum, erythrocytes and urine: Pharmacokinetics of broccoli sprout isothiocyanates in humans. Clin. Chim. Acta 2002, 316, 43–53. [Google Scholar] [CrossRef]
- Alumkal, J.J.; Slottke, R.; Schwartzman, J.; Cherala, G.; Munar, M.; Graff, J.N.; Beer, T.M.; Ryan, C.W.; Koop, D.R.; Gibbs, A.; et al. A phase II study of sulforaphane-rich broccoli sprout extracts in men with recurrent prostate cancer. Investig. New Drugs 2015, 33, 480–489. [Google Scholar] [CrossRef] [PubMed]
- Lozanovski, V.J.; Houben, P.; Hinz, U.; Hackert, T.; Herr, I.; Schemmer, P. Pilot study evaluating broccoli sprouts in advanced pancreatic cancer (POUDER trial)—Study protocol for a randomized controlled trial. Trials 2014, 15. [Google Scholar] [CrossRef] [PubMed]
- WHO (Ed.) WHO Traditional Medicine Strategy, 2014–2023; WHO Press: Geneva, Switzerland, 2013.
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ullah, M.F. Sulforaphane (SFN): An Isothiocyanate in a Cancer Chemoprevention Paradigm. Medicines 2015, 2, 141-156. https://doi.org/10.3390/medicines2030141
Ullah MF. Sulforaphane (SFN): An Isothiocyanate in a Cancer Chemoprevention Paradigm. Medicines. 2015; 2(3):141-156. https://doi.org/10.3390/medicines2030141
Chicago/Turabian StyleUllah, Mohammad Fahad. 2015. "Sulforaphane (SFN): An Isothiocyanate in a Cancer Chemoprevention Paradigm" Medicines 2, no. 3: 141-156. https://doi.org/10.3390/medicines2030141