Abstract
The salivary gland (SG) microvasculature constitutes a dynamic cellular organization instrumental to preserving tissue stability and homeostasis. The interplay between pericytes (PCs) and endothelial cells (ECs) culminates as a key ingredient that coordinates the development, maturation, and integrity of vessel building blocks. PCs, as a variety of mesenchymal stem cells, enthrall in the field of regenerative medicine, supporting the notion of regeneration and repair. PC-EC interconnections are pivotal in the kinetic and intricate process of angiogenesis during both embryological and post-natal development. The disruption of this complex interlinkage corresponds to SG pathogenesis, including inflammation, autoimmune disorders (Sjögren’s syndrome), and tumorigenesis. Here, we provided a global portrayal of major signaling pathways between PCs and ECs that cooperate to enhance vascular steadiness through the synergistic interchange. Additionally, we delineated how the crosstalk among molecular networks affiliate to contribute to a malignant context. Additionally, within SG microarchitecture, telocytes and myoepithelial cells assemble a labyrinthine companionship, which together with PCs appear to synchronize the regenerative potential of parenchymal constituents. By underscoring the intricacy of signaling cascades within cellular latticework, this review sketched a perceptive basis for target-selective drugs to safeguard SG function.
1. Introduction
The salivary gland (SG) microvasculature represents a kinetic cellular system, crucial for maintaining tissue vitality and homeostasis. One of the key components of this complex are pericytes (PCs), mural cells that ensheath the abluminal interface of the endothelial lining, while sharing the same basement membrane (BM) [1,2,3]. The mutual interplay between PCs and endothelial cells (ECs) has come into view as central teamwork which governs the blood vessels maturation, remodeling, development, generation, and stabilization of new vessel-building blocks [2,4,5,6]. As a variety of mesenchymal stem cells (MSCs), PCs attract attention in the field of regenerative medicine by their pivotal commission in the matter of tissue regeneration and repair [7,8]. Correspondingly, PCs play a well-documented role in the dynamic and intricate process of angiogenesis, defined as the proliferation of ECs, sprouting, constitution, and branching of new vessels from pre-existing ones in order to establish interlinking capillary networks by mechanical support and paracrine factors [3,6,9,10,11,12,13,14]. Dysfunction of this comprehensive interconnection parallels the pathogenesis of SG disorders [15,16]. The absence of PCs is a hallmark of the disruption of vascular integrity [17]. In accordance, several diseases and stress conditions, such as inflammation and ischemia, autoimmune background (Sjögren’s syndrome), and post-radiotherapy in neoplastic contexture result in the disorganizing of SG microarchitecture [3,18,19]. These phenomena are ascribed to acinar cells atrophy, apoptosis, and uncontrolled progression of fibrosis and lead to SG dysfunction (hyposalivation, xerostomia), thereby exacerbating some of the pathological processes [20,21]. Of note, these disorders all correspond to inflammatory status emerging from microvascular dysfunction [18,22]. Restoring and safeguarding vascular integrity as the primary target might represent a gear in the “master plan” for the treatment of the above-mentioned pathologies [15,22]. Moreover, recent studies revealed the embryological origin of PCs (neuroectoderm-derived neural crest cells) in the head and neck regions, as demonstrated in chick-quail chimeras [16,23].
With the intention of discerning the traditional immunophenotype (CD146+/endoglin+/Platelet-derived growth factor receptor beta (PDGFRB+)) of PCs from other mesenchymal cells, a myriad of biomarkers have been identified [3,24]. It is paramount to emphasize that PCs constitute a heterogenous population with no specific marker, so their identification is contingent on a coalescence of multiple markers. One of the most used membrane-bound markers is NG2 (neural/glial antigen 2) [25], together with alanine aminopeptidase (CD13) and CD90 [2,25]. Another cytosolic marker useful in PCs identification is alpha-smooth muscle actin (alpha SMA) [26], related to modulating the blood flow [1,3]. In fact, these markers vary between organs and typify in distinct stages of development and in their pathophysiological circumstances. Communication between PCs and ECs is indispensable for the balance, homeostasis, and formation of the SG vasculature, which is synchronized with an array of signaling molecules that acts in a coordinated manner to manage several biological processes. This review highlighted a global picture of a complex interaction network of crosstalk among signaling pathways between PCs and ECs, expressing synergistic reciprocity that delineates a rational basis for different pathogenetic elements as therapeutic targets in SG diseases.
2. Pericytes and Their Relationship with Endothelial Cells
The SG vasculature is an indispensable complex of an accurately organized hierarchical tree of diversified cellularity, decisive in establishing and preserving tissue health by virtue of blood perfusion and through the potent reciprocity of the cells and the extracellular matrix (ECM). The build-up added to regeneration and/or repair of effective blood vessels turn out by intussusceptive angiogenesis, the chief event involving proliferation and migration of ECs and equilibrium of the microvascular lattice via coverage with PCs, archetypal multipotent stem cells (SCs), as a stringent fundamental ingredient [25]. The processes entailing ECs differentiation and organization into distinct miniatures are organotypic and tissue-specific and attach a note of intricacy to vascular development, balanced by shear stress and gene profile [27,28].
Within the framework of this complex organization, PCs are closely associated with endothelial coating, act as co-regulators of ECs, and are phenotypically divergent, being contingent upon the anatomical region [29]. On account of their perivascular location, Rouget cells were coined as PCs by Zimmermann, as reported by Brown et al. [30]. Transmission electron microscopy (TEM) upholds the identification of PCs features: they generally possess a large, spherical nucleus with profuse heterochromatin, a small amount of cytoplasm, incorporating collagenous and noncollagenous protein-synthesizing organelles with plenty of rough endoplasmic reticulum, in addition to a small proportion of glycogen particles, liposomes, and Golgi apparatus [12,31]. Moreover, PCs exhibit primary cytoplasmic finger-like projections disposed along the long axis of the vessel that give rise to orthometric secondary projections, linking to ECs [2]. Furthermore, microtubules take up the main part of all cytoplasmic processes, while intermediate filaments, including vimentin and desmin, are accumulated mainly within the primary ones [2]. The cytomorphological characteristics of PCs depend upon their differentiation circumstances, and also upon the anatomical site: they range from stereotypical flat and elongated to stellate shaped and are distinct in different zones of microcirculation [6,12].
According to our understanding, PCs exhibit two types of physical contacts with ECs: “peg-and-socket” and adhesion plaques [23]. Within the spots lacking a BM, cytoplasmic projections of PCs (the presumed “pegs”) liaise with ECs ‘ infoldings (the sockets), comprising N-cadherin-adherens junctions and gap junctions [2,8]. Upon some contact points, the dot-like adhesion plaques include principally fibronectin, connecting the plasma membrane with the BM through the intermediacy of the integrins [24]. A plethora of studies have suggested that the bidirectional signals between ECs and PCs point out the finale of vessel pliability and mark the quiescent level of vascular networks [32]. We illustrated briefly the typical pericyte-endothelial interconnections in Figure 1 as a starting point for the rest of our manuscript. Undoubtedly, proper communications are required for the maturation and stabilization of the SG vasculatures both in embryogenesis and adulthood, coordinated by a multitude of candidates and signaling molecules, such as Vascular endothelial growth factor (VEGF), WNT, NOTCH, HEDGEHOG (HH), Platelet-derived growth factor (PDGF), Transforming growth factor beta (TGFB), etc. [33,34]. Besides these biological mediators, extracellular vesicles (EVs), namely exosomes (EXOs), derived not only from PCs but also from ECs, which operate as information emissaries and stick out as potential biomarkers with promising therapeutic effects [35,36].
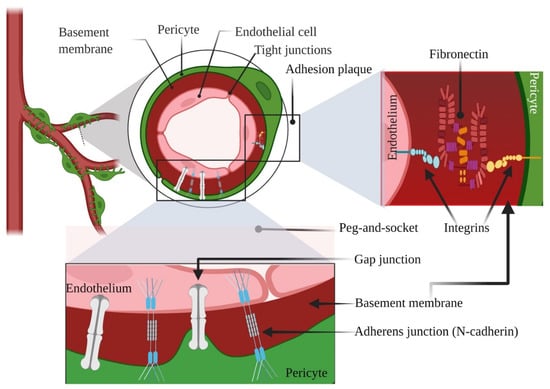
Figure 1.
Illustrative representation showing typical pericyte-endothelial interconnections. Created with BioRender.com, Agreement number YY238VSLPL. Retrieved 26 November 2021.
3. Changes in Salivary Glands Cell Repertoire
Although several pathological conditions including autoimmune diseases (Sjögren’s syndrome), irradiation, malignant tumors, and acute or chronic inflammation bring about disorders of the SGs, owing to the acinar cells hypofunction, and even apoptosis, all these pathologies exhibit corresponding microvascular dysfunction [37,38,39]. SG function is radically disrupted by radiotherapy (RT) as part of the multimodal treatment of head and neck cancer (HNC) [20]. Notwithstanding that SGs display slow-going proliferating tissue, the salivary acinar epithelial cells are highly sensitive to RT [40]. The emerging condition that evolves into hyposalivation, xerostomia/dry mouth, is accredited to monumental acinar cell irreversible loss, as well as to a lower likelihood of salivary gland progenitor cells (SGPCs) to differentiate into acinar cells [34,41,42].In an effort to explain the modus operandi of the radiation process with respect to puzzling, distinct radiosensitivity of salivary cells, Radfar et al. [21] noted that profiles of the irradiated parotid and submandibular gland were set apart by acinar atrophy, deprivation of translucent secretory granules, interstitial fibrosis, parenchymal loss, and duct proliferation. On the other hand, myoepithelial cells (MECs) engage in underpinning the morphogenesis and polarization of salivary acini [43]. Hakim et al. [44] found out that a significant post-irradiation (post-IR) loss of both alpha SMA and vimentin-positive MECs is coupled with a decrease of the proliferating rhythm in the acinar cells. Recently, the expression of CD44 was also reported as a marker of acinar cells in a MSCs population from the human parotid gland, along with PDGFRB and NG2, suggesting that these progenitor cell types could be PCs involved in the rescue of SG injury post-IR [3]. Along the same lines, the circulatory system is a requisite for conserving the viability and proper function of each adult organ. In this multiplex substructure, ECs are promptly subjected to death post-IR, and, therefore, targeting the disabled SG vasculature might be a successful appeal to prevent irreversible glandular damage [22,34]. Conditional upon slow turnover rate, within the vasculogenic zone, tissue-resident stromal cells, particularly PCs, hold up the matter of regeneration/repair [38]. There is a wealth of evidence illustrating that CD34-positive, CD31-negative adventitial SCs possess the capability to differentiate post-IR into PCs and encourage the new vessel generation by expressing WNT and PDGF genes, thereby supporting the idea that both blood vessel-framing ECs (CD34+CD31+ cells) and PCs may have a common mesenchymal precursor [45,46,47]. Interestingly, another recent study delineated that CD34-positive MSCs from the labial glands were virtually absent in patients suffering from Sjögren’s syndrome (SS) compared with healthy individuals [48].
Salivary gland tumors (SGTs) represent an assemble of histologically miscellaneous neoplasms that possess low prevalence among HNCs [49]. In the Hiroshima Tumor Tissue Registry, pleomorphic adenoma (PA) was the most frequent histotype and, subsequently, Warthin tumor and basal cell adenoma (BCA), whilst in the category of SG carcinomas are included adenoid cystic (ACC) and mucoepidermoid carcinoma [50]. Some SG carcinomas arise from dedifferentiation or else from benign tumors that develop into malignant ones [51]. Into the bargain, solitary fibrous tumor/hemangiopericytoma (SFT/HPC) constitutes a rare mesenchymal neoplasm; even so, in the WHO classification, the notion of HPC as a PC-derived tumor was cast aside on the side of fibroblast origin [52]. Even though the majority of SFTs follow a benign itinerary, a small percentage become malignant, or they can present zones of dedifferentiation of epithelial neoplasm mimicry [53]. De novo formation of SGTs, in addition to malignancy, is appointed to a plethora of signaling elements, the foremost hallmark being angiogenesis, evidenced through factors like VEGF and CD105 (endoglin) [54].
Crosstalks between PCs and ECs are acknowledged as culminate interlinkages in the labyrinthine marvelous process of angiogenesis. Straightforwardly, tumors surpass 2–3 mm3 and metastasize in the company of new vasculature that develops from five steps: (1) the increase of hypoxia-inducible factor 1-alpha HIF1Awhich attaches to hypoxia-response elements in the VEGF promoter that induces EC proliferation, followed by (2) the discharge of matrix metalloproteinases (MMPs) by both PCs and ECs, and degradation of the ECM/BM, (3) activation and migration of ECs, (4) formation of the capillary lumen, and ultimately (5) steadiness of tumor neovessels [6,55,56,57]. The stereotypical mechanism to assess the progression of angiogenesis is the analysis of microvessel density (MVD) within terms of units of vessels per high power fields (HPFs) employing immunohistochemical (IHC) methods to reveal certain EC-markers, aside from the well-known CD31 and CD34 and even greater CD105, the last one being substantiated as a fundamental co-receptor for the TGFB family, illustrating a pivotal role in angiogenesis [58,59].
4. Angiogenetic Behavior as a Consequence of PC/EC Crosstalk
Here, to make the landscape more complex, we reviewed the framework of elaborate protein-protein interrelatedness among decisive signaling pathways in a paracrine or autocrine manner. We underscored how the crosstalk intermingles to promote vascular development and the misregulations that can balance the homeostasis state into a tumorigenic program, as well as how the signaling components interface with each other to exert aversion to pharmacological approaches.
4.1. VEGF Signaling Pathway
VEGF is an assertive signaling factor that belongs to the PDGF supergene family and governs the angiogenic events during embryogenesis and adulthood in both pathological and physiological conditions [56,60]. The VEGF family is composed of five members: VEGF A-D and placenta growth factor (PGF) that interact with allied tyrosine kinases receptors (TKRs) VEGFR1-R3 [60,61,62]. VEGFA is the quintessential member of the VEGF family (referred to as VEGF henceforth), secreted by ECs in an autocrine loop and also produced by PCs as regards paracrine stimulation and binds to VEGFR1 and -2, expressed on the surface of ECs [10,60]. PCs also possess VEGFR1 on their surface, designated as a decoy receptor, and bind to VEGF, seizing it from ECs and deflecting the inauguration of angiogenesis, consequently promoting stabilization and quiescence in mature vessels [63]. In contrast, VEGFR1 knockout leads to ECs hyperplasia and ectopic vascular morphogenesis [64]. The signal transduction through VEGFR2 is propagated intracellularly by plenty of downstream signaling pathways networks. Studies show that, dissimilar to most of TKRs which operate on RAS-RAF-mitogen-activated protein kinase (MEK), extracellular signal-regulated kinase (ERK) pathway, or Phosphatidylinositol 3-kinase (PI3K)/AKT/mammalian target of rapamycin (mTOR) pathway, VEGF/VEGFR2 highly activates the PLC-gamma-PKC-MAPK pathway, employed as the key indicator for EC proliferation [65,66,67,68]. Essentially, Growth factor receptor-bound protein 2 (GRB2) binds to the VEGF/VEGFR2 complex and associates with the Son of sevenless (SOS) to turn on RAS, which thereafter activates RAF that is competent to phosphorylate MEK and the last one further phosphorylates ERK1/2 [69,70,71]. Targeted deletion of RAF, MEK1, and RAS-GAP causes defective angiogenesis during embryogenesis [72,73,74]. Numerous studies suggest that RAS/RAF and after all ERK1/2 are fundamental for proper angiogenesis, ECs proliferation, survival, and motility [68,70,75,76]. Instead, compared with normal SG parenchyma, phosphorylated ERK1/2 immunoreactivity was increased in mucoepidermoid carcinoma samples by IHC; so, possibly, it can represent a therapeutic target for novel antitumor drugs [77]. Meanwhile, when HIF1A is upregulated and VEGF binds to VEGFR2 on normal ECs, and the RAS and PI3K pathways are set in motion [67]. PI3K is a dominant downstream effector pathway of RAS, which is instrumental in the formation of the normal blood vessel and ECs migration during angiogenesis [78,79,80]. When activated, PI3K converts phosphatidylinositol (4,5)-bisphosphate (PIP2) into phosphatidylinositol (3,4,5)-trisphosphate (PIP3), which sequentially binds to AKT/PKB, is expressed on ECs, and regulates a vast range of cellular responses [78,79]. AKT manages cell growth by entailing the phosphorylation of mTOR [67]. Markedly, The PI3K/AKT signaling pathway is upregulated in SGTs and hyperactivates, in part, mTOR, as a chief regulator of manifold cellular events such as cancer cell survival and metastasis [81,82,83]. PI3K signaling is usually intensified by the loss of function of the negative regulation of PTEN, whose main substrate is PIP3, thus enhancing the activation of AKT [84,85]. Correspondingly, several studies denote VEGF in respect to the tumor microenvironment (TME) as a prognostic factor commensurate with tumor size, aggressive behavior and metastasis, and cell growth [85,86,87]. Basically, TME is characterized by heterogeneous, aberrant vasculature derived from an imbalance among pro- and non-angiogenic factors [88,89]. Intriguingly, mTOR composes networks of crosstalk with the signaling pathways within the PI3K/AKT pathway [90] and the inhibition of mTOR determines the minimization of MVD and suppresses the tumor growth [82]. In addition to the chemotherapeutic drugs, such as bevacizumab and temozolomide, which inhibit VEGF in a paracrine loop and [10,91,92,93] provide the rationale to inhibit tumor progression, sorafenib, a multi-Tyrosine Kinase Inhibitor (mTKI), has been shown to restrict the action of VEGFR2, RAS kinase, and PDGFR, [94] targeting twofold PCs and ECs through the hindrance of the autocrine VEGF signaling loop [10].
We brought together the interlinking latticework among VEGF, NOTCH, PDGF, TGFB, and downstream signaling pathways in a diagram (Figure 2). Briefly, VEGF is secreted by both PC and EC and binds to VEGFR1, expressed by the two cell types, and VEGFR2, expressed by EC. VEGFR1 can enter a competition with VEGFR2 to seize VEGF from VEGFR2. Following the attaching of VEGF to VEGFR2, RAS-RAF-MEK-ERK and/or PI3K/AKT/mTOR and/or PLC-gamma-PKC pathways are set in motion. When actuated, PI3K adjusts PIP2 to PIP3, which further activates AKT and mTOR. In contrast, PIP3 is inhibited by PTEN. RAPA, another negative regulator, inhibits AKT and mTOR. NOTCH also plays a pivotal role in the regulation of angiogenesis. Both PC and EC display NOTCH 1–3 receptors, but NOTCH4 is more restricted to EC. Jagged 1/Delta-like 1 (JAG1/DLL1) are mainly induced by PC, while DLL4/JAG1/2 are expressed by EC. After the generation of ligand-receptor complex, NOTCH receptor is susceptible to double cleavage by A-disintegrin and metalloprotease (ADAM) in the ECM and gamma-secretase within the cell, initiating the release of Notch intracellular domain (NICD), which translocates to the nucleus to associate with the CBF1/suppressor of hairless/LAG1 (CSL) and to switch on the transcriptional co-activator Mastermind-like (MAML), and further to activate the target genes. Instrumental for vascular homeostasis is PDGFB, chiefly secreted by EC to act on PDGFRB, expressed by PC. Sorafenib, an mTKI, inhibits VEGFR2, RAS, and PDGFRB. TGFB receptors, together with the co-receptor CD105 are evinced by the two cells, activin-like kinase 1 (ALK1) being more confined to EC. When mobilized by a ligand, type I receptors activate receptor-regulated Small Mothers Against Decapentaplegic proteins, SMADs (R-SMADs (SMAD-1, -5, and -8 for the BMP family and SMAD2 and -3 for the TGFB family)). R-SMADs connect with Co-SMAD (SMAD4) and advance to the nucleus to trigger the transcription of the genes. I-SMADs (SMAD6 for the BMP family and SMAD7 for the TGFB family) obstruct the interaction of R-SMADs with type I receptors. In ECs, ALK5 is inhibited by ALK1, so angiogenesis is promoted. The propelling of ALK5 in PC leads to the discharge of matrix metalloproteinases (MMPs) and ECM proteins like fibronectin and collagen.
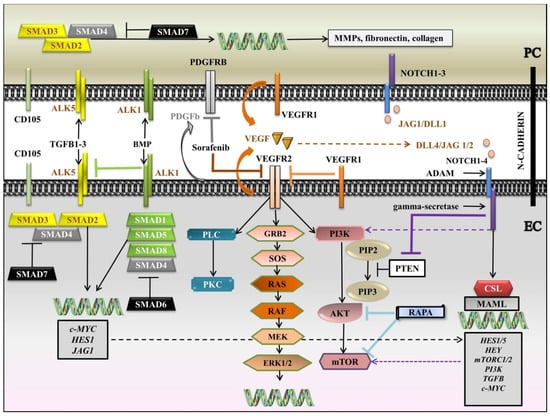
Figure 2.
Diagram of the interlinking latticework among VEGF, NOTCH, PDGF, TGFB, and downstream signaling pathways. ADAM: Adisintegrin and metalloprotease; ALK: Activin-like kinase; BMP: Bone morphogenetic protein; CSL: CBF1/suppressor of hairless/LAG1; DLL: Delta-like; EC: endothelial cell; ERK1/2: Extracellular signal-regulated kinases 1/2; GRB2: Growth factor receptor-binding protein 2; JAG: Jagged; MAML: Mastermind-like; MEK: Mitogen-activated protein kinase kinase; MMPs: matrix metalloproteinases; mTKI: multi-Tyrosine Kinase Inhibitor; mTOR: mammalian target of rapamycin; NICD: Notch intracellular domain; PC: pericyte; PDGFB: Platelet-Derived Growth Factor b; PDGFRB: Platelet-Derived Growth Factor Receptor B; PI3K: Phosphatidylinositol 3-kinase; PIP2: phosphatidylinositol (4,5)-bisphosphate; PIP3: phosphatidylinositol (3,4,5)-trisphosphate; PKC: Protein kinase C; PLC: Phospholipase C gamma; RAPA: rapamycin; R- SMAD: receptor-regulated SMAD; SMAD: Small Mothers Against Decapentaplegic protein; SOS: Son of sevenless; TGFB: Transforming growth factor beta; VEGF: Vascular endothelial growth factor; black arrows: main signaling pathways; blunt-ended lines: blockade/inhibition; dashed arrows: induction/activation. Segments of the figure were sketched by using artworks from Servier Medical Art (15 November 2021). Servier Medical Art by Servier is licensed under a Creative Commons Attribution 3.0 Unported License, https://creativecommons.org/licenses/by/3.0/ (access on 15 November 2021).
4.2. NOTCH Signaling Pathway
The NOTCH pathway represents an evolutionary thoroughly conserved pathway that regulates fundamental cellular processes, inclusive of cell-to-cell communication, tissue differentiation, SCs maintenance, proliferation, and development, as well as cell fate ascertainment of vascular ECs and the regulation of angiogenesis [89,95,96]. The family NOTCH receptors comprises four transmembrane proteins (NOTCH1–4), present also in normal SG tissue that interrelate with five distinct sets of ligands, Delta-like (DLL-1, -3, and -4) and (Serrate-like Jagged-1 and -2 (JAG1/2)) [33,97]. The activation of the canonical pathway is appointed to double concomitant proteolytic cleavages of NOTCH receptors by two enzymes: an A-disintegrin and metalloprotease (ADAM) and gamma-secretase in the sight of Presenilin 1 and 2 (PS1/2) [98]. These contingencies lead to the release of Notch intracellular domain (NICD) [97,99,100,101] which disentangles from the plasma membrane and goes ahead to the nucleus where it connects with the transcriptional repressor CSL (CBF1/Suppressor of hairless/LAG1 or RBPJ kappa (recombining binding protein suppressor of hairless J kappa)). The NICD–RBPJ complex cooperates with a member of transcriptional co-activators such as Mastermind-like (MAML1–3) ([102]) to inaugurate the transcription of target genes like HES1/5, HEY [99], mTORC1/2 [96,103], PI3K [96], TGFB [96], and c-MYC [85] to control ECs proliferation, differentiation, and apoptosis [104].
ECs mainly express DLL4, JAG1, and JAG2, in addition to NOTCH receptors 1–4, whilst PCs possess NOTCH receptors 1–3 on their surface and JAG1 and DLL1 [105,106,107,108,109]. The adjustment of cell fate commitment is mastered by NOTCH signaling and the tip/stalk cell phenotype is an emblem of the primary effect of NOTCH, too [110]. Upon ECs, VEGF brings on the generation of filopodia, attributing the supposed tip cells design [104]. The crosstalk between NOTCH and VEGF signaling are crucial for sprouting angiogenesis and for the configuration of ECs heterogeneity [111]. Guiding role of tip cells was previously reported in embryonic [112] but also adult tissues [113]. In essence, VEGF upregulates DLL4 in tip cells at the end of the sprout which further switches on Notch1 in the stalk cells, triggering the downregulation of VEGFR2 in tip cells and upregulation of VEGFR1 [104,111]. Notch signaling contributes to the prevention of excessive sprouting by the process denominated as “lateral inhibition” through activation and subduing branching of stalk cells [95,104], besides the third novel hybrid state derived from the unbalanced proportion of NICD, pivoting on divergent effects of DLL versus JAG, guiding to proliferation versus maintenance, a similar fate [114]. Although, JAG1 can contend with DLL4 through a negative feedback loop to regulate angiogenesis [95]. Moreover, the genetic deficiency of NOTCH3 correlates with the ongoing loss of PCs which associates with its performance in PCs differentiation and survival [115,116,117]. Accordingly, NOTCH3 gain-of-function and loss-of-function denote the fact that NOTCH3 is indispensable for PCs proliferation and limitation of blood vessel permeability [109]. The unsuccessful attempt to recruit PCs to the neovessels through NOTCH signaling during angiogenesis correlates with arteriovenous malformations (AVMs), ECs hyperplasia, microaneurysms, and edemas [109]. Regarding its pivotal role in the remodeling of the vascular tree and stabilization of junctional complexes [95,110], the importance of NOTCH signaling is also substantiated in the context of atypical angiogenesis in TME [118]. Thus, it is rational to speculate that a meticulous understanding of the NOTCH signaling mechanism may lead to a new departure in the formula of cancer therapy. A lot of studies have disclosed the expression of NOTCH components in tumor contexture, especially DLL4/NOTCH1, being upregulated by VEGF [108]. For instance, inhibition of VEGF in murine neoplasms promoted the decrease of DLL4 expression, inducing non-productive vessels formation within TME, since DLL4 is required for vascular organization; therefore, concomitantly, the blockade of dual VEGF and DLL4 can bring hope to have more potent effects in tumors than the solitary obstruction of either factor [95,119,120]. Furthermore, NOTCH signaling interacts with other pathways, including MEK/ERK1/2, TGFB, AKT, and mTOR pathways [103,121,122,123]. Other reported findings impart evidence that NOTCH1 and DLL4 were overexpressed within the tumor vasculature and upregulation of NOTCH/c-MYC activates the AKT pathway via PTEN phosphorylation [123]; consequently, the NOTCH activation intensifies PI3K/mTOR activity [85,123]. In concert, the blockade of AKT directly or with PI3K inhibitors or with rapamycin (RAPA) treatment dropped off JAG1, implicating the AKT/mTOR pathway as a feedback loop in ECs [122] (Figure 2).
4.3. PDGF Signaling Pathway
The PDGF family of chemokines and mitogens is known to possess four members which assemble into homo- or heterodimer forms: PDGF-a, -b, -c, -d that bind to two tyrosine kinase receptors, PDGFRA and PDGFRB [124,125]. PDGFb is mainly delivered by ECs from tip cells and acts on PDGFRB, expressed by PCs [2,6,126] (Figure 2). Subsequent to new blood vessel formation, the PDGFb-PDGFRB signaling axis is paramount in PCs recruitment into the new angiogenic sprouts and vascular homeostasis [125,127]. Activation of PDGFRB upholds PC proliferation and promotes the stabilization of developing vasculature, inhibiting angiogenesis in wholly formed mature vessels [127]. KO of the PGDFb or PDFGRB genes is lethal, assignable to vascular dysfunction, and caused by PCs deficiency [128,129]. Additionally, VEGF/VEGFR2 signaling decreases PCs’ oppressive response by stimulating the release of PDGFb, and the phosphorylation of the PDGFRB, inhibiting PCs migration to the vessels undergoing active angiogenesis [130]. The complexity of the picture is sharpened by the crosstalk between PDGF, NOTCH, VEGF, and PI3K/AKT/mTOR pathways [131,132]. Loss of NOTCH signaling leads to downregulation of PDGFRB levels and PC apoptosis, showing the NOTCH regulation of PC survival and proliferation via PDGFRB [109]. Activation of PDGF is associated with the stimulation of the PI3K/AKT/mTOR pathway, while the MAPK pathway is confirmed to be unimpressed by its activation [131,133]; even so, PI3K/AKT/mTOR communicates with the MAPK pathway through signaling crosstalk [134,135]. At the administration of an AKT inhibitor, the phosphorylation of AKT was increased and downregulated mTOR, PI3K, and ERK [131]. Interestingly, the inhibition of PI3K downregulated AKT and PDGFb [132] and upregulated ERK due to the discharge of the negative regulation of AKT on the MAPK pathway [131]. Additionally, PDGF and VEGF turned down the apoptosis and increased the range of living cells in the company of the AKT inhibitor, suggesting the anti-apoptotic, pro-proliferating, and cytoprotective potential of PDGF [131]. Intriguingly, the twofold inhibitor BEZ235 of PI3K and mTOR stimulated the phosphorylation of ERK by upregulation of RAS/RAF/MEK cascade [136]. Likewise, another study reported that the inhibition of mTOR with RAPA is associated with the downregulation of VEGF and with the decrease of ECs proliferation and tumor angiogenesis [137]; thus, future studies will be required to acknowledge more specific crosstalk between these pathways in order to coin novel medical useful approaches.
4.4. TGFB Signaling Pathway
The TGFB superfamily members comprise more than thirty constitutional related signaling molecules, counting TGFBs stricto sensu, Bone morphogenetic proteins (BMPs), activin, and nodals families [138,139] that bind to several categories of receptors: TGFB receptors (TGFRBs), BMP receptors (BMPRs), and Activin-like kinases (ALKs) [140]. TGFB family emulates as the key element with pleiotropic functions in angiogenesis, migration, cell proliferation, apoptosis, and differentiation [141]. The functions of TGFB signaling have been scrupulously studied and have demonstrated the protective role in the vascular media, as well as the homeostasis and integrity [142]. The TGFB family members (TGFB1, TGFB2, and TGFB3) and BMPs signal through two major classes of Serine/Threonine kinase receptors: type I and type II [121]. Typically, when activated by a ligand, the type II receptors (TGFRBII and BMPRII) encounter a conformational change, acceding them to phosphorylate and switching on the type I receptors (TGFRBI and BMPRI) [121,140]. The type I receptors, important in angiogenesis, include ALK5, expressed on both ECs and PCs and ALK1, more restricted to ECs [6,121,140,143], and are associated with the co-receptor Endoglin [59,121]. Once activated by a ligand, type I receptors phosphorylate and, in turn, a subgroup of Small Mothers Against Decapentaplegic proteins (SMADs), the receptor-regulated SMADs (R-SMADs), including SMAD2 and -3 for TGFB family and SMAD1, -5, and -8 for BMP family, as initial responders that transduce the signal from receptors, bind to Co-SMADs (SMAD4) [143,144]. Finally, the complex R-SMAD/Co-SMAD translocates to the nucleus and regulates the transcription genes: c-MYC, pointed to proliferation, HES1, and JAG1 [89,145,146,147]. In contrast, inhibitory SMADs (I-SMADs), including SMAD6 for the BMP family and SMAD7 for the TGFB family, are negative regulators that compete with R-SMADs to interact with activated type I receptors [148]. To dissect the specific roles of type I receptors, it should be underscored that they have opposing effects. Activation of ALK1 determines the phosphorylation of SMAD1/5/8 and promotes the proliferation of ECs and activates angiogenesis [2,149,150,151]. On the other hand, activation of ALK5 in PCs brings about phosphorylation of SMAD2/3, encouraging differentiation of PCs and the release of MMPs and ECM proteins, such as fibronectin and collagen from both PCs and ECs [152,153]. In ECs, ALK1 inhibits ALK5, suggesting the composite reciprocity, necessary for vessel development and stabilization [2] (Figure 2). Moreover, stimulation of ALK5 upregulates VEGFR1 and downregulates VEGFR2, inhibiting the proliferation of ECs [65,150]. Interestingly, it was demonstrated that TGFB collaborates with NOTCH signaling in the modulation of N-cadherin [154]. The KO of SMAD4 is related to reduced expression of N-cadherin and leads to the disruption of heterotypic contacts with PCs that further results in downregulation of TGFB signaling and disallows proliferation of PCs and promotes ECs hyperplasia [6,154]. Recent studies revealed that SMAD6, which regulates the inputs of SMAD1/5/8, as anti-angiogenic, is adjusted essentially upon NOTCH/DLL4 and VEGF levels regarding whether to promote sprouting angiogenesis or to broaden the original vasculature [110,155]. Another study delineated the alterations of TGFB1 signaling in SG pathogenesis [156]. The existence of all three isoforms of TGFB was confirmed upon ECs in patients going through SS [157]. Additionally, in mucoepidermoid carcinoma, TGFB1 was overexpressed on ECs and, of note, TGFRB2 was inversely proportional to tumor grade: low-grade tumors overexpressed TGFRBII, whereas neither high-grade tumor showed TGFRBII expression [158]. Of interest is that TGFB synchronizes ECM synthesis, along with ECs proliferation and migration, and as such TGFB1 induces PDGFb, instrumental to PCs recruitment to support stable vasculature [121].
4.5. HEDGEHOG Signaling Pathway
The HH signaling pathway plays an imperative role in a multiplicity of developmental and postnatal processes, including cell proliferation and differentiation, orchestrating the regulation of angiogenesis, blood vessel maturation, repair of normal tissues, and survival of normal/malignant SCs [159,160,161,162,163]. The actuating of the canonical HH pathway is denoted by the association of the three ligands-morphogens, Sonic hedgehog (SHH), Indian hedgehog (IHH), and Desert hedgehog (DHH) with the Patched1 receptor (PTCH1) and is regulated by assorted coreceptors, such as CDON, Brother of CDON (BOC), and Growth arrest specific 1 (GAS1) that promote ligand-receptor association; meanwhile, HH-interacting protein (HHIP) obstructs it [162,164]. In the absence of HH ligands, PTCH1 suppresses the activity of the transducer Smoothened (SMO) and the downstream transcription factors (TFs), GLI1, GLI2, and GLI3 are connected with Suppressor of fused (SUFU), a negative regulator of HH signaling and Kinesin family member 7 (KIF7) [162,165,166]. Notably, GLI1 constitutes the readout of the HH’s scheme, serving as the main downstream effector of the pathway, and also as a target gene [167,168,169], inasmuch as GLI2 and GLI3 organize into full-length (FL) as activator and as repressor (GLIR) configurations [170]. KIF 7 and SUFU sustain the phosphorylation of GLIFL by protein kinaseA (PKA), glycogen synthase kinase3 (GSK3), and casein kinase1(CK1) ([162,171]). Under basal conditions, the phosphorylated forms of GLI2 and GLI3 are controlled by proteasome degradation through E3 ubiquitin (UBE3) ligase complex and BTB/POZ protein/Cullin 3 (SPOP/CUL3) to induce GLI2R and GLI3R, the repressor configurations [162,172]. In contrast, the activation of HH signaling through the existence of HH ligand/receptor complex relieves the SMO inhibition that avoids the cleavage of GLI2 and GLI3 and activates the cascade of intracellular events [162], promoting the release of GLI from SUFU that translocates to the nucleus and activates HH target genes, by regulation of apoptosis (BCL2), cell cycle (CyclinD1(CCND1)), and N-MYC [162,167,173,174].
Adding to this puzzle, Shh represents the most significantly expressed Hh within the vasculature, along with Ihh, expressed by ECs, and also by cancer cells in Oral squamous cell carcinomas (OSCCs) [175,176,177]. A plurality of studies sympathize in reporting the proangiogenic properties of HH ligands, especially of SHH [178,179,180]. By contrast, a genetic deficiency in SHH in murine embryos causes lethality [181]. Of note, Nielsen and Dymecki [182] delineated the angiogenic portrayal of SHH in companionship with VEGF, whereby ECs from choroid plexus induce SHH and the signal is transduced by PCs, as they expressed PTCH1 rather than their ECs counterparts, suggesting that ECs are chiefly coordinated by PCs in the throughput of HH signaling. Once again, the proangiogenic and proliferative roles of Shh should be stressed, as they promote PTCH1, GLI2, NOTCH1, NOCTH3, BCL2 in ECs, whereas they upgrade PTCH1, GLI2, and NOTCH1 in PCs [162]. GLI1 upregulates VEGFR2, as the main effector of HH-promoting angiogenesis, and HHIP in mature vessels, while HHIP is downregulated in ECs engaged in angiogenesis and tumor neovessels [162,169]. As expected, most studies emphasize the hyperactivation of HH signaling to amplify tumor angiogenesis. In detail, the inhibition of Hh signaling with cyclopamine, a SMO antagonist, decreases VEGF and PTCH1 amounts and results in the reduction of MVD in OSCC [162,183]. Furthermore, it was shown that administration of erismodegib, another SMO inhibitor, restored the MVD and reduced the PCs coverage, increasing the measure of immature vasculature [184]. Additionally, pristimerin-administered has been shown to block SHH-induced ECs proliferation and PC recruitment into neovessels, therefore inhibiting MVD and tumor growth [185]. Notably, HH signaling can integrate with elements of other major signaling pathways, including NOTCH [95], VEGF/VEGFR2 [186], and CUL3-SPOP-DAXX axis [187,188]. The crosstalk between NOTCH and SHH within the retinal microvascular compartment has been reported in vivo [175]. SHH interceded the upregulation of the NOTCH1 receptor in both PCs and ECs but was diminished after cyclopamine treatment. It was shown that high blood flow rates are accompanied by inhibition of HH and NOTCH signaling constituents that dictate the apoptosis of PCs while, interestingly, decreasing the apoptotic signals in ECs. Nonetheless, absorbing for the induction of angiogenesis is the complicity of the novel regulatory axis CUL3-SPOP-DAXX. Sakaue and colleagues [187] determined that conjugation of CUL3-based UBE3 with NEDD8, a process denominated as neddylation [189] (Figure 3), a pivotal post-translational modification (PTM) besides ubiquitination, was instrumental for the upregulation of VEGFR2, in addition to NOTCH1 and DLL4. By contrast, the knockdown of SPOP- the CUL3 substrate adaptor plus repressor of DAXX- and CUL3 generated the upregulation of Death-domain associated protein (DAXX) and downregulation of VEGFR2 levels. Likewise, SPOP constitutes a transcriptional target of HIFs and hypoxia determines the accumulation of SPOP into the cytosol, which is satisfactory for the instauration of the tumorigenic program. The tumorigenesis materializes via ubiquitination and degradation of tumor suppressors like PTEN, ERK phosphatases, DAXX, and GLI2 [188]. Finally, by sketching the impact of HH on other pathways and by understanding the molecular mechanisms within cellular networks, a novel blueprint for the disclosure of target-discriminatory “quick-witted” drugs would revolutionize the development of medical therapy for preserving the SG function.
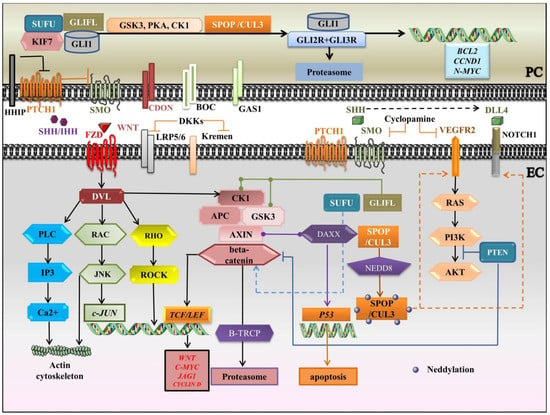
Figure 3.
A schematic portrayal of crosstalk between HH, WNT, VEGF, and NOTCH signaling pathways. APC: Adenomatous polyposis coli; BOC: Brother of CDON; CK1: casein kinase1; CUL3: Cullin 3; DAXX: Death-domain associated protein; DKKs: Dickkopfs; DLL: Delta-like; DVL: Dishevelled; EC: endothelial cell; FZD: Frizzled receptor; GAS1: Growth arrest specific 1; GLIFL: Gli full-length; GSK3: glycogen synthase kinase-3; HHIP: HH interacting protein; HH: Indian hedgehog; IP3: Inositol 1, 4, 5-trisphosphate; JAG: Jagged; JNK: c-JUN N-terminal kinase; KIF7: Kinesin family member 7; LRP5/6: Lipoprotein receptor-related proteins 5/6; PC: pericyte; PCP: planar cellpolarity; PI3K: Phosphatidylinositol 3-kinase; PKA: protein kinaseA; PLC: Phospholipase C gamma; PTCH1: Patched1 receptor; PTM: post-translational modification; ROCK: RHO-associated protein kinase; SHH: Sonic hedgehog; SMO: Smoothened; SPOP: BTB/POZ protein; SUFU: Suppressor of fused; TCF/LEF: T-Cell factor/Lymphoid enhancing factor; UBE3: E3 ubiquitin ligase; VEGFR2: Vascular Endothelial Growth Factor Receptor 2; black arrows: main signaling pathways; blunt-ended lines: blockade/inhibition, dashed arrows: induction/activation, round-ended lines: association. Segments of the figure were sketched by using artworks from Servier Medical Art (15 November 2021). Servier Medical Art by Servier is licensed under a Creative Commons Attribution 3.0 Unported License, https://creativecommons.org/licenses/by/3.0/ (access on 15 November 2021).
We summarize the crosstalk between HH, WNT, VEGF, and NOTCH signaling pathways in Figure 3. Briefly, in the HH canonical pathway, SHH/IHH-widely involved in angiogenesis- binds to PTCH1 and the inhibition of SMO is relieved, which avoids the cleavage of GLI2 and GLI3 and induces the release of GLI from SUFU and KIF7. The associated coreceptors, CDON, BOC, and GAS1 enhance ligand-receptor association, whereas HHIP inhibits it. In the absence of HH ligands, GLI1, GLI2, and GLI3 are phosphorylated by PKA, GSK3, and CK1, and are supervised by the UBE3 ligase complex, SPOP/CUL3, to induce the repressor forms, targeted for proteasome degradation. After HH ligand/receptor complex formation, the signal is mainly transduced through GLI1, serving as both downstream effector and target gene. Cyclopamine, which is a SMO antagonist, also inhibits VEGF and PTCH1 in EC. SHH upregulates the NOTCH1 receptor in both PC and EC. The neddylation (conjugation with NEDD8- a PTM) of CUL3-based UBE3 increases VEGFR2 (the main effector of HH), NOTCH1, and DLL4 levels. The WNT signaling is a key ingredient in cell proliferation/apoptosis, and vessel remodeling. WNTs associate with Frizzled (FZD), linked to co-receptors LRP5/6. Dishevelled (DVL) mediates the signal throughput to canonical and non-canonical pathways. The non-canonical WNT signaling is divided into WNT/PCP and WNT/Ca2+ pathways and coordinates actin cytoskeletal rearrangements. In WNT/PCP signaling, upon RAC GTPase actuating, JNK settles c-JUN transcription. In turn, RHO GTPase activates ROCK. Besides, the WNT/Ca2+ pathway turns on PLC, which activates IP3 to release Ca2+. DKKs, as WNT antagonists, associate with LRP5/6 and Kremen receptors and dictate the withdrawal of LRPs from the plasma membrane. In the canonical WNT pathway, if a WNT ligand is absent, beta catenin is phosphorylated by a destruction complex (APC, AXIN, CK1, GSK3), which is discerned by UBE3 ligase B-TRCP and targeted for proteasomal degradation, so the target genes are repressed by TCF/LEF. Once the pathway is activated by a ligand, the stabilization of beta-catenin is promoted, and it moves to the nucleus where it activates TCF/LEF and transcribes the target genes. WNT/beta-catenin collaborates with HH signaling in a positive feedback loop. The two pathways are mediated by GSK3, CK1, SUFU, PTEN, and SMO. SUFU negatively regulates GLI signaling and beta-catenin. Additionally, the loss of PTEN could switch on both beta catenin and GLI. Prominently, DAXX associates with AXIN to stimulate the tumor suppressor P53 to prompt apoptosis.
4.6. WNT Signaling Pathway
The WNT signaling pathway dictates a plethora of cellular events, including formation and remodeling of vessels, cell fate specification, proliferation, survival, and apoptosis [190,191]. The WNT family of glycoproteins bind to the Frizzled receptor (FZD), linked to the co-receptors Lipoprotein receptor-related proteins 5/6 (LRP5/6), and transduce the cellular signals to cytoplasmic phosphoprotein Dishevelled (DVL). On the level of DVL, the WNT signal splits up into substantial cascades: the canonical WNT/-beta catenin dependent pathway and the non-canonical or -beta catenin-independent pathway [190]. The non-canonical pathway is driven apart into the Planar cell polarity (PCP) and WNT/Ca2+ pathways [192]. The non-canonical PCP pathway switches on the small GTPases RHO and RAC and harmonizes cytoskeletal rearrangements [190]. The transduction of signal is settled through RAC activation of the c-JUN N-terminal kinase (JNK) pathway to mediate c-JUN transcription or RHO actuating of RHO-associated protein kinase (ROCK) [193,194,195]. The non-canonical WNT/Ca2+ pathway turns on the Phospholipase C (PLC), which in turn activates Inositol 1, 4, 5-trisphosphate (IP3) to increase intracellular Ca2+ [196,197,198]. Moreover, there are endogenous WNT antagonists, counting secreted rizzled-related proteins (SFRP1-5), the Dickkopfs (DKKs), the WNT inhibitory-factors (WIFs), and Cerberus [199]. DKKs connect with LRP5/6 and high-affinity receptors of the Kremen family, generating the withdrawal of LRPs from the plasma membrane [200].
The canonical WNT/- beta catenin signaling pathway governs multiple developmental events, including renewal and regeneration processes, and also the regulation of ECs growth and angiogenesis [190,201]. In the absence of WNT ligands, the cytoplasmic beta cateninis degraded via a beta catenin destruction complex which incorporates the Adenomatous polyposis coli (APC), the scaffolding protein AXIN, the Casein kinase 1 (CK1), and Glycogen synthase kinase 3 (GSK3) [196]. The phosphorylation of beta catenin by this complex induces PTMs, which is recognized by the UBE3 ligase B-TRCP and is targeted for destruction by the proteasome [202]. Together, these episodes avert the translocation of beta catenin to the nucleus and the target genes are therefore suppressed by the TFs, t-cell factor/lymphoid-enhancing factor (TCF/LEF) [196]. The pathway is activated when a canonical ligand like WNT3a, WNT4, or WNT7a/7b links to FZD and employs the DVL, which disrupts the action of the destruction complex, thereby promoting the stabilization beta catenin which moves to the nucleus [190,203]. Once there, beta catenin commutes the TCF/LEF repressor composite into a transcriptional activator system that facilitates the transcription of WNT target genes, including WNT constituents [204], c-MYC [196,205], JAG1 [206], and CCND [196,207]. Into adulthood, WNT4 is highly expressed into SG, whereas during SG murine development, FZD-6 is upregulated constantly [201]. Notably, beta catenin interacts with N-cadherins, thus preserving their interaction with the cytoskeleton and tissue integrity [208,209]. Furthermore, in murine, the loss of beta catenin is associated with altered epithelial-mesenchymal transition (EMT) and impedes the development of the endocardial cushion [210]. The WNT/beta-catenin signaling plays a pivotal role in harmonic vascular development since deletion of beta catenin causes defective vascular remodeling and results in early lethality in utero [211]. Additionally, within the microvasculature, the WNT/beta catenin signaling controls PC recruitment [212]. WNT5a is crucial for the maintenance of post-natal homeostasis and principally activates the beta-catenin-independent WNT signaling cascade [195]. Of note, Yuan et al. [213] demonstrated that the production of non-canonical WNT5a [214] by ECs is crucial for migration of PCs toward neovessels, while WNT5a KO corresponds to reduced PCs coverage and disruption of vascular stability. Another recent study reported the crosstalk between CCN1 and PC-derived WNT5a in ECs-PCs cocultures [215]. The WNT5a signaling evoked by PCs suppresses the CCN1 gene-a negative regulator of VEGF, in ECs, enhancing proliferation and EC hyperplasia. Prominently, WNT5a signals through FZD-ROR-RAC receptors and regulates the angiogenesis, the vascular morphogenesis via PCP, and ECs proliferation, being overexpressed in HPC/SFT [57,216]. To attest its intricacy and collusion in the vascular generation, WNT/beta-catenin cooperates with HH signaling in a positive feedback loop. Fundamentally, both pathways are adjusted by GSK3, CK1, SUFU, PTEN, and SMO [217]. SUFU represents not only a negative regulator of GLI signaling, but also it connects with β-catenin to supervise their nuclear–cytoplasmic disseminations [218]. Accordingly, loss of PTEN could activate both beta catenin and GLI [217]. Several studies have designated GSK3 as a convergent element among WNT/beta catenin and PI3K/PTEN signaling [219,220,221]. The accumulation of beta catenin is complemented via PTEN KO that further increases the activation of the PI3K/AKT pathway [221]. Intriguingly, SMO KO downregulates beta catenin levels, which is autonomous of the GLI effect [222]. On the other hand, the inhibition of HH signaling by cyclopamine reduces beta catenin [223]. Moreover, WNT/catenincatenin cooperates with NOTCH and VEGF/VEGFR2 signaling cascades. The gain-of-function of beta catenin upregulates DLL-4 expression [191]. The depletion of betacatenin or VEGFR2 from ECs leads to hypovascularization and rescinds angiogenesis following DLL4 downregulation in tip cells [224]. The crosstalks between NOTCH and WNT pathways are interceded by the regulation of GSK3. WNT1 countermanded the phosphorylation of NOTCH2 by GSK3 which, finally led to the upregulation of HES1 [225]. Notwithstanding, another study shed light upon the mechanistic role of DAXX in regulating tumorigenicity in correlation with the beta catenin pathway. It was shown that DAXX firmly cooperates with Axin to stimulate the tumor suppressor P53 to induce apoptosis [226] (Figure 2). Thus, acknowledging the interplay between all these molecular networks can provide a platform to identify novel anti-angiogenic/tumorigenic therapies.
4.7. Extracellular Vesicles/Exosomes
EVs constitute a miscellaneous population of bilayer membrane nanostructures, accommodating transmembrane proteins and incorporating assorted messenger nucleic acids, counting mRNAs, microRNAs (miRNAs), also other non-coding RNAs (ncRNAs), and signaling molecules for interchanging information with recipient cells [227,228,229]. Based on their subcellular origin, EVs are organized into three subgroups: apoptotic bodies, microvesicles (MVs), and EXOs [230]. Notably, EXOs, which are the minutest subgroup of EVs, assemble as Intraluminal Vesicles (ILVs) within the endosomal compartments called Multivesicular Bodies (MVBs) and are delivered to the extracellular environment after melding of MVBs with the plasmalemma [231,232]. EXOs represent key intermediaries of cell-to-cell communication that give knowledge about the prototypical cellular background via enclosed biomolecules and surface markers [233], being available in all body fluids, including saliva [234,235]. The stereotypical detected proteins include tetraspanins (CD63, CD9, and CD81), membrane transporters (RAB GTPases), and heat shock proteins (HSP70 and HSP90) [236]. Furthermore, EXOs appear like a “double-edged sword”, given their role in manifold physiological and pathological aspects [233]. As a prognostic and diagnostic tool, salivary EXOs not only could be employed as drug conveyance vehicles, but also tumor-derived EXOs have been reported to enhance the formation of TME, accelerating angiogenesis and generating the “premetastatic niche” [237,238].
There is substantial therapeutic potential of EXOs from EC-PC communication. It was shown that activation of the HIF pathway induces an angiogenic response from PCs through the exosomal bidirectional interconnection between both cell types [36]. Of note, PC-derived EXOs (PC-EXOs) via the PTEN/AKT pathway could also improve EC potentiality to regulate blood flow and decrease HIF1A and MMP2 levels [239]. PCs inhibited PTEN expression and promoted AKT levels, reducing apoptosis of ECs [239]. The study by Yuan and colleagues [213] indicated that in a PC-EC coculture, WNT5a from EC-derived EXOs (EC-EXOs) is crucial to trigger WNT/PCP in PCs and to recruit them to pulmonary blood vessels. In contrast, WNT5a EC KO was related to pulmonary arterial hypertension and right ventricular failure, corresponding to reduced PC coverage of microvasculature. Along the same line, EC-EXOs promoted by inflammatory impetuses convey particular miRNAs that mediate responses in PCs to amplify VEGFb expression, a specific ligand of VEGFR1 [240]. Additionally, there are plenty of studies to support the crosstalk among ECs and tumor cells via EVs. OSCC-derived EVs (OSCC-EVs), containing miRNA-142-3p, can intensify TGFBRI labor in ECs, advocating angiogenesis and tumor growth [241]. Additionally, ACC-derived EXOs can downregulate beta catenin in ECs to enhance the hematogenous metastasis of ACC cells [238]. Further, epiregulin-enriched ACC-derived EXOs promote EMT by upregulating N-cadherin and downregulating E-cadherin and and GLI1 [242]. Moreover, HNC-EXOs can bolster the malignant behavior of tumor cells by the distribution of SHH, initiating the non-canonical RHO/ROCK signaling cascade, enhancing the expression of MMP9, and being positively affiliated with MVD [243]. Isolated CD146+ CK7+ alpha-SMA stromal cells in the Schneiderian membrane may be involved in EMT-related regenerative processes [244]. Considering their role as potential disease biomarkers, SGT-EXOs present dissimilarities compared with healthy individuals [245]. In detail, SGT-EXOs are greater on atomic force microscopy [245] and remarkably increase in the expression of CD63, whereas CD9 and CD81 are reduced, congruent with the standpoint that the last two surface markers can impede the neoplasm metastasis [246,247,248]. Therefore, a sympathetic comprehension of the accurate function of EVs/EXOs would supplement the prognosis appraisal and may provide novel treatment approaches for HNCs.
5. Salivary Pericytes, Telocytes, and Myoepithelial Cells—Putative Therapeutical Local Aids in a Brighter Future?
Congruous with the desideratum for novel therapeutic perspectives to reimpose SG function and along with the knowledge that radiation therapy brings about deleterious side effects [38], the goal is to pinpoint the regenerative potentiality of parenchymal components via self-renewal and to identify progenitor/SC populations which synchronize tissue homeostasis and regeneration [249,250].
Although several approaches aim at fundamentally regenerating the duct and acinar cell lineages, it is attractive to speculate that, in fact, restoration of microvasculature after SG damage (e.g., post-IR) is the primary target [18,22], inasmuch as blood vessels govern the homeostasis, development, metabolism, and tissue microenvironment, so angio-targeted therapeutics may rehabilitate hitherto SG disorders [251]. Within this composite microarchitecture, the performance of PCs is unassailable not only in angiogenesis but also in early development and tissue regeneration [252,253]. Intriguingly, the damaged environment draws the distinction operation as to whether PCs undergo transdifferentiation or dedifferentiation [8]. As putative multipotent SCs, PCs are considerably believed to contribute to SG restoration post-IR, in addition to the adjustment of saliva secretion both in the physiological and radio-damaged model (see [3] and references therein). Furthermore, telocytes (TCs), archetypal interstitial cells, stabilize labyrinthine companionship with both PCs and ECs through direct (nano)contacts and EXOs, along with secretory acini, exocrine epithelial ducts, nerve fibers, and SCs [254,255,256,257,258]. Additionally, TCs establish an intricate three-dimensional cellular meshwork that mediates homeostasis, remodeling, and SC activity, interestingly through electrical cytoskeletal events [259]. Conspicuously, TCs can be designated as “rulers” in supervising SCs proliferation and differentiation, regardless of their location [260]. The role of TCs in angiogenesis is well-documented in several organs during development and tissue repair [261,262]. In this regard, TCs induce VEGF and release MMP9, as well as secretory vesicles to enhance EC proliferation and directed migration [257]. Intriguingly, it has been reported that TCs mediate skeletal muscle regeneration by invading the niche of PAX7+ satellite cells and secreting VEGF [260], critical for myoblast proliferation/differentiation [263,264]. Noteworthy, in addition to double expression of PDGFRA/CD34, pivotal for TC phenotyping [265,266], they also are PDGFRB immunopositive in context-dependent localization [267,268], therefore TCs may be engaged in PC recruitment and vessel stabilization [267]. Moreover, another recent study demonstrated that treatment with miRNA-21-5p-enriched TCs-EXOs inhibited EC apoptosis and promoted the regeneration of myocardial infarction [269]. Once again, it should be stressed the regenerative potential of TCs via SC niche modulation and intercellular signaling [270,271,272]. Given these features, it has been suggested that TCs are highly involved in SG homeostasis and local immune surveillance [273] since, within minor SGs distressed by SS, TCs are preserved in periacinar areas, and are not affected by the inflammatory status [274]. Of note, a study conducted by Shoshkes-Carmel et al. [275] delineated that a TC-FOXL1+ population is thoroughly essential for SC proliferation and maintenance by induction of WNT proteins. Critically, Halpern and colleagues [276] portrayed a highly preserved population of LGR5+ TCs from intestinal villus tip niche, as a source of BMP ligands and WNT5a that orchestrates the gene expression scheme. Furthermore, it has been revealed that myoepithelial cells (MECs) act as a reserve of SCs that can proliferate and transdifferentiate to enhance regeneration, following damage of resident SCs expressing cellular plasticity [277]. Additionally, MECs are preserved through self-duplication [278]. In the adult SG, K5 expression is confined to MECs and intercalated/excretory ducts [279]. The SG plasticity may assume a changeover in cellular identity from a lineage-restricted cell to another type of differentiated cell [280]. The majority of regenerated acini in a model injury have derived from differentiated MECs and KIT+ ductal cells by retrograding toward a progenitor-like state, and thereafter re-differentiating to acinar cells [281]. Although most studies exhibit that regeneration of acinar elements, following duct ligation from an atrophic state [281], turn out mainly by self-duplication of surviving acini [282,283], it should be denoted that distinct progenitors/SCs contribute to secretory cells renewal and maintenance, too [278,284,285,286]. Interestingly, LGR5 (leucine-rich repeat-containing G-protein coupled receptor 5) represents a WNT target gene [287], and a putative SC marker [201]; thereafter, LGR5+ cells have been suggested to be part of SG SCs repertoire [288]. In adult SG, WNT/beta catenin signaling is weakly expressed but is notably turned on during effective regeneration [201]. Indeed, K5- and WNT-receptive duct cells are designated as bipotent SCs, and are able to generate both duct and acinar cells [287,289]. In contrast, K5/AXIN 2-responsive intercalated duct cells are lineage-restricted progenitor cells [279]. However, assuming that beta catenin gain-of-function promotes belligerent SG Squamous Cell Carcinomas, the proper expansion of regeneration without tumorigenesis necessitates an exquisite balance. Finally, the interplay between PCs, TCs, and MECs appears like an intricate organization with pleiotropic functions which governs SG microarchitecture, and it would be erudite to consider them as putative therapeutical targets.
6. Conclusions
Salivary gland (SG) microvasculature constitutes an indispensable cellular organization that possesses specialized features to maintain tissue stability and homeostasis. Pericyte-endotelial cell (PC-EC) interconnections are instrumental for vascular development, maturation, and remodeling in both physiological and pathological conditions.
As mesenchymal stem cells (MSCs), PCs are widely explored in the field of regenerative medicine, as they represent an impregnable candidate ingredient not only for enhancing vascular integrity and angiogenesis but also to reinstitute SG function after damage, thereby accomplishing tissue regeneration and repair. The molecular events appertaining to PC-EC sophisticated interconnections were meticulously characterized to unravel the phenomena that bring about SG disorders. Biological operations, including cell proliferation, SC renewal, and differentiation, are orchestrated by a plethora of signaling pathways that cooperate with each other to harmonize the developmental and postnatal equilibrium status. Consequently, an exquisite modulation of these molecular pathways can provide a plan of action to develop novel target-selective drugs to overcome SG dysfunction.
Author Contributions
Conceptualization, I.C. and M.I.N.; software, I.C.; validation, M.I.N.; writing—original draft preparation, I.C.; writing—review and editing, I.C. and M.I.N.; supervision, M.I.N. All authors have read and agreed to the published version of the manuscript.
Funding
This research was partially funded by the Ministry of Research, Innovation, and digitization, under the Nucleu Programme, grant PN 19.29.02.02.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Psaltis, P.J.; Simari, R.D. Vascular Wall Progenitor Cells in Health and Disease. Circ. Res. 2015, 116, 1392–1412. [Google Scholar] [CrossRef] [PubMed]
- Armulik, A.; Genové, G.; Betsholtz, C. Pericytes: Developmental, Physiological, and Pathological Perspectives, Problems, and Promises. Dev. Cell 2011, 21, 193–215. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y. Pericytes in Irradiated Salivary Gland Repair. Master’s Thesis, McGill University, Montreal, QC, Canada, 2019. [Google Scholar]
- Armulik, A.; Abramsson, A.; Betsholtz, C. Endothelial/pericyte interactions. Circ. Res. 2005, 97, 512–523. [Google Scholar] [CrossRef]
- Keswani, S.G.; Balaji, S.; Le, L.D.; Leung, A.; Parvadia, J.K.; Frischer, J.; Yamano, S.; Taichman, N.; Crombleholme, T.M. Role of salivary vascular endothelial growth factor (VEGF) in palatal mucosal wound healing. Wound Repair Regen. 2013, 21, 554–562. [Google Scholar] [CrossRef] [PubMed]
- Sweeney, M.; Foldes, G. It Takes Two: Endothelial-Perivascular Cell Cross-Talk in Vascular Development and Disease. Front. Cardiovasc. Med. 2018, 5, 154. [Google Scholar] [CrossRef] [PubMed]
- Sharpe, P.T. Dental mesenchymal stem cells. Development 2016, 143, 2273–2280. [Google Scholar] [CrossRef]
- Wong, S.P.; Rowley, J.E.; Redpath, A.N.; Tilman, J.D.; Fellous, T.G.; Johnson, J.R. Pericytes, mesenchymal stem cells and their contributions to tissue repair. Pharmacol. Ther. 2015, 151, 107–120. [Google Scholar] [CrossRef]
- Stapor, P.C.; Sweat, R.S.; Dashti, D.C.; Betancourt, A.M. Pericyte Dynamics during Angiogenesis: New Insights from New Identities. J. Vasc. Res. 2014, 5698, 163–174. [Google Scholar] [CrossRef]
- Franco, M.; Roswall, P.; Cortez, E.; Hanahan, D.; Pietras, K. Pericytes promote endothelial cell survival through induction of autocrine VEGF-Asignaling and Bcl-w expression. Blood 2011, 118, 2906–2917. [Google Scholar] [CrossRef]
- Parthiban, S.P.; He, W.; Monteiro, N.; Athirasala, A.; França, C.M.; Bertassoni, L.E. Engineering pericyte-supported microvascular capillaries in cell-laden hydrogels using stem cells from the bone marrow, dental pulp and dental apical papilla. Sci. Rep. 2020, 10, 21579. [Google Scholar] [CrossRef]
- Zhang, Z.-S.; Zhou, H.-N.; He, S.-S.; Xue, M.-Y.; Li, T.; Liu, L.-M. Research advances in pericyte function and their roles in diseases. Chin. J. Traumatol.—Engl. Ed. 2020, 23, 89–95. [Google Scholar] [CrossRef]
- Chiaverina, G.; di Blasio, L.; Monica, V.; Accardo, M.; Palmiero, M.; Peracino, B.; Vara-Messler, M.; Puliafito, A.; Primo, L. Dynamic Interplay between Pericytes and Endothelial Cells during Sprouting Angiogenesis. Cells 2019, 8, 1109. [Google Scholar] [CrossRef]
- Traktuev, D.O.; Merfeld-clauss, S.; Li, J.; Kolonin, M.; Arap, W.; Pasqualini, R.; Johnstone, B.H.; March, K.L. A Population of Multipotent CD34-Positive Adipose Stromal Cells Share Pericyte and Mesenchymal Surface Markers, Reside in a Periendothelial Location, and Stabilize Endothelial Networks. Circ. Res. 2008, 102, 77–85. [Google Scholar] [CrossRef] [PubMed]
- Uhl, B.; Braun, C.; Dominik, J.; Luft, J.; Canis, M.; Reichel, C.A. A Novel Experimental Approach for In Vivo Analyses of the Salivary Gland Microvasculature. Front. Immunol. 2021, 11, 604470. [Google Scholar] [CrossRef] [PubMed]
- Etchevers, H.C. Pericyte Ontogeny: The Use of Chimeras to Track a Cell Lineage of Diverse Germ Line Origins. In Methods in Molecular Biology; Péault, B.M., Ed.; Humana Press Inc.: New York, NY, USA, 2021; Volume 2235, pp. 61–87. [Google Scholar]
- Hammes, H.-P.; Lin, J.; Renner, O.; Shani, M.; Lundqvist, A.; Betsholtz, C.; Brownlee, M.; Deutsch, U. Pericytes and the Pathogenesis of Diabetic Retinopathy. Diabetes 2002, 51, 3107–3112. [Google Scholar] [CrossRef] [PubMed]
- Mizrachi, A.; Cotrim, A.P.; Katabi, N.; Mitchell, J.B.; Haimovitz-Friedman, A. Radiation-Induced Microvascular Injury as a Mechanism of Salivary Gland Hypofunction and Potential Target for Radioprotectors. Radiat. Res. 2016, 195, 189–195. [Google Scholar] [CrossRef]
- Bartoloni, E.; Alunno, A.; Cafaro, G.; Valentini, V.; Bistoni, O.; Bonifacio, A.F.; Gerli, R. Subclinical atherosclerosis in primary Sjögren’s syndrome: Does inflammation matter? Front. Immunol. 2019, 10, 69. [Google Scholar] [CrossRef]
- Jasmer, K.J.; Gilman, K.E.; Forti, K.M.; Weisman, G.A.; Limesand, K.H. Radiation-induced salivary gland dysfunction: Mechanisms, therapeutics and future directions. J. Clin. Med. 2020, 9, 4095. [Google Scholar] [CrossRef]
- Radfar, L.; Sirois, D.A. Structural and functional injury in minipig salivary glands following fractionated exposure to 70 Gy of ionizing radiation: An animal model for human radiation-induced salivary gland injury. Oral Surg. Oral Med. Oral Pathol. Oral Radiol. Endodontol. 2003, 96, 267–274. [Google Scholar] [CrossRef]
- Cotrim, A.P.; Sowers, A.; Mitchell, J.B.; Baum, B.J. Prevention of Irradiation-induced Salivary Hypofunction by Microvessel Protection in Mouse Salivary Glands. Mol. Ther. 2007, 15, 2101–2106. [Google Scholar] [CrossRef]
- Bergwerff, M.; Verberne, M.E.; DeRuiter, M.C.; Poelmann, R.E.; Gittenberger-de Groot, A.C. Neural crest cell contribution to the developing circulatory system implications for vascular morphology? Circ. Res. 1998, 82, 221–231. [Google Scholar] [CrossRef]
- Yianni, V.; Sharpe, P.T. Transcriptomic Profiling of Dental Pulp Pericytes: An RNAseq Approach. Front. Dent. Med. 2020, 1, 6. [Google Scholar] [CrossRef]
- Crisan, M.; Yap, S.; Casteilla, L.; Chen, C.; Corselli, M.; Park, T.S.; Andriolo, G.; Sun, B.; Zheng, B.; Zhang, L.; et al. Article A Perivascular Origin for Mesenchymal Stem Cells in Multiple Human Organs. Cell Stem Cell 2008, 3, 301–313. [Google Scholar] [CrossRef] [PubMed]
- Zyrianova, T.; Basova, L.V.; Makarenkova, H. Isolation of myoepithelial cells from adult murine lacrimal and submandibular glands. J. Vis. Exp. 2019, 148, e59602. [Google Scholar] [CrossRef] [PubMed]
- Marcu, R.; Choi, Y.J.; Xue, J.; Fortin, C.L.; Wang, Y.; Nagao, R.J.; Xu, J.; MacDonald, J.W.; Bammler, T.K.; Murry, C.E.; et al. Human Organ-Specific Endothelial Cell Heterogeneity. iScience 2018, 4, 20–35. [Google Scholar] [CrossRef] [PubMed]
- Betsholtz, C. Cell–cell signaling in blood vessel development and function. EMBO Mol. Med. 2018, 10, 2–5. [Google Scholar] [CrossRef]
- Bergers, G.; Song, S. The role of pericytes in blood-vessel formation and maintenance. Neuro-Oncology 2005, 7, 452–464. [Google Scholar] [CrossRef]
- Brown, L.S.; Foster, C.G.; Courtney, J.-M.; King, N.E.; Howells, D.W.; Sutherland, B.A. Pericytes and Neurovascular Function in the Healthy and Diseased Brain. Front. Cell. Neurosci. 2019, 13, 282. [Google Scholar] [CrossRef]
- Yousif, L.F.; Di Russo, J.; Sorokin, L. Laminin isoforms in endothelial and perivascular basement membranes. Cell Adhes. Migr. 2013, 7, 101–110. [Google Scholar] [CrossRef]
- Hoffmann, J.; Feng, Y.; Hillenbrand, A.; Lin, J.; Erber, R.; Vajkoczy, P.; Gourzoulidou, E.; Waldmann, H.; Giannis, A.; Wolburg, H.; et al. Endothelial survival factors and spatial completion, but not pericyte coverage of retinal capillaries, determine vessel plasticity. FASEB J. 2005, 19, 2035–2036. [Google Scholar] [CrossRef]
- Porcheri, C.; Meisel, C.T.; Mitsiadis, T. Multifactorial contribution of notch signaling in head and neck squamous cell carcinoma. Int. J. Mol. Sci. 2019, 20, 1520. [Google Scholar] [CrossRef] [PubMed]
- Kwon, H.R.; Nelson, D.A.; Desantis, K.A.; Morrissey, J.M.; Larsen, M. Endothelial cell regulation of salivary gland epithelial patterning. Development 2017, 144, 211–220. [Google Scholar] [CrossRef] [PubMed]
- Weder, N.; Zhang, H.; Jensen, K.; Yang, B.Z.; Simen, A.; Jackowski, A.; Lipschitz, D.; Douglas-Palumberi, H.; Ge, M.; Perepletchikova, F.; et al. Child abuse, depression, and methylation in genes involved with stress, neural plasticity, and brain circuitry. J. Am. Acad. Child Adolesc. Psychiatry 2014, 53, 1173–1178. [Google Scholar] [CrossRef] [PubMed]
- Mayo, J.N.; Bearden, S.E. Driving the Hypoxia Inducible Pathway in Human Pericytes Promotes Vascular Density in an Exosome Dependent Manner. Microcirculation 2016, 22, 711–723. [Google Scholar] [CrossRef]
- Wilson, K.F.; Meier, J.D.; Ward, P.D. Salivary gland disorders. Am. Fam. Physician 2014, 89, 882–888. [Google Scholar]
- Takashi, I.; Ueda, Y.; Wörsdörfer, P.; Sumita, Y.; Asahina, I.; Ergün, S. Resident CD34-positive cells contribute to peri-endothelial cells and vascular morphogenesis in salivary gland after irradiation. J. Neural Transm. 2020, 127, 1467–1479. [Google Scholar] [CrossRef]
- Nam, K.; Dean, S.M.; Brown, C.T.; Smith, R.J.; Lei, P.; Andreadis, S.T.; Baker, O.J. Synergistic effects of laminin-1 peptides, VEGF and FGF9 on salivary gland regeneration. Acta Biomater. 2019, 91, 186–194. [Google Scholar] [CrossRef]
- Cheng, S.C.H.; Wu, V.W.C.; Kwong, D.L.W.; Ying, M.T.C. Assessment of post-radiotherapy salivary glands. Br. J. Radiol. 2011, 84, 393–402. [Google Scholar] [CrossRef]
- Vissink, A.; Mitchell, J.B.; Baum, B.J.; Limesand, K.H.; Jensen, S.B.; Fox, P.C.; Elting, L.S.; Langendijk, J.A.; Coppes, R.P.; Reyland, M.E. Clinical management of salivary gland hypofunction and xerostomia in head-and-neck cancer patients: Successes and barriers. Int. J. Radiat. Oncol. Biol. Phys. 2010, 78, 983–991. [Google Scholar] [CrossRef]
- Baharvand, M.; Khodadoustan, A.; Mohammadi, M.; Mortazavi, H.; Movahhedian, A. Xerostomia due to systemic disease: A review of 20 conditions and mechanisms. Ann. Med. Health Sci. Res. 2014, 4, 503–510. [Google Scholar] [CrossRef]
- Kujan, O.; Othman, R.; Alshehri, M.; Iqbal, F.; Kochaji, N. Proliferative Activity of Myoepithelial Cells in Irradiated Rabbit Parotid and Submandibular Salivary Glands. J. Int. Oral Health 2015, 7, 1–5. [Google Scholar]
- Hakim, S.G.; Kosmehl, H.; Lauer, I.; Nadrowitz, R.; Wedel, T.; Sieg, P. The role of myoepithelial cells in the short-term radiogenic impairment of salivary glands. An immunohistochemical, ultrastructural and scintigraphic study. Anticancer Res. 2002, 22, 4121–4128. [Google Scholar] [PubMed]
- Togarrati, P.P.; Sasaki, R.T.; Abdel-Mohsen, M.; Dinglasan, N.; Deng, X.; Desai, S.; Emmerson, E.; Yee, E.; Ryan, W.R.; da Silva, M.C.P.; et al. Identification and characterization of a rich population of CD34+ mesenchymal stem/stromal cells in human parotid, sublingual and submandibular glands. Sci. Rep. 2017, 7, 3484. [Google Scholar] [CrossRef] [PubMed]
- Invernici, G.; Emanueli, C.; Madeddu, P.; Cristini, S.; Gadau, S.; Benetti, A.; Ciusani, E.; Stassi, G.; Siragusa, M.; Nicosia, R.; et al. Human Fetal Aorta Contains Vascular Progenitor Cells Capable of Inducing Vasculogenesis, Angiogenesis, and Myogenesis in Vitro and in a Murine Model of Peripheral Ischemia. Am. J. Pathol. 2007, 170, 1879–1892. [Google Scholar] [CrossRef]
- Kramann, R.; Goettsch, C.; Wongboonsin, J.; Jain, S.; Aikawa, E.; Humphreys, B.D.; Kramann, R.; Goettsch, C.; Wongboonsin, J.; Iwata, H.; et al. Adventitial MSC-like Cells Are Progenitors of Vascular Smooth Muscle Cells and Drive Vascular Calcification in Chronic Kidney Disease Article Adventitial MSC-like Cells Are Progenitors of Vascular Smooth Muscle Cells and Drive Vascular Calcification in Ch. Stem Cell 2016, 19, 628–642. [Google Scholar] [CrossRef]
- Wang, S.-Q.; Wang, Y.-X.; Hua, H. Characteristics of Labial Gland Mesenchymal Stem Cells of Healthy Individuals and Patients with Sjögren’s Syndrome: A Preliminary Study. Stem Cells Dev. 2017, 26, 1171–1185. [Google Scholar] [CrossRef]
- Stryjewska-Makuch, G.; Kolebacz, B.; Janik, M.A.; Wolnik, A. Increase in the incidence of parotid gland tumors in the years 2005–2014. Otolaryngol. Pol. 2017, 71, 29–34. [Google Scholar] [CrossRef] [PubMed]
- Sentani, K.; Ogawa, I.; Ozasa, K.; Sadakane, A.; Utada, M.; Tsuya, T.; Kajihara, H.; Yonehara, S.; Takeshima, Y.; Yasui, W. Characteristics of 5015 salivary gland neoplasms registered in the hiroshima tumor tissue registry over a period of 39 years. J. Clin. Med. 2019, 8, 566. [Google Scholar] [CrossRef]
- Young, A.; Okuyemi, O.T. Malignant Salivary Gland Tumors. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2021. [Google Scholar]
- Fletcher, C.D.M. The evolving classification of soft tissue tumours—An update based on the new 2013 WHO classification. Histopathology 2014, 64, 2–11. [Google Scholar] [CrossRef]
- Lee, C.K.; Liu, K.L.; Huang, S.K. A dedifferentiated solitary fibrous tumor of the parotid gland: A case report with Cytopathologic findings and review of the literature. Diagn. Pathol. 2019, 14, 20. [Google Scholar] [CrossRef]
- Sowa, P.; Goroszkiewicz, K.; Szydelko, J.; Chechlinska, J.; Pluta, K.; Domka, W.; Misiolek, M.; Scierski, W. A Review of Selected Factors of Salivary Gland Tumour Formation and Malignant Transformation. Biomed. Res. Int. 2018, 2018, 2897827. [Google Scholar] [CrossRef]
- Yoo, S.Y.; Kwon, S.M. Angiogenesis and its therapeutic opportunities. Mediat. Inflamm. 2013, 2013, 127170. [Google Scholar] [CrossRef] [PubMed]
- Kondo, S.; Mukudai, Y.; Soga, D.; Nishida, T.; Takigawa, M.; Shirota, T. Differential expression of vascular endothelial growth factor in high- and low-metastasis cell lines of salivary gland adenoid cystic carcinoma. Anticancer Res. 2014, 34, 671–677. [Google Scholar]
- Hong, J.; Noh, M.; Akanda, M.R.; Kim, Y.J.; Kim, S.H.; Jung, T.-Y.; Jung, S.; Lee, J.-H.; Rhee, J.H.; Kim, K.-K.; et al. Solitary Fibrous Tumor/Hemangiopericytoma Metastasizes Extracranially, Associated with Altered Expression of WNT5A and MMP9. Cancers 2021, 13, 1142. [Google Scholar] [CrossRef] [PubMed]
- Cardoso, S.V.; Souza, K.C.N.; Faria, P.R.; Eisenberg, A.L.A.; Dias, F.L.; Loyola, A.M. Assessment of angiogenesis by CD105 antigen in epithelial salivary gland neoplasms with diverse metastatic behavior. BMC Cancer 2009, 9, 391. [Google Scholar] [CrossRef]
- Rossi, E.; Bernabeu, C.; Smadja, D.M. Endoglin as an Adhesion Molecule in Mature and Progenitor Endothelial Cells: A Function Beyond TGF-β. Front. Med. 2019, 6, 10. [Google Scholar] [CrossRef] [PubMed]
- Shibuya, M. Vascular Endothelial Growth Factor (VEGF) and Its Receptor (VEGFR) Signaling in Angiogenesis: A Crucial Target for Anti- and Pro-Angiogenic Therapies. Genes Cancer 2011, 2, 1097–1105. [Google Scholar] [CrossRef]
- Karamysheva, A.F. Mechanisms of angiogenesis. Biochemistry 2008, 73, 751–762. [Google Scholar] [CrossRef]
- Yancopoulos, G.D.; Davis, S.; Gale, N.W.; Rudge, J.S.; Wiegand, S.J.; Holash, J. Vascular-specific growth factors and blood vessel formation. Nature 2000, 407, 242–248. [Google Scholar] [CrossRef] [PubMed]
- Eilken, H.M.; Diéguez-Hurtado, R.; Schmidt, I.; Nakayama, M.; Jeong, H.; Arf, H.; Adams, S.; Ferrara, N.; Adams, R.H. Pericytes regulate VEGF-induced endothelial sprouting through VEGFR1. Nat. Commun. 2017, 8, 1574. [Google Scholar] [CrossRef]
- Otowa, Y.; Moriwaki, K.; Sano, K.; Shirakabe, M. Flt1/VEGFR1 heterozygosity causes transient embryonic edema. Sci. Rep. 2016, 6, 27186. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, T.; Yamaguchi, S.; Chida, K.; Shibuya, M. A single autophosphorylation site on KDR/Flk-1 is essential for VEGF-A-dependent activation of PLC-gamma and DNA synthesis in vascular endothelial cells. EMBO J. 2001, 20, 2768–2778. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, T.; Ueno, H.; Shibuya, M. VEGF activates protein kinase C-dependent, but Ras-independent Raf-MEK-MAP kinase pathway for DNA synthesis in primary endothelial cells. Oncogene 1999, 18, 2221–2230. [Google Scholar] [CrossRef] [PubMed]
- Maity, A. PI3K/AKT/mTOR pathway in angiogenesis. Front. Mol. Neurosci. 2011, 4, 51. [Google Scholar] [CrossRef]
- Murphy, D.A.; Makonnen, S.; Lassoued, W.; Feldman, M.D.; Carter, C.; Lee, W.M.F. Inhibition of tumor endothelial ERK activation, angiogenesis, and tumor growth by sorafenib (BAY43-9006). Am. J. Pathol. 2006, 169, 1875–1885. [Google Scholar] [CrossRef]
- Song, M.; Finley, S.D. Mechanistic insight into activation of MAPK signaling by pro-angiogenic factors. BMC Syst. Biol. 2018, 12, 145. [Google Scholar] [CrossRef]
- Srinivasan, R.; Zabuawala, T.; Huang, H.; Zhang, J.; Gulati, P.; Fernandez, S.; Karlo, J.C.; Landreth, G.E.; Leone, G.; Ostrowski, M.C. Erk1 and Erk2 regulate endothelial cell proliferation and migration during mouse embryonic angiogenesis. PLoS ONE 2009, 4, e8283. [Google Scholar] [CrossRef]
- Guo, Y.J.; Pan, W.W.; Liu, S.B.; Shen, Z.F.; Xu, Y.; Hu, L.L. ERK/MAPK signalling pathway and tumorigenesis (Review). Exp. Ther. Med. 2020, 19, 1997–2007. [Google Scholar] [CrossRef]
- Galabova-Kovacs, G.; Matzen, D.; Piazzolla, D.; Meissl, K.; Plyushch, T.; Chen, A.P.; Silva, A.; Baccarini, M. Essential role of B-Raf in ERK activation during extraembryonic development. Proc. Natl. Acad. Sci. USA 2006, 103, 1325–1330. [Google Scholar] [CrossRef]
- Giroux, S.; Tremblay, M.; Bernard, D.; Cardin-Girard, J.F.; Aubry, S.; Larouche, L.; Rousseau, S.; Huot, J.; Landry, J.; Jeannotte, L.; et al. Embryonic death of Mek1-deficient mice reveals a role for this kinase in angiogenesis in the labyrinthine region of the placenta. Curr. Biol. 1999, 9, 369–372. [Google Scholar] [CrossRef]
- Wojnowski, L.; Zimmer, A.M.; Beck, T.W.; Hahn, H.; Bernal, R.; Rapp, U.R.; Zimmer, A. Endothelial apoptosis in Braf-deficient mice. Nat. Genet. 1997, 16, 293–297. [Google Scholar] [CrossRef]
- Klemke, R.L.; Cai, S.; Giannini, A.L.; Gallagher, P.J.; de Lanerolle, P.; Cheresh, D.A. Regulation of cell motility by mitogen-activated protein kinase. J. Cell Biol. 1997, 137, 481–492. [Google Scholar] [CrossRef] [PubMed]
- Mavria, G.; Vercoulen, Y.; Yeo, M.; Paterson, H.; Karasarides, M.; Marais, R.; Bird, D.; Marshall, C.J. ERK-MAPK signaling opposes Rho-kinase to promote endothelial cell survival and sprouting during angiogenesis. Cancer Cell 2006, 9, 33–44. [Google Scholar] [CrossRef]
- Handra-Luca, A.; Bilal, H.; Bertrand, J.-C.; Fouret, P. Extra-cellular signal-regulated ERK-1/ERK-2 pathway activation in human salivary gland mucoepidermoid carcinoma: Association to aggressive tumor behavior and tumor cell proliferation. Am. J. Pathol. 2003, 163, 957–967. [Google Scholar] [CrossRef]
- Castellano, E.; Downward, J. RAS Interaction with PI3K: More Than Just Another Effector Pathway. Genes Cancer 2011, 2, 261–274. [Google Scholar] [CrossRef] [PubMed]
- Ha, J.M.; Baek, S.H.; Kim, Y.H.; Jin, S.Y.; Lee, H.S.; Kim, S.J.; Shin, H.K.; Lee, D.H.; Song, S.H.; Kim, C.D.; et al. Regulation of retinal angiogenesis by phospholipase C-β3 signaling pathway. Exp. Mol. Med. 2016, 48, e240. [Google Scholar] [CrossRef] [PubMed]
- Serban, D.; Leng, J.; Cheresh, D. H-Ras Regulates Angiogenesis and Vascular Permeability by Activation of Distinct Downstream Effectors. Circ. Res. 2008, 1350–1358. [Google Scholar] [CrossRef]
- Sabatini, D.M. mTOR and cancer: Insights into a complex relationship. Nat. Rev. Cancer 2006, 6, 729–734. [Google Scholar] [CrossRef]
- Andrade, N.P.; Warner, K.A.; Zhang, Z.; Pearson, A.T.; Mantesso, A.; Guimaraēs, D.M.; Altemani, A.; Mariano, F.V.; Nunes, F.D.; Nör, J.E. Survival of salivary gland cancer stem cells requires mTOR signaling. Cell Death Dis. 2021, 12, 108. [Google Scholar] [CrossRef]
- Mayer, I.A.; Arteaga, C.L. The PI3K/AKT Pathway as a Target for Cancer Treatment. Annu. Rev. Med. 2016, 67, 11–28. [Google Scholar] [CrossRef]
- Blanco-Aparicio, C.; Renner, O.; Leal, J.F.M.; Carnero, A. PTEN, more than the AKT pathway. Carcinogenesis 2007, 28, 1379–1386. [Google Scholar] [CrossRef]
- Shepherd, C.; Banerjee, L.; Cheung, C.W.; Mansour, M.R.; Jenkinson, S.; Gale, R.E.; Khwaja, A. PI3K/mTOR inhibition upregulates NOTCH-MYC signalling leading to an impaired cytotoxic response. Leukemia 2013, 27, 650–660. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Peng, B.; Chen, X. Expressions of Nuclear Factor kB, Inducible Nitric Oxide Synthase, and Vascular Endothelial Growth Factor in Adenoid Cystic Carcinoma of Salivary Glands: Correlations with the Angiogenesis and Clinical Outcome. Clin. Cancer Res. 2005, 11, 7334–7344. [Google Scholar] [CrossRef] [PubMed]
- Pouloudi, D.; Sotiriadis, A.; Theodorakidou, M.; Sarantis, P.; Pergaris, A.; Karamouzis, M.V.; Theocharis, S. The Impact of Angiogenesis in the Most Common Salivary Gland Malignant Tumors. Int. J. Mol. Sci. 2020, 21, 9335. [Google Scholar] [CrossRef] [PubMed]
- Folkman, J. Role of angiogenesis in tumor growth and metastasis. Semin. Oncol. 2002, 29, 15–18. [Google Scholar] [CrossRef]
- Pelullo, M.; Zema, S.; Nardozza, F.; Checquolo, S.; Screpanti, I.; Bellavia, D. Wnt, Notch, and TGF-β pathways impinge on hedgehog signaling complexity: An open window on cancer. Front. Genet. 2019, 10, 711. [Google Scholar] [CrossRef]
- Guertin, D.A.; Sabatini, D.M. Defining the role of mTOR in cancer. Cancer Cell 2007, 12, 9–22. [Google Scholar] [CrossRef]
- Park, M.S.; Patel, S.R.; Ludwig, J.A.; Trent, J.C.; Conrad, C.A.; Lazar, A.J.; Wang, W.-L.; Boonsirikamchai, P.; Choi, H.; Wang, X.; et al. Activity of temozolomide and bevacizumab in the treatment of locally advanced, recurrent, and metastatic hemangiopericytoma and malignant solitary fibrous tumor. Cancer 2011, 117, 4939–4947. [Google Scholar] [CrossRef]
- Maeda, O.; Ohka, F.; Maesawa, S.; Matsuoka, A.; Shimokata, T.; Mitsuma, A.; Urakawa, H.; Nakamura, S.; Shimoyama, Y.; Nakaguro, M.; et al. Solitary fibrous tumor/hemangiopericytoma treated with temozolomide plus bevacizumab: A report of four cases and literature review. Nagoya J. Med. Sci. 2020, 82, 631–644. [Google Scholar] [CrossRef]
- Nieder, C.; Wiedenmann, N.; Andratschke, N.; Molls, M. Current status of angiogenesis inhibitors combined with radiation therapy. Cancer Treat. Rev. 2006, 32, 348–364. [Google Scholar] [CrossRef]
- Dammrich, D.J.; Santos, E.S.; Raez, L.E. Efficacy of sorafenib, a multi-tyrosine kinase inhibitor, in an adenoid cystic carcinoma metastatic to the lung: Case report and review of literature. J. Med. Case Rep. 2011, 5, 483. [Google Scholar] [CrossRef]
- Akil, A.; Gutiérrez-García, A.K.; Guenter, R.; Rose, J.B.; Beck, A.W.; Chen, H.; Ren, B. Notch Signaling in Vascular Endothelial Cells, Angiogenesis, and Tumor Progression: An Update and Prospective. Front. Cell Dev. Biol. 2021, 9, 177. [Google Scholar] [CrossRef] [PubMed]
- Janghorban, M.; Xin, L.; Rosen, J.M.; Zhang, X.H.F. Notch signaling as a regulator of the tumor immune response: To target or not to target? Front. Immunol. 2018, 9, 1649. [Google Scholar] [CrossRef] [PubMed]
- Kopan, R.; Ilagan, M.X.G. The canonical Notch signaling pathway: Unfolding the activation mechanism. Cell 2009, 137, 216–233. [Google Scholar] [CrossRef]
- Azimi, M.; Le, T.T.; Brown, N.L. Presenilin gene function and Notch signaling feedback regulation in the developing mouse lens. Differentiation 2018, 102, 40–52. [Google Scholar] [CrossRef]
- Bellavia, D.; Palermo, R.; Felli, M.P.; Screpanti, I.; Checquolo, S. Notch signaling as a therapeutic target for acute lymphoblastic leukemia. Expert Opin. Ther. Targets 2018, 22, 331–342. [Google Scholar] [CrossRef] [PubMed]
- Mumm, J.S.; Kopan, R. Notch signaling: From the outside in. Dev. Biol. 2000, 228, 151–165. [Google Scholar] [CrossRef]
- Fortini, M.E. Notch signaling: The core pathway and its posttranslational regulation. Dev. Cell 2009, 16, 633–647. [Google Scholar] [CrossRef]
- Wu, L.; Aster, J.C.; Blacklow, S.C.; Lake, R.; Artavanis-Tsakonas, S.; Griffin, J.D. MAML1, a human homologue of Drosophila mastermind, is a transcriptional co-activator for NOTCH receptors. Nat. Genet. 2000, 26, 484–489. [Google Scholar] [CrossRef]
- Hibdon, E.S.; Razumilava, N.; Keeley, T.M.; Wong, G.; Solanki, S.; Shah, Y.M.; Samuelson, L.C. Notch and mTOR Signaling Pathways Promote Human Gastric Cancer Cell Proliferation. Neoplasia 2019, 21, 702–712. [Google Scholar] [CrossRef]
- Caliceti, C.; Nigro, P.; Rizzo, P.; Ferrari, R. ROS, Notch, and Wnt signaling pathways: Crosstalk between three major regulators of cardiovascular biology. Biomed. Res. Int. 2014, 2014, 318714. [Google Scholar] [CrossRef]
- Tian, D.-Y.; Jin, X.-R.; Zeng, X.; Wang, Y. Notch Signaling in Endothelial Cells: Is It the Therapeutic Target for Vascular Neointimal Hyperplasia? Int. J. Mol. Sci. 2017, 18, 1615. [Google Scholar] [CrossRef] [PubMed]
- Lovschall, H.; Mitsiadis, T.A.; Poulsen, K.; Jensen, K.H.; Kjeldsen, A.L. Coexpression of Notch3 and Rgs5 in the pericyte-vascular smooth muscle cell axis in response to pulp injury. Int. J. Dev. Biol. 2007, 51, 715–721. [Google Scholar] [CrossRef] [PubMed]
- Liu, H. Evaluation of Notch3 Deficiency in Diabetes-Induced Pericyte Loss in the Retina. J. Vasc. Res. 2018, 55, 308–318. [Google Scholar] [CrossRef] [PubMed]
- Thurston, G.; Kitajewski, J. VEGF and Delta-Notch: Interacting signalling pathways in tumour angiogenesis. Br. J. Cancer 2008, 99, 1204–1209. [Google Scholar] [CrossRef]
- Nadeem, T.; Bogue, W.; Bigit, B.; Cuervo, H. Deficiency of Notch signaling in pericytes results in arteriovenous malformations. JCI Insight 2020, 5, e125940. [Google Scholar] [CrossRef]
- Mack, J.J.; Iruela-Arispe, M.L. NOTCH regulation of the endothelial cell phenotype. Curr. Opin. Hematol. 2018, 25, 212–218. [Google Scholar] [CrossRef]
- Blanco, R.; Gerhardt, H. VEGF and Notch in tip and stalk cell selection. Cold Spring Harb. Perspect. Med. 2013, 3, a006569. [Google Scholar] [CrossRef]
- Rusu, M.C.; Poalelungi, C.V.; Vrapciu, A.D.; Nicolescu, M.I.; Hostiuc, S.; Mogoanta, L.; Taranu, T. Endocardial tip cells in the human embryo-facts and hypotheses. PLoS ONE 2015, 10, e0115853. [Google Scholar] [CrossRef]
- Dallinga, M.G.; Boas, S.E.; Klaassen, I.; Merks, R.H.; van Noorden, C.J.; Schlingemann, R.O. Tip Cells in Angiogenesis. In eLS; Zheng, Y., Ed.; John Wiley & Sons, Ltd.: Chichester, UK, 2015; pp. 1–10. [Google Scholar]
- Boareto, M.; Jolly, M.K.; Lu, M.; Onuchic, J.N.; Clementi, C.; Ben-Jacob, E. Jagged-delta asymmetry in Notch signaling can give rise to a sender/receiver hybrid phenotype. Proc. Natl. Acad. Sci. USA 2015, 112, E402–E409. [Google Scholar] [CrossRef]
- Kofler, N.M.; Cuervo, H.; Uh, M.K.; Murtomäki, A.; Kitajewski, J. Combined deficiency of Notch1 and Notch3 causes pericyte dysfunction, models CADASIL, and results in arteriovenous malformations. Sci. Rep. 2015, 5, 16449. [Google Scholar] [CrossRef]
- Hofmann, J.J.; Luisa Iruela-Arispe, M. Notch expression patterns in the retina: An eye on receptor-ligand distribution during angiogenesis. Gene Expr. Patterns 2007, 7, 461–470. [Google Scholar] [CrossRef]
- Liu, H.; Zhang, W.; Kennard, S.; Caldwell, R.B.; Lilly, B. Notch3 is critical for proper angiogenesis and mural cell investment. Circ. Res. 2010, 107, 860–870. [Google Scholar] [CrossRef] [PubMed]
- Carmeliet, P. Angiogenesis in life, disease and medicine. Nature 2005, 438, 932–936. [Google Scholar] [CrossRef] [PubMed]
- Ridgway, J.; Zhang, G.; Wu, Y.; Stawicki, S.; Liang, W.-C.; Chanthery, Y.; Kowalski, J.; Watts, R.J.; Callahan, C.; Kasman, I.; et al. Inhibition of Dll4 signalling inhibits tumour growth by deregulating angiogenesis. Nature 2006, 444, 1083–1087. [Google Scholar] [CrossRef]
- Noguera-Troise, I.; Daly, C.; Papadopoulos, N.J.; Coetzee, S.; Boland, P.; Gale, N.W.; Lin, H.C.; Yancopoulos, G.D.; Thurston, G. Blockade of Dll4 inhibits tumour growth by promoting non-productive angiogenesis. Nature 2006, 444, 1032–1037. [Google Scholar] [CrossRef] [PubMed]
- Holderfield, M.T.; Hughes, C.C.W. Crosstalk between Vascular Endothelial Growth Factor, Notch, and Transforming Growth Factor-β in Vascular Morphogenesis. Circ. Res. 2008, 102, 637–652. [Google Scholar] [CrossRef] [PubMed]
- Kerr, B.A.; West, X.Z.; Kim, Y.-W.; Zhao, Y.; Tischenko, M.; Cull, R.M.; Phares, T.W.; Peng, X.-D.; Bernier-Latmani, J.; Petrova, T.V.; et al. Stability and function of adult vasculature is sustained by Akt/Jagged1 signalling axis in endothelium. Nat. Commun. 2016, 7, 10960. [Google Scholar] [CrossRef]
- Vo, K.; Amarasinghe, B.; Washington, K.; Gonzalez, A.; Berlin, J.; Dang, T.P. Targeting notch pathway enhances rapamycin antitumor activity in pancreas cancers through PTEN phosphorylation. Mol. Cancer 2011, 10, 138. [Google Scholar] [CrossRef]
- Yamamoto, S.; Fukumoto, E.; Yoshizaki, K.; Iwamoto, T.; Yamada, A.; Tanaka, K.; Suzuki, H.; Aizawa, S.; Arakaki, M.; Yuasa, K.; et al. Platelet-derived growth factor receptor regulates salivary gland morphogenesis via fibroblast growth factor expression. J. Biol. Chem. 2008, 283, 23139–23149. [Google Scholar] [CrossRef]
- Xiang, D.; Feng, Y.; Wang, J.; Zhang, X.; Shen, J.; Zou, R.; Yuan, Y. Platelet-derived growth factor-BB promotes proliferation and migration of retinal microvascular pericytes by up-regulating the expression of C-X-C chemokine receptor types 4. Exp. Ther. Med. 2019, 18, 4022–4030. [Google Scholar] [CrossRef] [PubMed]
- Lindahl, P.; Johansson, B.R.; Levéen, P.; Betsholtz, C. Pericyte loss and microaneurysm formation in PDGF-B-deficient mice. Science 1997, 277, 242–245. [Google Scholar] [CrossRef] [PubMed]
- Gerhardt, H.; Betsholtz, C. Endothelial-pericyte interactions in angiogenesis. Cell Tissue Res. 2003, 314, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Soriano, P. Abnormal kidney development and hematological disorders in PDGF beta-receptor mutant mice. Genes Dev. 1994, 8, 1888–1896. [Google Scholar] [CrossRef] [PubMed]
- Levéen, P.; Pekny, M.; Gebre-Medhin, S.; Swolin, B.; Larsson, E.; Betsholtz, C. Mice deficient for PDGF B show renal, cardiovascular, and hematological abnormalities. Genes Dev. 1994, 8, 1875–1887. [Google Scholar] [CrossRef] [PubMed]
- Greenberg, J.I.; Shields, D.J.; Barillas, S.G.; Acevedo, L.M.; Murphy, E.; Huang, J.; Scheppke, L.; Stockmann, C.; Johnson, R.S.; Angle, N.; et al. A role for VEGF as a negative regulator of pericyte function and vessel maturation. Nature 2008, 456, 809–813. [Google Scholar] [CrossRef]
- Moench, R.; Grimmig, T.; Kannen, V.; Tripathi, S.; Faber, M.; Moll, E.M.; Chandraker, A.; Lissner, R.; Germer, C.T.; Waaga-Gasser, A.M.; et al. Exclusive inhibition of PI3K/Akt/mTOR signaling is not sufficient to prevent PDGF-mediated effects on glycolysis and proliferation in colorectal cancer. Oncotarget 2016, 7, 68749–68767. [Google Scholar] [CrossRef]
- Razmara, M.; Heldin, C.-H.; Lennartsson, J. Platelet-derived growth factor-induced Akt phosphorylation requires mTOR/Rictor and phospholipase C-γ1, whereas S6 phosphorylation depends on mTOR/Raptor and phospholipase D. Cell Commun. Signal. 2013, 11, 3. [Google Scholar] [CrossRef]
- Kaulfuß, S.; Seemann, H.; Kampe, R.; Meyer, J.; Dressel, R.; König, B.; Scharf, J.G.; Burfeind, P. Blockade of the PDGFR family together with SRC leads to diminished proliferation of colorectal cancer cells. Oncotarget 2013, 4, 1037–1049. [Google Scholar] [CrossRef][Green Version]
- Mendoza, M.C.; Er, E.E.; Blenis, J. The Ras-ERK and PI3K-mTOR pathways: Cross-talk and compensation. Trends Biochem. Sci. 2011, 36, 320–328. [Google Scholar] [CrossRef]
- Aksamitiene, E.; Kiyatkin, A.; Kholodenko, B.N. Cross-talk between mitogenic Ras/MAPK and survival PI3K/Akt pathways: A fine balance. Biochem. Soc. Trans. 2012, 40, 139–146. [Google Scholar] [CrossRef]
- Matkar, S.; An, C.; Hua, X. Kinase inhibitors of HER2/AKT pathway induce ERK phosphorylation via a FOXO-dependent feedback loop. Am. J. Cancer Res. 2017, 7, 1476–1485. [Google Scholar] [PubMed]
- Wang, S.; Lu, J.; You, Q.; Huang, H.; Chen, Y.; Liu, K. The mTOR/AP-1/VEGF signaling pathway regulates vascular endothelial cell growth. Oncotarget 2016, 7, 53269–53276. [Google Scholar] [CrossRef] [PubMed]
- Zi, Z.; Chapnick, D.A.; Liu, X. Dynamics of TGF-β/Smad signaling. FEBS Lett. 2012, 586, 1921–1928. [Google Scholar] [CrossRef]
- Saha, S.; Ji, L.; De Pablo, J.J.; Palecek, S.P. TGFβ/activin/nodal pathway in inhibition of human embryonic stem cell differentiation by mechanical strain. Biophys. J. 2008, 94, 4123–4133. [Google Scholar] [CrossRef] [PubMed]
- Heldin, C.-H.; Moustakas, A. Signaling Receptors for TGF-β Family Members. Cold Spring Harb. Perspect. Biol. 2016, 8, a022053. [Google Scholar] [CrossRef]
- Gonzalo-Gil, E.; Galindo-Izquierdo, M. Role of Transforming Growth Factor-Beta (TGF) Beta in the Physiopathology of Rheumatoid Arthritis. Reumatol. Clín. 2014, 10, 174–179. [Google Scholar] [CrossRef]
- Walshe, T.E.; Saint-Geniez, M.; Maharaj, A.S.R.; Sekiyama, E.; Maldonado, A.E.; D’Amore, P.A. TGF-β Is Required for Vascular Barrier Function, Endothelial Survival and Homeostasis of the Adult Microvasculature. PLoS ONE 2009, 4, e5149. [Google Scholar] [CrossRef]
- Nickel, J.; Ten Dijke, P.; Mueller, T.D. TGF-β family co-receptor function and signaling. Acta Biochim. Biophys. Sin. 2018, 50, 12–36. [Google Scholar] [CrossRef]
- Nickel, J.; Mueller, T.D. Specification of BMP Signaling. Cells 2019, 8, 1579. [Google Scholar] [CrossRef]
- Hata, A.; Chen, Y.-G. TGF-β Signaling from Receptors to Smads. Cold Spring Harb. Perspect. Biol. 2016, 8, a022061. [Google Scholar] [CrossRef]
- Feng, X.-H.; Liang, Y.-Y.; Liang, M.; Zhai, W.; Lin, X. Direct interaction of c-Myc with Smad2 and Smad3 to inhibit TGF-beta-mediated induction of the CDK inhibitor p15(Ink4B). Mol. Cell 2002, 9, 133–143. [Google Scholar] [CrossRef]
- Xu, Y.; Xue, S.; Zhou, J.; Voorhees, J.J.; Fisher, G.J. Notch and TGF-β pathways cooperatively regulate receptor protein tyrosine phosphatase-κ (PTPRK) gene expression in human primary keratinocytes. Mol. Biol. Cell 2015, 26, 1199–1206. [Google Scholar] [CrossRef]
- Aimaiti, Y.; Jin, X.; Wang, W.; Chen, Z.; Li, D. TGF-β1 signaling regulates mouse hepatic stellate cell differentiation via the Jagged1/Notch pathway. Life Sci. 2018, 192, 221–230. [Google Scholar] [CrossRef] [PubMed]
- Goumans, M.-J.; Valdimarsdottir, G.; Itoh, S.; Rosendahl, A.; Sideras, P.; ten Dijke, P. Balancing the activation state of the endothelium via two distinct TGF-beta type I receptors. EMBO J. 2002, 21, 1743–1753. [Google Scholar] [CrossRef] [PubMed]
- Jarad, M.; Kuczynski, E.A.; Morrison, J.; Viloria-Petit, A.M.; Coomber, B.L. Release of endothelial cell associated VEGFR2 during TGF-β modulated angiogenesis in vitro. BMC Cell Biol. 2017, 18, 10. [Google Scholar] [CrossRef]
- Oh, S.P.; Seki, T.; Goss, K.A.; Imamura, T.; Yi, Y.; Donahoe, P.K.; Li, L.; Miyazono, K.; ten Dijke, P.; Kim, S.; et al. Activin receptor-like kinase 1 modulates transforming growth factor-beta 1 signaling in the regulation of angiogenesis. Proc. Natl. Acad. Sci. USA 2000, 97, 2626–2631. [Google Scholar] [CrossRef]
- Schor, A.M.; Canfield, A.E.; Sloan, P.; Schor, S.L. Differentiation of pericytes in culture is accompanied by changes in the extracellular matrix. Vitr. Cell. Dev. Biol. J. Tissue Cult. Assoc. 1991, 27A, 651–659. [Google Scholar] [CrossRef]
- Davis, G.E.; Senger, D.R. Endothelial Extracellular Matrix. Circ. Res. 2005, 97, 1093–1107. [Google Scholar] [CrossRef]
- Li, F.; Lan, Y.; Wang, Y.; Wang, J.; Yang, G.; Meng, F.; Han, H.; Meng, A.; Wang, Y.; Yang, X. Endothelial Smad4 maintains cerebrovascular integrity by activating N-cadherin through cooperation with Notch. Dev. Cell 2011, 20, 291–302. [Google Scholar] [CrossRef]
- Mouillesseaux, K.P.; Wiley, D.S.; Saunders, L.M.; Wylie, L.A.; Kushner, E.J.; Chong, D.C.; Citrin, K.M.; Barber, A.T.; Park, Y.; Kim, J.; et al. branching in vessel networks via SMAD6. Nat. Commun. 2016, 7, 13247. [Google Scholar] [CrossRef]
- Janebodin, K.; Buranaphatthana, W.; Ieronimakis, N.; Hays, A.L.; Reyes, M. An in vitro culture system for long-term expansion of epithelial and mesenchymal salivary gland cells: Role of TGF-β1 in salivary gland epithelial and mesenchymal differentiation. Biomed. Res. Int. 2013, 2013, 815895. [Google Scholar] [CrossRef]
- Mason, G.I.; Hamburger, J.; Bowman, S.; Matthews, J.B. Salivary gland expression of transforming growth factor beta isoforms in Sjogren’s syndrome and benign lymphoepithelial lesions. Mol. Pathol. 2003, 56, 52–59. [Google Scholar] [CrossRef] [PubMed]
- Dillard, D.G.; Muller, S.; Cohen, C.; Bloch, D.; Del Gaudio, J.M.; Gal, A.A. High tumor grade in salivary gland mucoepidermoid carcinomas and loss of expression of transforming growth factor beta receptor type II. Arch. Otolaryngol.-Head Neck Surg. 2001, 127, 683–686. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Nüsslein-Volhard, C.; Wieschaus, E. Mutations affecting segment number and polarity in Drosophila. Nature 1980, 287, 795–801. [Google Scholar] [CrossRef]
- Napolitano, M.; Marfia, G.A.; Vacca, A.; Centonze, D.; Bellavia, D.; Di Marcotullio, L.; Frati, L.; Bernardi, G.; Gulino, A.; Calabresi, P. Modulation of gene expression following long-term synaptic depression in the striatum. Brain Res. Mol. Brain Res. 1999, 72, 89–96. [Google Scholar] [CrossRef]
- Varjosalo, M.; Taipale, J. Hedgehog: Functions and mechanisms. Genes Dev. 2008, 22, 2454–2472. [Google Scholar] [CrossRef] [PubMed]
- Chapouly, C.; Guimbal, S.; Hollier, P.-L.; Renault, M.-A. Role of Hedgehog Signaling in Vasculature Development, Differentiation, and Maintenance. Int. J. Mol. Sci. 2019, 20, 3076. [Google Scholar] [CrossRef] [PubMed]
- Yao, Q.; Renault, M.-A.; Chapouly, C.; Vandierdonck, S.; Belloc, I.; Jaspard-Vinassa, B.; Daniel-Lamazière, J.-M.; Laffargue, M.; Merched, A.; Desgranges, C.; et al. Sonic hedgehog mediates a novel pathway of PDGF-BB-dependent vessel maturation. Blood 2014, 123, 2429–2437. [Google Scholar] [CrossRef] [PubMed]
- Robbins, D.J.; Fei, D.L.; Riobo, N.A. The Hedgehog signal transduction network. Sci. Signal. 2012, 5, re6. [Google Scholar] [CrossRef] [PubMed]
- Burns, M.A.; Liao, Z.W.; Yamagata, N.; Pouliot, G.P.; Stevenson, K.E.; Neuberg, D.S.; Thorner, A.R.; Ducar, M.; Silverman, E.A.; Hunger, S.P.; et al. Hedgehog pathway mutations drive oncogenic transformation in high-risk T-cell acute lymphoblastic leukemia. Leukemia 2018, 32, 2126–2137. [Google Scholar] [CrossRef] [PubMed]
- Niewiadomski, P.; Niedziółka, S.M.; Markiewicz, Ł.; Uśpieński, T.; Baran, B.; Chojnowska, K. Gli Proteins: Regulation in Development and Cancer. Cells 2019, 8, 147. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, H.; Nishizaki, Y.; Hui, C.; Nakafuku, M.; Kondoh, H. Regulation of Gli2 and Gli3 activities by an amino-terminal repression domain: Implication of Gli2 and Gli3 as primary mediators of Shh signaling. Development 1999, 126, 3915–3924. [Google Scholar] [CrossRef] [PubMed]
- Regl, G.; Neill, G.W.; Eichberger, T.; Kasper, M.; Ikram, M.S.; Koller, J.; Hintner, H.; Quinn, A.G.; Frischauf, A.-M.; Aberger, F. Human GLI2 and GLI1 are part of a positive feedback mechanism in Basal Cell Carcinoma. Oncogene 2002, 21, 5529–5539. [Google Scholar] [CrossRef] [PubMed]
- Di Mauro, C.; Rosa, R.; D’Amato, V.; Ciciola, P.; Servetto, A.; Marciano, R.; Orsini, R.C.; Formisano, L.; De Falco, S.; Cicatiello, V.; et al. Hedgehog signalling pathway orchestrates angiogenesis in triple-negative breast cancers. Br. J. Cancer 2017, 116, 1425–1435. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Wang, C.; Pan, Y.; Bai, Z.; Wang, B. Increased proteolytic processing of full-length Gli2 transcription factor reduces the hedgehog pathway activity in vivo. Dev. Dyn. 2011, 240, 766–774. [Google Scholar] [CrossRef]
- Price, M.A.; Kalderon, D. Proteolysis of the Hedgehog signaling effector Cubitus interruptus requires phosphorylation by Glycogen Synthase Kinase 3 and Casein Kinase 1. Cell 2002, 108, 823–835. [Google Scholar] [CrossRef]
- Zeng, C.; Wang, Y.; Lu, Q.; Chen, J.; Zhang, J.; Liu, T.; Lv, N.; Luo, S. SPOP suppresses tumorigenesis by regulating Hedgehog/Gli2 signaling pathway in gastric cancer. J. Exp. Clin. Cancer Res. 2014, 33, 75. [Google Scholar] [CrossRef]
- Ryan, K.E.; Chiang, C. Hedgehog secretion and signal transduction in vertebrates. J. Biol. Chem. 2012, 287, 17905–17913. [Google Scholar] [CrossRef]
- Bigelow, R.L.H.; Chari, N.S.; Undén, A.B.; Spurgers, K.B.; Lee, S.; Roop, D.R.; Toftgård, R.; McDonnell, T.J. Transcriptional Regulation of bcl-2 Mediated by the Sonic Hedgehog Signaling Pathway through gli-1. J. Biol. Chem. 2004, 279, 1197–1205. [Google Scholar] [CrossRef]
- Walshe, T.E.; Connell, P.; Cryan, L.; Ferguson, G.; Gardiner, T.; Morrow, D.; Redmond, E.M.; O’Brien, C.; Cahill, P.A. Microvascular Retinal Endothelial and Pericyte Cell Apoptosis In Vitro: Role of Hedgehog and Notch Signaling. Investig. Opthalmol. Vis. Sci. 2011, 52, 4472–4483. [Google Scholar] [CrossRef]
- Valverde, L.D.F.; Pereira, T.D.A.; Dias, R.B.; Guimarães, V.S.N.; Ramos, E.A.G.; Santos, J.; Rocha, C.A.G. Macrophages and endothelial cells orchestrate tumor-associated angiogenesis in oral cancer via hedgehog pathway activation. Tumour Biol. 2016, 37, 9233–9241. [Google Scholar] [CrossRef]
- Yan, G.; Yang, L.; Lv, Y.; Shi, Y.; Shen, L.; Yao, X.; Guo, Q.; Zhang, P.; Cui, Y.; Zhang, X.; et al. Endothelial cells promote stem-like phenotype of glioma cells through activating the Hedgehog pathway. J. Pathol. 2014, 234, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.; Barnett, A.; Zhang, Y.; Yu, X.; Luo, Y. Poststroke Sonic Hedgehog Agonist Treatment Improves Functional Recovery by Enhancing Neurogenesis and Angiogenesis. Stroke 2017, 48, 1636–1645. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, R.P.H.; Haider, K.H.; Shujia, J.; Afzal, M.R.; Ashraf, M. Sonic Hedgehog gene delivery to the rodent heart promotes angiogenesis via iNOS/netrin-1/PKC pathway. PLoS ONE 2010, 5, e8576. [Google Scholar] [CrossRef] [PubMed]
- Kusano, K.F.; Pola, R.; Murayama, T.; Curry, C.; Kawamoto, A.; Iwakura, A.; Shintani, S.; Ii, M.; Asai, J.; Tkebuchava, T.; et al. Sonic hedgehog myocardial gene therapy: Tissue repair through transient reconstitution of embryonic signaling. Nat. Med. 2005, 11, 1197–1204. [Google Scholar] [CrossRef]
- Le Bras, A.; Vijayaraj, P.; Oettgen, P. Molecular mechanisms of endothelial differentiation. Vasc. Med. 2010, 15, 321–331. [Google Scholar] [CrossRef]
- Nielsen, C.M.; Dymecki, S.M. Sonic hedgehog is required for vascular outgrowth in the hindbrain choroid plexus. Dev. Biol. 2010, 340, 430–437. [Google Scholar] [CrossRef]
- Kuroda, H.; Kurio, N.; Shimo, T.; Matsumoto, K.; Masui, M.; Takabatake, K.; Okui, T.; Ibaragi, S.; Kunisada, Y.; Obata, K.; et al. Oral Squamous Cell Carcinoma-derived Sonic Hedgehog Promotes Angiogenesis. Anticancer Res. 2017, 37, 6731–6737. [Google Scholar] [CrossRef]
- Chaudhuri, T.R.; Straubinger, N.L.; Pitoniak, R.F.; Hylander, B.L.; Repasky, E.A.; Ma, W.W.; Straubinger, R.M. Tumor-Priming Smoothened Inhibitor Enhances Deposition and Efficacy of Cytotoxic Nanoparticles in a Pancreatic Cancer Model. Mol. Cancer Ther. 2016, 15, 84–93. [Google Scholar] [CrossRef]
- Lei, X.; Zhong, Y.; Huang, L.; Li, S.; Fu, J.; Zhang, L.; Zhang, Y.; Deng, Q.; Yu, X. Identification of a novel tumor angiogenesis inhibitor targeting Shh/Gli1 signaling pathway in Non-small cell lung cancer. Cell Death Dis. 2020, 11, 232. [Google Scholar] [CrossRef]
- Chen, W.; Tang, T.; Eastham-Anderson, J.; Dunlap, D.; Alicke, B.; Nannini, M.; Gould, S.; Yauch, R.; Modrusan, Z.; DuPree, K.J.; et al. Canonical hedgehog signaling augments tumor angiogenesis by induction of VEGF-A in stromal perivascular cells. Proc. Natl. Acad. Sci. USA 2011, 108, 9589–9594. [Google Scholar] [CrossRef]
- Sakaue, T.; Sakakibara, I.; Uesugi, T.; Fujisaki, A.; Nakashiro, K.; Hamakawa, H.; Kubota, E.; Joh, T.; Imai, Y.; Izutani, H.; et al. The CUL3-SPOP-DAXX axis is a novel regulator of VEGFR2 expression in vascular endothelial cells. Sci. Rep. 2017, 7, 42845. [Google Scholar] [CrossRef]
- Li, G.; Ci, W.; Karmakar, S.; Chen, K.; Dhar, R.; Fan, Z.; Guo, Z.; Zhang, J.; Ke, Y.; Wang, L.; et al. SPOP Promotes Tumorigenesis by Acting as a Key Regulatory Hub in Kidney Cancer. Cancer Cell 2014, 25, 455–468. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, M.; Calabrese, M.F.; Liu, J.; Waddell, M.B.; Nourse, A.; Hammel, M.; Miller, D.J.; Walden, H.; Duda, D.M.; Seyedin, S.N.; et al. Structures of SPOP-substrate complexes: Insights into molecular architectures of BTB-Cul3 ubiquitin ligases. Mol. Cell 2009, 36, 39–50. [Google Scholar] [CrossRef] [PubMed]
- Kuo, A.; Lee, M.Y.; Sessa, W.C. Lipid Droplet Biogenesis and Function in the Endothelium. Circ. Res. 2017, 120, 1289–1297. [Google Scholar] [CrossRef] [PubMed]
- Corada, M.; Nyqvist, D.; Orsenigo, F.; Caprini, A.; Giampietro, C.; Taketo, M.M.; Iruela-Arispe, M.L.; Adams, R.H.; Dejana, E. The Wnt/β-catenin pathway modulates vascular remodeling and specification by upregulating Dll4/notch signaling. Dev. Cell 2010, 18, 938–949. [Google Scholar] [CrossRef]
- Kohn, A.D.; Moon, R.T. Wnt and calcium signaling: Beta-catenin-independent pathways. Cell Calcium 2005, 38, 439–446. [Google Scholar] [CrossRef]
- Rosso, S.B.; Sussman, D.; Wynshaw-Boris, A.; Salinas, P.C. Wnt signaling through Dishevelled, Rac and JNK regulates dendritic development. Nat. Neurosci. 2005, 8, 34–42. [Google Scholar] [CrossRef]
- Habas, R.; Dawid, I.B.; He, X. Coactivation of Rac and Rho by Wnt/Frizzled signaling is required for vertebrate gastrulation. Genes Dev. 2003, 17, 295–309. [Google Scholar] [CrossRef]
- Kumawat, K.; Gosens, R. WNT-5A: Signaling and functions in health and disease. Cell. Mol. Life Sci. 2016, 73, 567–587. [Google Scholar] [CrossRef]
- Komiya, Y.; Habas, R. Wnt signal transduction pathways. Organogenesis 2008, 4, 68–75. [Google Scholar] [CrossRef]
- Dejana, E. The role of wnt signaling in physiological and pathological angiogenesis. Circ. Res. 2010, 107, 943–952. [Google Scholar] [CrossRef]
- Song, Z.; Wang, Y.; Zhang, F.; Yao, F.; Sun, C. Calcium Signaling Pathways: Key Pathways in the Regulation of Obesity. Int. J. Mol. Sci. 2019, 20, 2768. [Google Scholar] [CrossRef] [PubMed]
- Goodwin, A.M.; D’Amore, P.A. Wnt signaling in the vasculature. Angiogenesis 2002, 5, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, T.; Nakamura, T.; Matsumoto, K. The functions and possible significance of Kremen as the gatekeeper of Wnt signalling in development and pathology. J. Cell. Mol. Med. 2008, 12, 391–408. [Google Scholar] [CrossRef] [PubMed]
- Hai, B.; Yang, Z.; Millar, S.E.; Choi, Y.S.; Taketo, M.M.; Nagy, A.; Liu, F. Wnt/β-catenin signaling regulates postnatal development and regeneration of the salivary gland. Stem Cells Dev. 2010, 19, 1793–1801. [Google Scholar] [CrossRef]
- He, X.; Semenov, M.; Tamai, K.; Zeng, X. LDL receptor-related proteins 5 and 6 in Wnt/beta-catenin signaling: Arrows point the way. Development 2004, 131, 1663–1677. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, J.K.; Takano, M.; Hiraoka-Kanie, M.; Shimazu, C.; Peishi, Y.; Yanagi, K.; Nakano, A.; Inoue, E.; Kita, F.; Nishikawa, S.-I. Prospective identification of cardiac progenitors by a novel single cell-based cardiomyocyte induction. FASEB J. 2005, 19, 1534–1536. [Google Scholar] [CrossRef]
- Macdonald, B.T.; Tamai, K.; He, X. Review Wnt/b-Catenin Signaling: Components, Mechanisms, and Diseases. Dev. Cell 2009, 17, 9–26. [Google Scholar] [CrossRef]
- He, T.C.; Sparks, A.B.; Rago, C.; Hermeking, H.; Zawel, L.; da Costa, L.T.; Morin, P.J.; Vogelstein, B.; Kinzler, K.W. Identification of c-MYC as a target of the APC pathway. Science 1998, 281, 1509–1512. [Google Scholar] [CrossRef] [PubMed]
- Katoh, M.; Katoh, M. Notch ligand, JAG1, is evolutionarily conserved target of canonical WNT signaling pathway in progenitor cells. Int. J. Mol. Med. 2006, 17, 681–685. [Google Scholar] [CrossRef] [PubMed]
- Tetsu, O.; McCormick, F. Beta-catenin regulates expression of cyclin D1 in colon carcinoma cells. Nature 1999, 398, 422–426. [Google Scholar] [CrossRef] [PubMed]
- Meng, W.; Takeichi, M. Adherens junction: Molecular architecture and regulation. Cold Spring Harb. Perspect. Biol. 2009, 1, a002899. [Google Scholar] [CrossRef]
- Stepniak, E.; Radice, G.L.; Vasioukhin, V. Adhesive and signaling functions of cadherins and catenins in vertebrate development. Cold Spring Harb. Perspect. Biol. 2009, 1, a002949. [Google Scholar] [CrossRef]
- Liebner, S.; Cattelino, A.; Gallini, R.; Rudini, N.; Iurlaro, M.; Piccolo, S.; Dejana, E. Beta-catenin is required for endothelial-mesenchymal transformation during heart cushion development in the mouse. J. Cell Biol. 2004, 166, 359–367. [Google Scholar] [CrossRef]
- Cattelino, A.; Liebner, S.; Gallini, R.; Zanetti, A.; Balconi, G.; Corsi, A.; Bianco, P.; Wolburg, H.; Moore, R.; Oreda, B.; et al. The conditional inactivation of the beta-catenin gene in endothelial cells causes a defective vascular pattern and increased vascular fragility. J. Cell Biol. 2003, 162, 1111–1122. [Google Scholar] [CrossRef]
- Birdsey, G.M.; Shah, A.V.; Dufton, N.; Reynolds, L.E.; Osuna Almagro, L.; Yang, Y.; Aspalter, I.M.; Khan, S.T.; Mason, J.C.; Dejana, E.; et al. The Endothelial Transcription Factor ERG Promotes Vascular Stability and Growth through Wnt/β-Catenin Signaling. Dev. Cell 2015, 32, 82–96. [Google Scholar] [CrossRef]
- Yuan, K.; Shamskhou, E.A.; Orcholski, M.E.; Nathan, A.; Reddy, S.; Honda, H.; Mani, V.; Zeng, Y.; Ozen, M.O.; Wang, L.; et al. Loss of Endothelium-Derived Wnt5a Is Associated With Reduced Pericyte Recruitment and Small Vessel Loss in Pulmonary Arterial Hypertension. Circulation 2019, 139, 1710–1724. [Google Scholar] [CrossRef]
- Korn, C.; Scholz, B.; Hu, J.; Srivastava, K.; Wojtarowicz, J.; Arnsperger, T.; Adams, R.H.; Boutros, M.; Augustin, H.G.; Augustin, I. Endothelial cell-derived non-canonical Wnt ligands control vascular pruning in angiogenesis. Development 2014, 141, 1757–1766. [Google Scholar] [CrossRef]
- Lee, S.; Elaskandrany, M.; Lau, L.F.; Lazzaro, D.; Grant, M.B.; Chaqour, B. Interplay between CCN1 and Wnt5a in endothelial cells and pericytes determines the angiogenic outcome in a model of ischemic retinopathy. Sci. Rep. 2017, 7, 1405. [Google Scholar] [CrossRef]
- Sato, A.; Yamamoto, H.; Sakane, H.; Koyama, H.; Kikuchi, A. Wnt5a regulates distinct signalling pathways by binding to Frizzled2. EMBO J. 2010, 29, 41–54. [Google Scholar] [CrossRef]
- Song, L.; Li, Z.-Y.; Liu, W.-P.; Zhao, M.-R. Crosstalk between Wnt/β-catenin and Hedgehog/Gli signaling pathways in colon cancer and implications for therapy. Cancer Biol. Ther. 2015, 16, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.; Poon, R.; Zhang, X.; Cheah, A.; Ding, Q.; Hui, C.C.; Alman, B. Suppressor of fused negatively regulates beta-catenin signaling. J. Biol. Chem. 2001, 276, 40113–40119. [Google Scholar] [CrossRef]
- Naito, A.T.; Akazawa, H.; Takano, H.; Minamino, T.; Nagai, T.; Aburatani, H.; Komuro, I. Phosphatidylinositol 3-kinase-Akt pathway plays a critical role in early cardiomyogenesis by regulating canonical Wnt signaling. Circ. Res. 2005, 97, 144–151. [Google Scholar] [CrossRef] [PubMed]
- Katoh, M.; Katoh, M. Cross-talk of WNT and FGF signaling pathways at GSK3beta to regulate beta-catenin and SNAIL signaling cascades. Cancer Biol. Ther. 2006, 5, 1059–1064. [Google Scholar] [CrossRef] [PubMed]
- Rychahou, P.G.; Kang, J.; Gulhati, P.; Doan, H.Q.; Chen, L.A.; Xiao, S.-Y.; Chung, D.H.; Evers, B.M. Akt2 overexpression plays a critical role in the establishment of colorectal cancer metastasis. Proc. Natl. Acad. Sci. USA 2008, 105, 20315–20320. [Google Scholar] [CrossRef] [PubMed]
- Arimura, S.; Matsunaga, A.; Kitamura, T.; Aoki, K.; Aoki, M.; Taketo, M.M. Reduced Level of Smoothened Suppresses Intestinal Tumorigenesis by Down-Regulation of Wnt Signaling. Gastroenterology 2009, 137, 629–638. [Google Scholar] [CrossRef]
- Qualtrough, D.; Rees, P.; Speight, B.; Williams, A.C.; Paraskeva, C. The Hedgehog Inhibitor Cyclopamine Reduces β-Catenin-Tcf Transcriptional Activity, Induces E-Cadherin Expression, and Reduces Invasion in Colorectal Cancer Cells. Cancers 2015, 7, 1885–1899. [Google Scholar] [CrossRef]
- Zarkada, G.; Heinolainen, K.; Makinen, T.; Kubota, Y.; Alitalo, K. VEGFR3 does not sustain retinal angiogenesis without VEGFR2. Proc. Natl. Acad. Sci. USA 2015, 112, 761–766. [Google Scholar] [CrossRef]
- Espinosa, L.; Inglés-Esteve, J.; Aguilera, C.; Bigas, A. Phosphorylation by Glycogen Synthase Kinase-3β Down-regulates Notch Activity, a Link for Notch and Wnt Pathways. J. Biol. Chem. 2003, 278, 32227–32235. [Google Scholar] [CrossRef]
- Li, Q.; Wang, X.; Wu, X.; Rui, Y.; Liu, W.; Wang, J.; Wang, X.; Liou, Y.; Ye, Z.; Lin, S. Daxx Cooperates with the Axin/HIPK2/p53 Complex to Induce Cell Death. Cancer Res. 2007, 67, 66–74. [Google Scholar] [CrossRef] [PubMed]
- Valadi, H.; Ekström, K.; Bossios, A.; Sjöstrand, M.; Lee, J.J.; Lötvall, J.O. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat. Cell Biol. 2007, 9, 654–659. [Google Scholar] [CrossRef]
- Shurtleff, M.J.; Yao, J.; Qin, Y.; Nottingham, R.M.; Temoche-Diaz, M.M.; Schekman, R.; Lambowitz, A.M. Broad role for YBX1 in defining the small noncoding RNA composition of exosomes. Proc. Natl. Acad. Sci. USA 2017, 114, E8987–E8995. [Google Scholar] [CrossRef]
- Tkach, M.; Théry, C. Communication by Extracellular Vesicles: Where We Are and Where We Need to Go. Cell 2016, 164, 1226–1232. [Google Scholar] [CrossRef] [PubMed]
- Yáñez-Mó, M.; Siljander, P.R.-M.; Andreu, Z.; Zavec, A.B.; Borràs, F.E.; Buzas, E.I.; Buzas, K.; Casal, E.; Cappello, F.; Carvalho, J.; et al. Biological properties of extracellular vesicles and their physiological functions. J. Extracell. Vesicles 2015, 4, 27066. [Google Scholar] [CrossRef]
- Maas, S.L.N.; Breakefield, X.O.; Weaver, A.M. Extracellular Vesicles: Unique Intercellular Delivery Vehicles. Trends Cell Biol. 2017, 27, 172–188. [Google Scholar] [CrossRef]
- Théry, C.; Zitvogel, L.; Amigorena, S. Exosomes: Composition, biogenesis and function. Nat. Rev. Immunol. 2002, 2, 569–579. [Google Scholar] [CrossRef]
- Kalluri, R.; LeBleu, V.S. The biology, function, and biomedical applications of exosomes. Science 2020, 367, eaau6977. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Liu, Y.; Liu, H.; Tang, W.H. Exosomes: Biogenesis, biologic function and clinical potential. Cell Biosci. 2019, 9, 19. [Google Scholar] [CrossRef]
- Han, Y.; Jia, L.; Zheng, Y.; Li, W. Salivary exosomes: Emerging roles in systemic disease. Int. J. Biol. Sci. 2018, 14, 633–643. [Google Scholar] [CrossRef]
- Nonaka, T.; Wong, D.T.W. Saliva-Exosomics in Cancer: Molecular Characterization of Cancer-Derived Exosomes in Saliva. Enzymes 2017, 42, 125–151. [Google Scholar] [CrossRef] [PubMed]
- Psaila, B.; Lyden, D. The metastatic niche: Adapting the foreign soil. Nat. Rev. Cancer 2009, 9, 285–293. [Google Scholar] [CrossRef] [PubMed]
- Zhan, C.; Yang, X.; Yin, X.; Hou, J. Exosomes and other extracellular vesicles in oral and salivary gland cancers. Oral Dis. 2020, 26, 865–875. [Google Scholar] [CrossRef] [PubMed]
- Yuan, X.; Wu, Q.; Wang, P.; Jing, Y.; Yao, H.; Tang, Y.; Li, Z.; Zhang, H.; Xiu, R. Exosomes Derived From Pericytes Improve Microcirculation and Protect Blood–Spinal Cord Barrier After Spinal Cord Injury in Mice. Front. Neurosci. 2019, 13, 319. [Google Scholar] [CrossRef]
- Yamamoto, S.; Niida, S.; Azuma, E.; Yanagibashi, T.; Muramatsu, M.; Huang, T.T.; Sagara, H.; Higaki, S.; Ikutani, M.; Nagai, Y.; et al. Inflammation-induced endothelial cell-derived extracellular vesicles modulate the cellular status of pericytes. Sci. Rep. 2015, 5, 8505. [Google Scholar] [CrossRef]
- Dickman, C.T.D.; Lawson, J.; Jabalee, J.; MacLellan, S.A.; LePard, N.E.; Bennewith, K.L.; Garnis, C. Selective extracellular vesicle exclusion of miR-142-3p by oral cancer cells promotes both internal and extracellular malignant phenotypes. Oncotarget 2017, 8, 15252–15266. [Google Scholar] [CrossRef]
- Yang, W.-W.; Yang, L.-Q.; Zhao, F.; Chen, C.-W.; Xu, L.-H.; Fu, J.; Li, S.-L.; Ge, X.-Y. Epiregulin Promotes Lung Metastasis of Salivary Adenoid Cystic Carcinoma. Theranostics 2017, 7, 3700–3714. [Google Scholar] [CrossRef]
- Huaitong, X.; Yuanyong, F.; Yueqin, T.; Peng, Z.; Wei, S.; Kai, S. Microvesicles releasing by oral cancer cells enhance endothelial cell angiogenesis via Shh/RhoA signaling pathway. Cancer Biol. Ther. 2017, 18, 783–791. [Google Scholar] [CrossRef]
- Derjac-Aramă, A.I.; Sarafoleanu, C.; Manea, C.M.; Nicolescu, M.I.; Vrapciu, A.D.; Rusu, M.C. Regenerative Potential of Human Schneiderian Membrane: Progenitor Cells and Epithelial-Mesenchymal Transition. Anat. Rec. Adv. Integr. Anat. Evol. Biol. 2015, 298, 2132–2140. [Google Scholar] [CrossRef]
- Sharma, S.; Gillespie, B.M.; Palanisamy, V.; Gimzewski, J.K. Quantitative nanostructural and single-molecule force spectroscopy biomolecular analysis of human-saliva-derived exosomes. Langmuir 2011, 27, 14394–14400. [Google Scholar] [CrossRef] [PubMed]
- Zlotogorski-Hurvitz, A.; Dayan, D.; Chaushu, G.; Salo, T.; Vered, M. Morphological and molecular features of oral fluid-derived exosomes: Oral cancer patients versus healthy individuals. J. Cancer Res. Clin. Oncol. 2016, 142, 101–110. [Google Scholar] [CrossRef] [PubMed]
- Xie, C.; Ji, N.; Tang, Z.; Li, J.; Chen, Q. The role of extracellular vesicles from different origin in the microenvironment of head and neck cancers. Mol. Cancer 2019, 18, 83. [Google Scholar] [CrossRef]
- Zöller, M. Tetraspanins: Push and pull in suppressing and promoting metastasis. Nat. Rev. Cancer 2009, 9, 40–55. [Google Scholar] [CrossRef] [PubMed]
- Takata, N.; Eiraku, M. Stem cells and genome editing: Approaches to tissue regeneration and regenerative medicine. J. Hum. Genet. 2018, 63, 165–178. [Google Scholar] [CrossRef]
- Voog, J.; Jones, D.L. Stem cells and the niche: A dynamic duo. Cell Stem Cell 2010, 6, 103–115. [Google Scholar] [CrossRef] [PubMed]
- Augustin, H.G.; Koh, G.Y. Organotypic vasculature: From descriptive heterogeneity to functional pathophysiology. Science 2017, 357, eaal2379. [Google Scholar] [CrossRef]
- Crisan, M.; Corselli, M.; Chen, C.-W.; Péault, B. Multilineage stem cells in the adult: A perivascular legacy? Organogenesis 2011, 7, 101–104. [Google Scholar] [CrossRef]
- Cui, Z.; Li, C.; Jiang, N.; Zhang, C.; Wang, Y.; Gao, H.; Zhou, Y. Isolation and characterization of minipig perivascular stem cells for bone tissue engineering. Mol. Med. Rep. 2018, 18, 3555–3562. [Google Scholar] [CrossRef]
- Nicolescu, M.I.; Bucur, A.; Dinca, O.; Rusu, M.C.; Popescu, L.M. Telocytes in parotid glands. Anat. Rec. 2012, 295, 378–385. [Google Scholar] [CrossRef]
- El Maadawi, Z.M. A Tale of Two Cells: Telocyte and Stem Cell Unique Relationship. Adv. Exp. Med. Biol. 2016, 913, 359–376. [Google Scholar] [CrossRef] [PubMed]
- Gherghiceanu, M.; Popescu, L.M. Cardiac telocytes—Their junctions and functional implications. Cell Tissue Res. 2012, 348, 265–279. [Google Scholar] [CrossRef] [PubMed]
- Soliman, S.A. Telocytes are major constituents of the angiogenic apparatus. Sci. Rep. 2021, 11, 5775. [Google Scholar] [CrossRef] [PubMed]
- Nicolescu, M.I. Telocytes in Exocrine Glands Stroma. Adv. Exp. Med. Biol. 2016, 913, 163–176. [Google Scholar]
- Smythies, J.; Edelstein, L. Telocytes, exosomes, gap junctions and the cytoskeleton: The makings of a primitive nervous system? Front. Cell. Neurosci. 2014, 7, 278. [Google Scholar] [CrossRef]
- Cretoiu, D.; Vannucchi, M.G.; Bei, Y.; Manetti, M.; Faussone-Pellegrini, M.S.; Ibba-Manneschi, L.; Xiao, J.; Maria Cretoiu, S. Telocytes: New Connecting Devices in the Stromal Space of Organs. In Innovations in Cell Research and Therapy; Loewy, Z., Ed.; IntechOpen: Rijeka, Croatia, 2020; pp. 1–25. [Google Scholar]
- Kucybala, I.; Janas, P.; Ciuk, S.; Cholopiak, W.; Klimek-Piotrowska, W.; Holda, M.K. A comprehensive guide to telocytes and their great potential in cardiovascular system. Bratisl. Lek. Listy 2017, 118, 302–309. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Chen, X.; Qian, M.; Zhang, M.; Zhang, D.; Bai, C.; Wang, Q.; Wang, X. Human lung telocytes could promote the proliferation and angiogenesis of human pulmonary microvascular endothelial cells in vitro. Mol. Cell. Ther. 2014, 2, 3. [Google Scholar] [CrossRef]
- Nishisho, T.; Yukata, K.; Matsui, Y.; Matsuura, T.; Higashino, K.; Suganuma, K.; Nikawa, T.; Yasui, N. Angiogenesis and myogenesis in mouse tibialis anterior muscles during distraction osteogenesis: VEGF, its receptors, and myogenin genes expression. J. Orthop. Res. 2012, 30, 1767–1773. [Google Scholar] [CrossRef]
- Deasy, B.M.; Feduska, J.M.; Payne, T.R.; Li, Y.; Ambrosio, F.; Huard, J. Effect of VEGF on the Regenerative Capacity of Muscle Stem Cells in Dystrophic Skeletal Muscle. Mol. Ther. 2009, 17, 1788–1798. [Google Scholar] [CrossRef]
- Díaz-Flores, L.; Gutiérrez, R.; García, M.P.; Gayoso, S.; Gutiérrez, E.; Díaz-Flores, L.; Carrasco, J.L. Telocytes in the Normal and Pathological Peripheral Nervous System. Int. J. Mol. Sci. 2020, 21, 4320. [Google Scholar] [CrossRef]
- Zhou, Q.; Wei, L.; Zhong, C.; Fu, S.; Bei, Y.; Huică, R.-I.; Wang, F.; Xiao, J. Cardiac telocytes are double positive for CD34/PDGFR-α. J. Cell. Mol. Med. 2015, 19, 2036–2042. [Google Scholar] [CrossRef] [PubMed]
- Suciu, L.C.; Popescu, B.O.; Kostin, S.; Popescu, L.M. Platelet-derived growth factor receptor-β-positive telocytes in skeletal muscle interstitium. J. Cell. Mol. Med. 2012, 16, 701–707. [Google Scholar] [CrossRef] [PubMed]
- Varga, I.; Kyselovič, J.; Danišovič, Ľ.; Gálfiová, P.; Kachlík, D.; Polák, Š.; Klein, M. Recently discovered interstitial cells termed telocytes: Distinguishing cell-biological and histological facts from fictions. Biologia 2019, 74, 195–203. [Google Scholar] [CrossRef]
- Liao, Z.; Chen, Y.; Duan, C.; Zhu, K.; Huang, R.; Zhao, H.; Hintze, M.; Pu, Q.; Yuan, Z.; Lv, L.; et al. Cardiac telocytes inhibit cardiac microvascular endothelial cell apoptosis through exosomal miRNA-21-5p-targeted cdip1 silencing to improve angiogenesis following myocardial infarction. Theranostics 2021, 11, 268–291. [Google Scholar] [CrossRef]
- Ceafalan, L.C.; Popescu, B.O.; Hinescu, M.E. Cellular Players in Skeletal Muscle Regeneration. Biomed. Res. Int. 2014, 2014, 957014. [Google Scholar] [CrossRef]
- Horch, R.E.; Kneser, U.; Polykandriotis, E.; Schmidt, V.J.; Sun, J.; Arkudas, A. Tissue engineering and regenerative medicine -where do we stand? J. Cell. Mol. Med. 2012, 16, 1157–1165. [Google Scholar] [CrossRef]
- Boos, A.M.; Weigand, A.; Brodbeck, R.; Beier, J.P.; Arkudas, A.; Horch, R.E. The potential role of telocytes in Tissue Engineering and Regenerative Medicine. Semin. Cell Dev. Biol. 2016, 55, 70–78. [Google Scholar] [CrossRef]
- Cretoiu, S.M.; Popescu, L.M. Telocytes revisited. Biomol. Concepts 2014, 5, 353–369. [Google Scholar] [CrossRef] [PubMed]
- Alunno, A.; Ibba-Manneschi, L.; Bistoni, O.; Rosa, I.; Caterbi, S.; Gerli, R.; Manetti, M. Telocytes in minor salivary glands of primary Sjögren’s syndrome: Association with the extent of inflammation and ectopic lymphoid neogenesis. J. Cell. Mol. Med. 2015, 19, 1689–1696. [Google Scholar] [CrossRef] [PubMed]
- Shoshkes-Carmel, M.; Wang, Y.J.; Wangensteen, K.J.; Tóth, B.; Kondo, A.; Massasa, E.E.; Itzkovitz, S.; Kaestner, K.H. Subepithelial telocytes are an important source of Wnts that supports intestinal crypts. Nature 2018, 557, 242–246. [Google Scholar] [CrossRef] [PubMed]
- Halpern, K.B.; Massalha, H.; Zwick, R.K.; Moor, A.E.; Castillo-Azofeifa, D.; Rozenberg, M.; Farack, L.; Egozi, A.; Miller, D.R.; Averbukh, I.; et al. Lgr5+ telocytes are a signaling source at the intestinal villus tip. Nat. Commun. 2020, 11, 1936. [Google Scholar] [CrossRef]
- Tata, A.; Kobayashi, Y.; Chow, R.D.; Tran, J.; Desai, A.; Massri, A.J.; McCord, T.J.; Gunn, M.D.; Tata, P.R. Myoepithelial Cells of Submucosal Glands Can Function as Reserve Stem Cells to Regenerate Airways after Injury. Cell Stem Cell 2018, 22, 668–683. [Google Scholar] [CrossRef] [PubMed]
- May, A.J.; Cruz-Pacheco, N.; Emmerson, E.; Gaylord, E.A.; Seidel, K.; Nathan, S.; Muench, M.O.; Klein, O.D.; Knox, S.M. Diverse progenitor cells preserve salivary gland ductal architecture after radiation-induced damage. Development 2018, 145, dev166363. [Google Scholar] [CrossRef] [PubMed]
- Weng, P.L.; Aure, M.H.; Maruyama, T.; Ovitt, C.E. Limited Regeneration of Adult Salivary Glands after Severe Injury Involves Cellular Plasticity. Cell Rep. 2018, 24, 1464–1470.e3. [Google Scholar] [CrossRef] [PubMed]
- Rocchi, C.; Emmerson, E. Mouth-Watering Results: Clinical Need, Current Approaches, and Future Directions for Salivary Gland Regeneration. Trends Mol. Med. 2020, 26, 649–669. [Google Scholar] [CrossRef]
- Ninche, N.; Kwak, M.; Ghazizadeh, S. Diverse epithelial cell populations contribute to the regeneration of secretory units in injured salivary glands. Development 2020, 147, dev192807. [Google Scholar] [CrossRef]
- Aure, M.H.; Konieczny, S.F.; Ovitt, C.E. Salivary gland homeostasis is maintained through acinar cell self-duplication. Dev. Cell 2015, 33, 231–237. [Google Scholar] [CrossRef]
- Aure, M.H.; Arany, S.; Ovitt, C.E. Salivary glands: Stem cells, self-duplication, or both? J. Dent. Res. 2015, 94, 1502–1507. [Google Scholar] [CrossRef]
- Oyelakin, A.; Song, E.A.C.; Min, S.; Bard, J.E.; Kann, J.V.; Horeth, E.; Smalley, K.; Kramer, J.M.; Sinha, S.; Romano, R.A. Transcriptomic and Single-Cell Analysis of the Murine Parotid Gland. J. Dent. Res. 2019, 98, 1539–1547. [Google Scholar] [CrossRef]
- Bullard, T.; Koek, L.; Roztocil, E.; Kingsley, P.D.; Mirels, L.; Ovitt, C.E. Ascl3 expression marks a progenitor population of both acinar and ductal cells in mouse salivary glands. Dev. Biol. 2008, 320, 72–78. [Google Scholar] [CrossRef]
- Rocchi, C.; Barazzuol, L.; Coppes, R.P. The evolving definition of salivary gland stem cells. NPJ Regen. Med. 2021, 6, 4. [Google Scholar] [CrossRef]
- Maimets, M.; Rocchi, C.; Bron, R.; Pringle, S.; Kuipers, J.; Giepmans, B.N.G.; Vries, R.G.J.; Clevers, H.; De Haan, G.; Van Os, R.; et al. Long-Term In Vitro Expansion of Salivary Gland Stem Cells Driven by Wnt Signals. Stem Cell Rep. 2016, 6, 150–162. [Google Scholar] [CrossRef] [PubMed]
- Yi, T.; Lee, S.; Choi, N.; Shin, H.-S.; Kim, J.; Lim, J.-Y. Single Cell Clones Purified from Human Parotid Glands Display Features of Multipotent Epitheliomesenchymal Stem Cells. Sci. Rep. 2016, 6, 36303. [Google Scholar] [CrossRef] [PubMed]
- Knox, S.M.; Lombaert, I.M.A.; Reed, X.; Vitale-Cross, L.; Gutkind, J.S.; Hoffman, M.P. Parasympathetic innervation maintains epithelial progenitor cells during salivary organogenesis. Science 2010, 329, 1645–1647. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).