
Highly Specific L-Type Amino Acid Transporter 1 Inhibition by JPH203 as a Potential Pan-Cancer Treatment

 ,
,  , ,
, ,
Abstract
:1. Introduction
2. Targeted Drugs and Targeting Abnormal Metabolism of Cancer
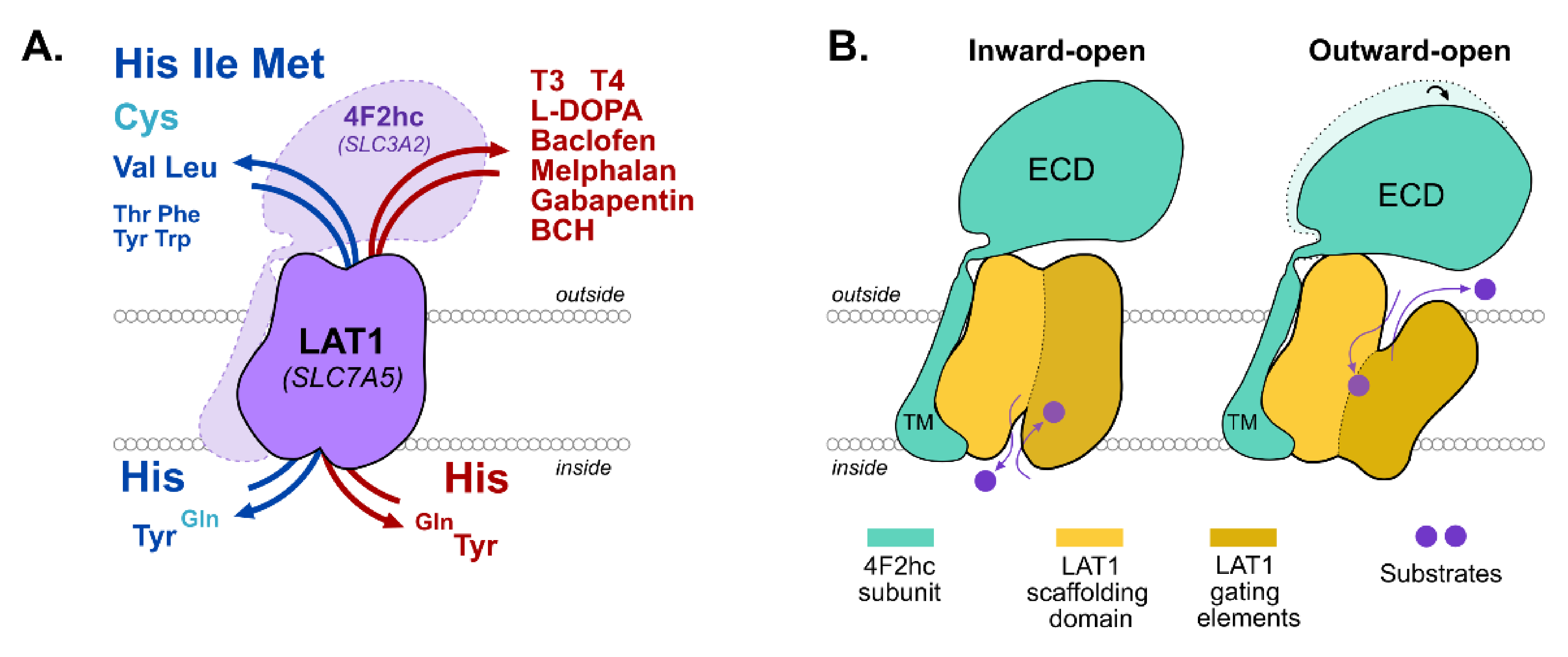
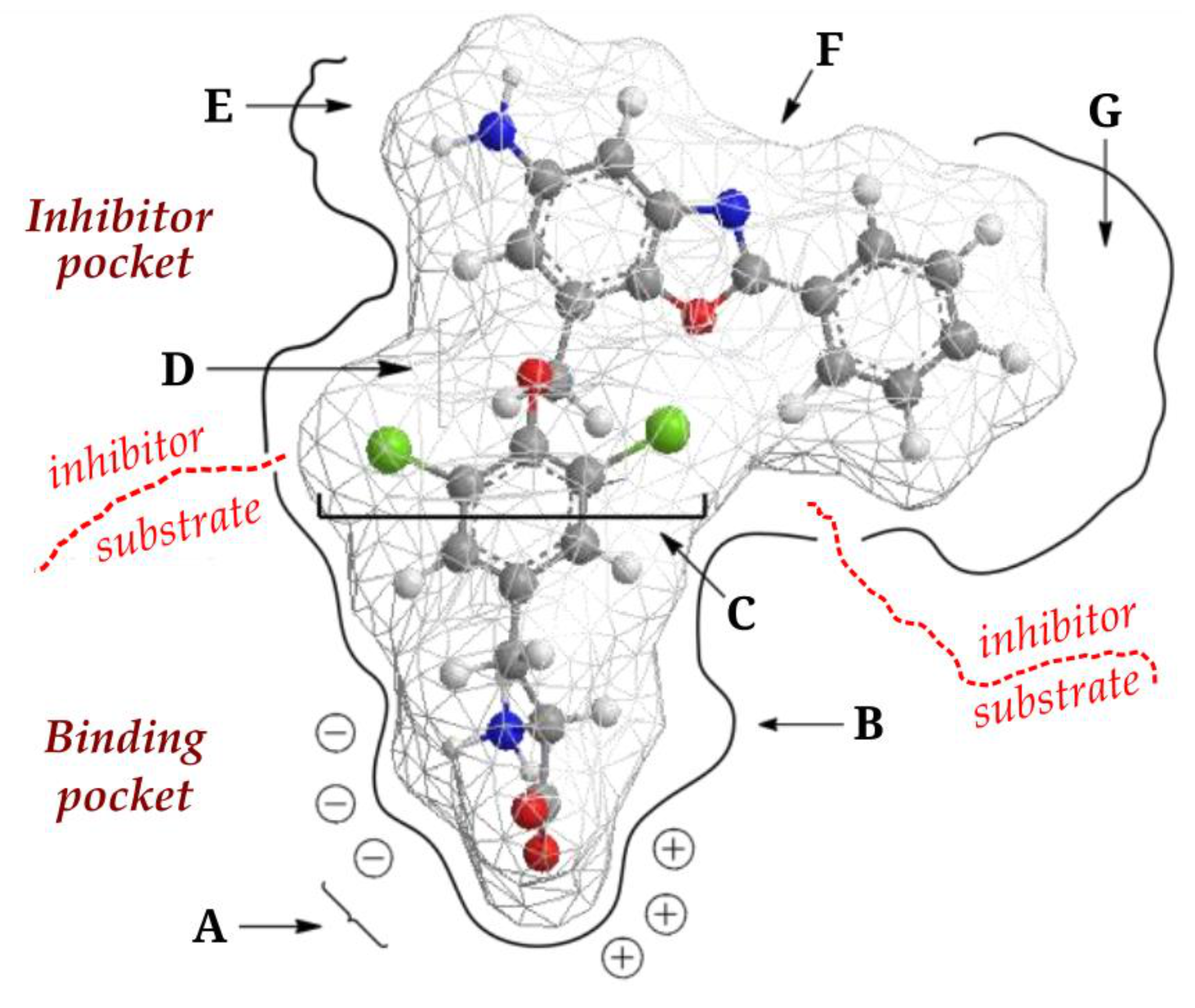
3. LAT1
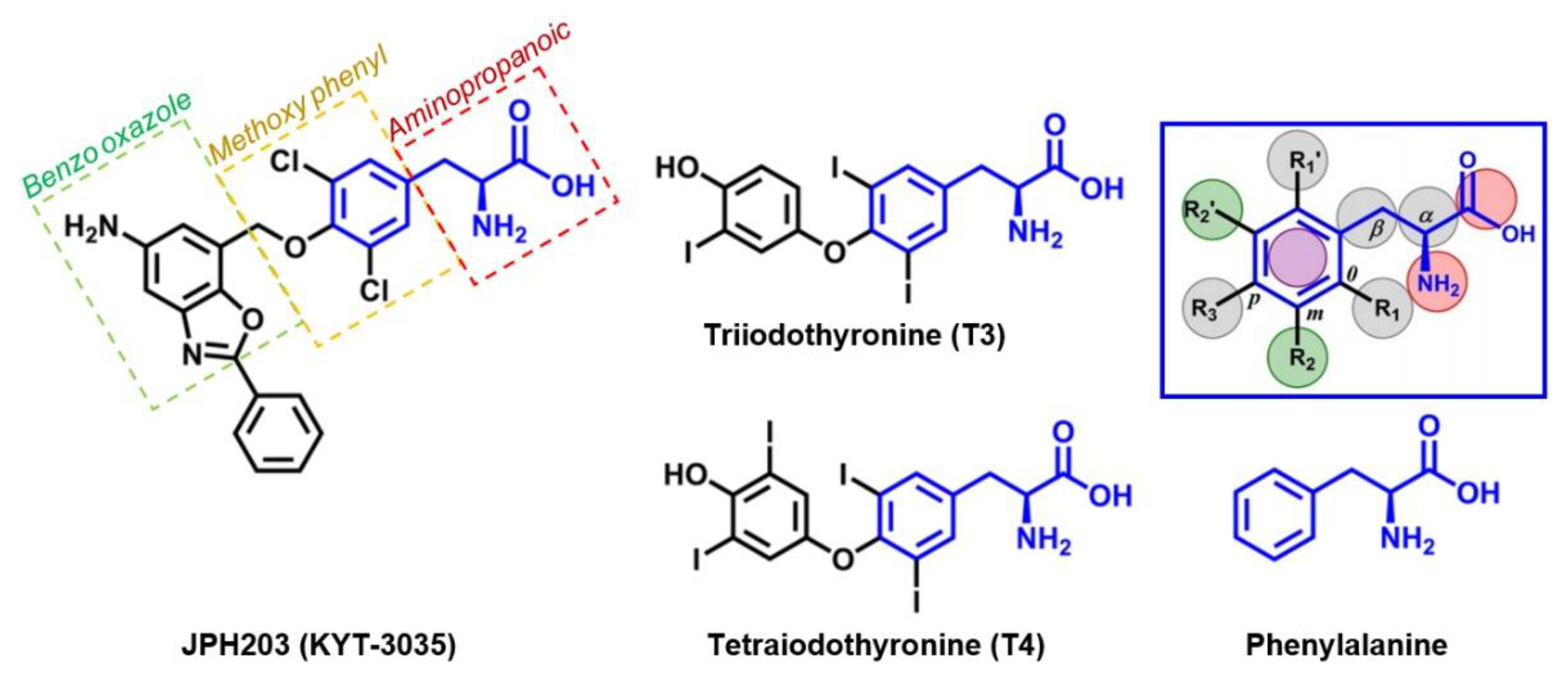
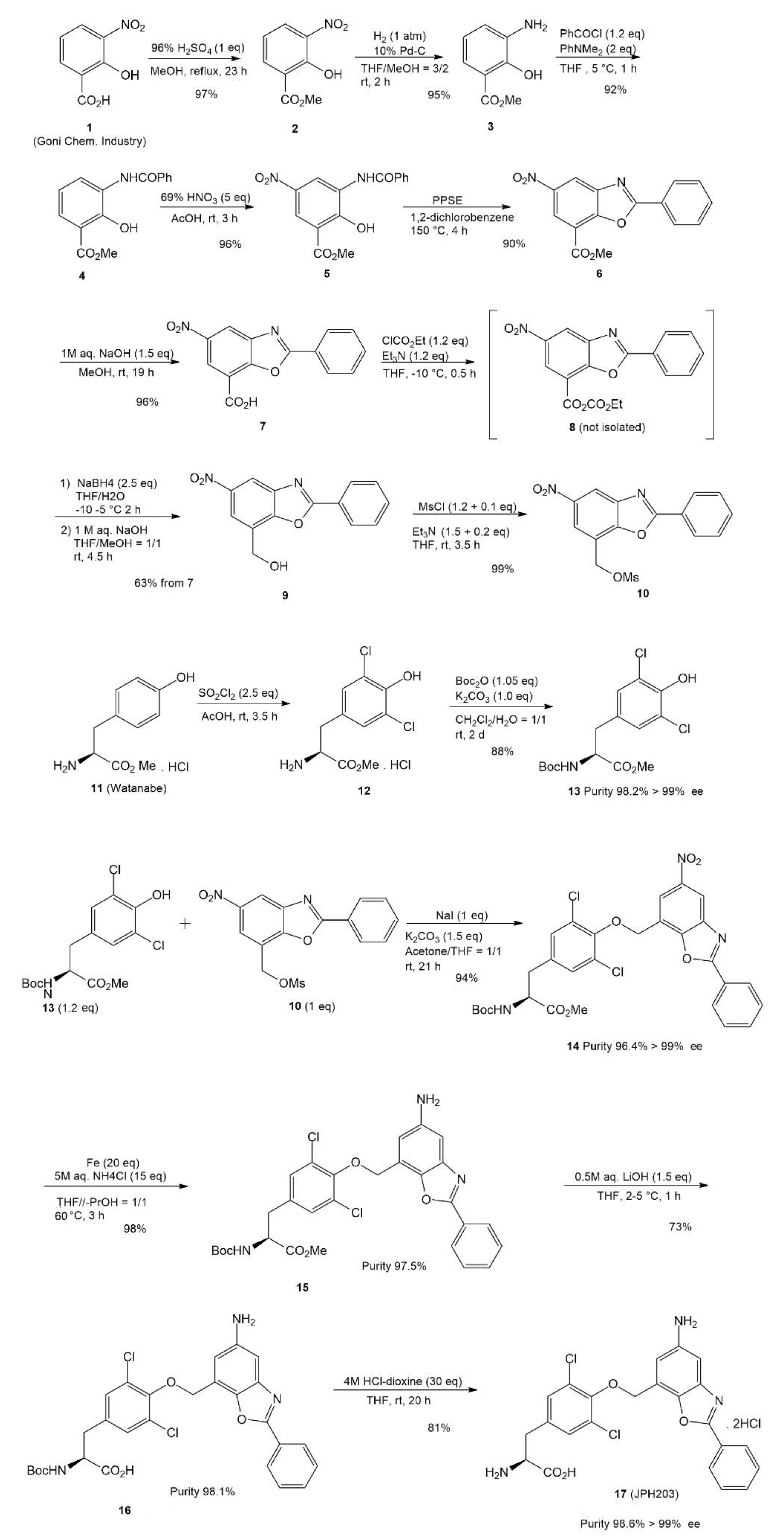
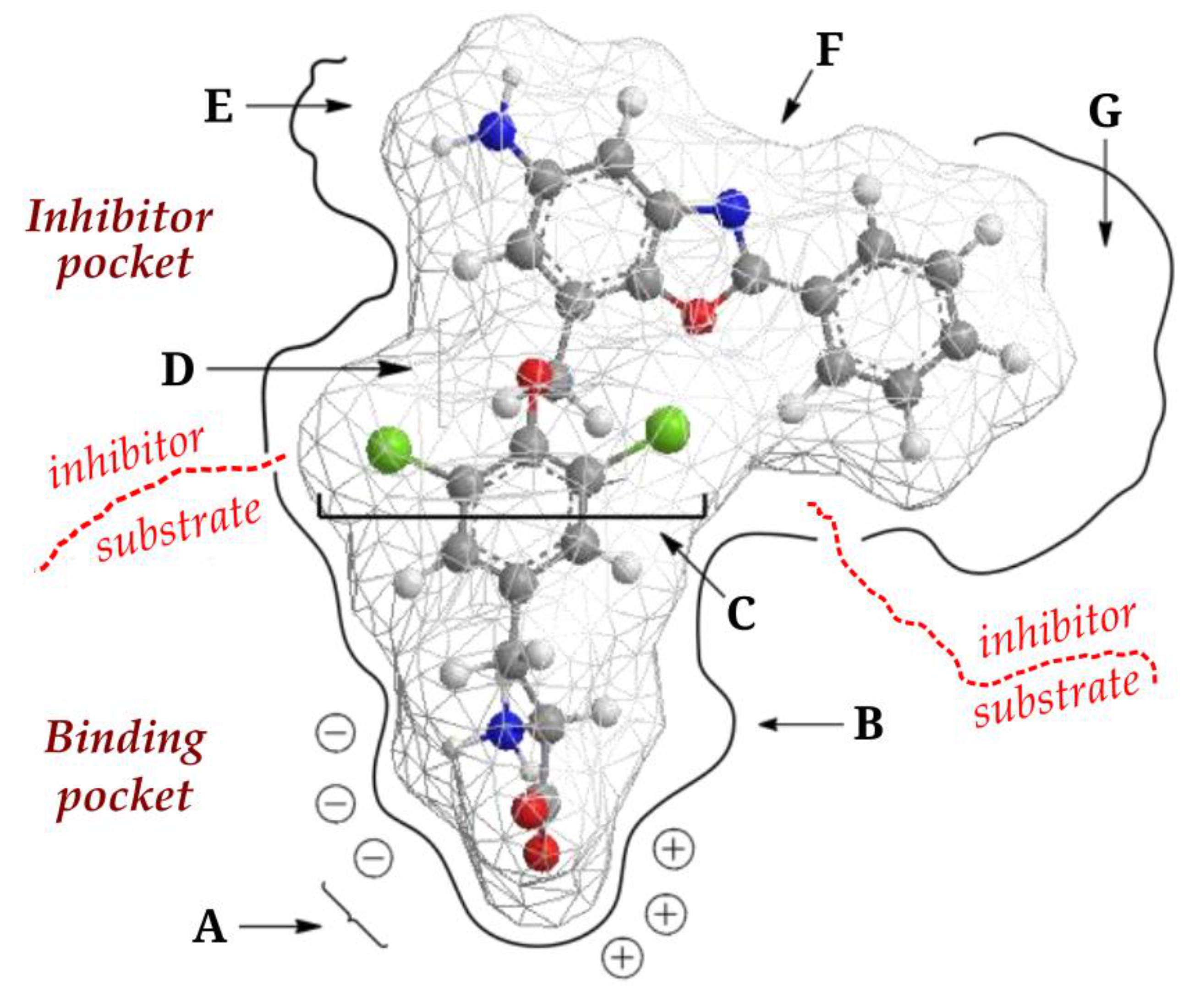
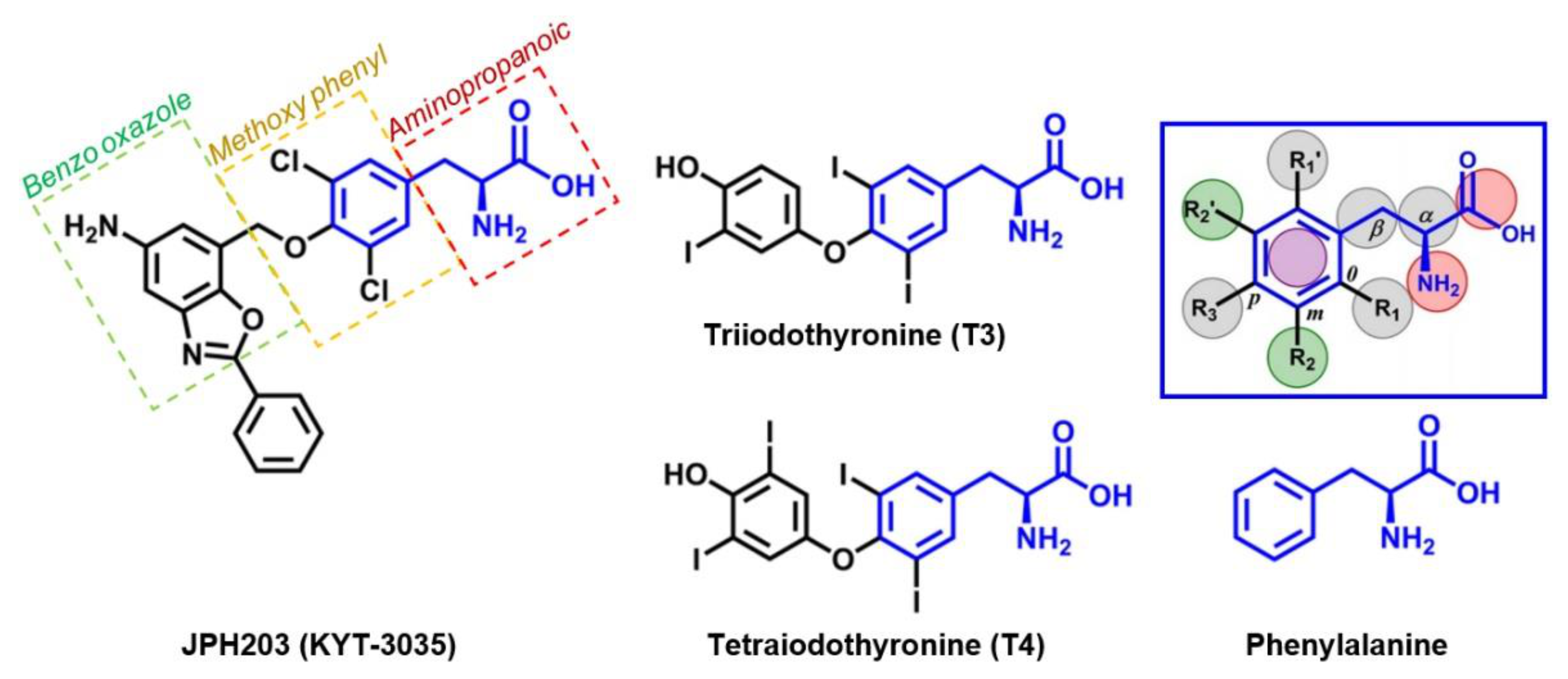
4. JPH203
5. In Vitro and In Vivo JPH203 Studies
- Cell culture in suitable media (37 °C incubation in 95% air and 5% CO2 atmosphere);
- LAT1 and 4F2hc/CD98 protein subunit expression analysis using immunohistochemistry, quantitative polymerase chain reaction (qPCR) or quantitative reverse transcription-polymerase chain reaction (qRT-PCR), and confirmed by a Western blot study;
- Cell viability study;
- l-leucine competitive uptake study; and
- Cell growth inhibition study.
5.1. Oral Cancer

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cancer Type [Ref] | Cell Type | LAT1 Expression | JPH203 Activities |
|---|---|---|---|
| Bladder [27] | Cell T24 and 5637 | LAT1 expression was significantly higher in cancerous tissue than in the surrounding normal tissue (p = 0.0051). LAT1 cell expression 5637 is higher than T24 cells. | Inhibit the proliferation of T24 and 5637 cells with increasing concentration (20 μM). |
| Anaplastic thyroid [51] | 3 human ATC cells: cell 8505C, OCUT-2, OCUT-6 | LAT1 immunoreactivity was detected in anaplastic thyroid cancer tissue (78%: 11/14 cases) | Inhibit 87.0% in 8505C cells, 78.6% in OCUT-2 cells, and 75.0% in OCUT-6 cells. |
| Differentiated thyroid cancer [52] | Papillary thyroid cancer cells: K1, KTC, TPC-1; ATC cells: Hth104, SW1736, 8505C | LAT1 was expressed in 6 thyroid cancer cells tested. | Block LAT1 and reduce the proliferation of 5 of 6 thyroid cancer cells (relative IC50 from 1.3 μM to 6.8 μM). PTC cells are less sensitive than K1 (16.9 μM). |
| Renal cells carcinoma [27] | Caki-1 cells and ACHN | LAT1 expression of 97.8% (90/92 cases) | Reduce cell viability by IC50 values of Caki-1 cells and ACHN of 2.5 and 2.7 μM, respectively. |
| Medulloblastoma (MB) [53] | Medulloblastoma cells are independent of subgroup 3 (HD-MB03) and Shh (DAOY) | LAT1 expression was significantly higher in cancerous tissue than in adjacent normal tissue. | Interfere with amino acid homeostasis, mTORC1 activity, proliferation, and survival of medulloblastoma cells. |
| Stomach [54] | MKN1 and MKN45 | LAT1 was expressed in MKN1 and MKN45 cells. | Reduce cancer cells growth (IC50 41.7 ± 2.3 µM in MKN1 cells and 4.6 ± 1.0 µM in MKN45 cells). |
| Colorectal [54] | LoVo and HT-29 | LAT1 was expressed in LoVo and HT29 cells. | Reduce cancer cells growth (IC50 2.3 ± 0.3 µM on LoVo and 30.0 ± 6.4 µM on HT29 cells) |
| Colorectal [47] | HT-29 | LAT1 was expressed in HT-29 cells. | Inhibit 14C-leucine uptake and cell growth (IC50 0.06 µM and 4.1 µM, respectively). |
| Bone (osteosarcoma) [55] | Human osteosarcoma cells Saos2 and human osteoblastic cells | LAT1 was detected and weakly expressed in Saos2 and FOB cells. | Antiproliferative effects (on Saos2 cells, IC50 1st day 4.09 ± 0.53 μM and 4th day 0.09 ± 0.01 μM; on FOB cells, IC50 1st day 24.1 ± 4.1 μM and 4th day 2.8 ± 0.3 μM. |
| Biliary Duct (cholangiocarcinoma) [56] | KKU-055, KKU-213, and KKU-100. | LAT1 was detected in all cells studied and was the main transporter of cholangiocarcinoma cells | IC50 values (mean ± SD) for leucine uptake inhibition: 0.20 ± 0.03 µM for KKU-055, 0.12 ± 0.02 µM for KKU-213 cells, and 0.25 ± 0.04 µM for KKU-100. IC50 values for cell growth inhibition on day 1 for KKU-055 cells, KKU-213, KKU-100, respectively, 31.95 ± 1.15 µM, 32.95 ± 1.16 µM, 48.74 ± 1.22 µM, and for the day 3 were 5.78 ± 1.15 µM, 2.47 ± 1.19 µM, 3.00 ± 1.28 µM. |
| Oral [50] | YD-38 and NHOKs | YD-38 cells express LAT1 but do not express LAT2. NHOKs cells express LAT1 and LAT2, with very weak LAT1 expression. | Inhibit l-leucine in YD-38 cells (IC50 value: 0.79 µM) and NHOK (IC50 value: > 100 µM). However, it is not enough to suppress the growth of YD-38 cells (IC50 value: 69 µM). |
| Cancer Type [Ref] | Tumor Model | LAT1 Expression | JPH203 Activities |
|---|---|---|---|
| Anaplastic Thyroid [51] | Mice xenograft of 8505C cell line with BRAF, PI3K3R1/2, and p53 mutations. | Excessive expression of LAT1 in human ATC (78%: 11/14 cases of ATC) | Reduce the growth ratio of xenograft tumors and also reduce tumor size. |
| Biliary duct (cholangiocarcinoma) [56] | KKA-213 CCA cell xenograft | - | On days 18 and 21, JPH203 inhibited dose-related tumor growth in the JPH203 group 12.5 mg/kg (on day 18, p < 0.05, day 21, p < 0.01) and 25 mg/kg (on days 18 and 21, p < 0.001) compared to the control group. |
| Colorectal [47] | HT-29 cell xenograft | - | Inhibit 14C-leucine absorption and cell growth (IC50 0.14 μM and 16.4 μM). |
5.2. Gastric and Colorectal Cancer
5.3. Anaplastic Thyroid Cancer
5.4. Osteosarcoma
5.5. Medulloblastoma
5.6. Renal Cell Carcinoma
5.7. Bladder Carcinoma
5.8. Biliary Duct Cancer
6. JPH203 in Phase I Clinical Trial
7. Insights for Radiotheranostic Purpose
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fitzmaurice, C.; Abate, D.; Abbasi, N.; Abbastabar, H.; Abd-Allah, F.; Abdel-Rahman, O.; Abdelalim, A.; Abdoli, A.; Abdollahpour, I.; Abdulle, A.S. Global, regional, and national cancer incidence, mortality, years of life lost, years lived with disability, and disability-adjusted life-years for 29 cancer groups, 1990 to 2017: A systematic analysis for the global burden of disease study. JAMA Oncol. 2019, 5, 1749–1768. [Google Scholar] [PubMed] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Chabner, B.A.; Roberts, T.G. Chemotherapy and the war on cancer. Nat. Rev. Cancer 2005, 5, 65–72. [Google Scholar] [CrossRef] [PubMed]
- Schirrmacher, V. From chemotherapy to biological therapy: A review of novel concepts to reduce the side effects of systemic cancer treatment. Int. J. Oncol. 2019, 54, 407–419. [Google Scholar] [PubMed]
- Lee, Y.T.; Tan, Y.J.; Oon, C.E. Molecular targeted therapy: Treating cancer with specificity. Eur. J. Pharmacol. 2018, 834, 188–196. [Google Scholar] [CrossRef] [PubMed]
- Falzone, L.; Salomone, S.; Libra, M. Evolution of Cancer Pharmacological Treatments at the Turn of the Third Millennium. Front. Pharmacol. 2018, 9, 1300. [Google Scholar] [CrossRef] [Green Version]
- Martinez-Outschoorn, U.E.; Peiris-Pages, M.; Pestell, R.G.; Sotgia, F.; Lisanti, M.P. Cancer metabolism: A therapeutic perspective. Nat. Rev. Clin. Oncol. 2017, 14, 11. [Google Scholar] [CrossRef]
- Wei, Z.; Liu, X.; Cheng, C.; Yu, W.; Yi, P. Metabolism of Amino Acids in Cancer. Front. Cell Dev. Biol. 2020, 8, 1628. [Google Scholar]
- Scalise, M.; Console, L.; Rovella, F.; Galluccio, M.; Pochini, L.; Indiveri, C. Membrane Transporters for Amino Acids as Players of Cancer Metabolic Rewiring. Cells 2020, 9, 2028. [Google Scholar] [CrossRef]
- Häfliger, P.; Charles, R.-P. The L-Type Amino Acid Transporter LAT1—An Emerging Target in Cancer. Int. J. Mol. Sci. 2019, 20, 2428. [Google Scholar] [CrossRef] [Green Version]
- Lopes, C.; Pereira, C.; Medeiros, R. ASCT2 and LAT1 Contribution to the Hallmarks of Cancer: From a Molecular Perspective to Clinical Translation. Cancers 2021, 13, 203. [Google Scholar] [CrossRef]
- Okano, N.; Naruge, D.; Kawai, K.; Kobayashi, T.; Nagashima, F.; Endou, H.; Furuse, J. First-in-human phase I study of JPH203, an L-type amino acid transporter 1 inhibitor, in patients with advanced solid tumors. Investig. New Drugs 2020, 38, 1495–1506. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Sun, Z.; Lu, L. Targeted Engineering of Medicinal Chemistry for Cancer Therapy: Recent Advances and Perspectives. Angew. Chem. Int. Ed. 2021, 60, 5626–5643. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.F.A.P.; Sadelain, M. The Journey from Discoveries in Fundamental Immunology to Cancer Immunotherapy. Cancer Cell 2015, 27, 439–449. [Google Scholar] [CrossRef] [Green Version]
- Sharma, P.; Hu-Lieskovan, S.; Wargo, J.A.; Ribas, A. Primary, Adaptive, and Acquired Resistance to Cancer Immunotherapy. Cell 2017, 168, 707–723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boumahdi, S.; de Sauvage, F.J. The great escape: Tumour cell plasticity in resistance to targeted therapy. Nat. Rev. Drug Discov. 2020, 19, 39–56. [Google Scholar] [CrossRef]
- Pavlova, N.N.; Thompson, C.B. The Emerging Hallmarks of Cancer Metabolism. Cell Metab. 2016, 23, 27–47. [Google Scholar] [CrossRef] [Green Version]
- Kandasamy, P.; Gyimesi, G.; Kanai, Y.; Hediger, M.A. Amino acid transporters revisited: New views in health and disease. Trends Biochem. Sci. 2018, 43, 752–789. [Google Scholar] [CrossRef]
- Hosios, A.M.; Hecht, V.C.; Danai, L.V.; Johnson, M.O.; Rathmell, J.C.; Steinhauser, M.L.; Manalis, S.R.; Vander Heiden, M.G. Amino Acids Rather than Glucose Account for the Majority of Cell Mass in Proliferating Mammalian Cells. Dev. Cell 2016, 36, 540–549. [Google Scholar] [CrossRef] [Green Version]
- Scalise, M.; Galluccio, M.; Console, L.; Pochini, L.; Indiveri, C. The Human SLC7A5 (LAT1): The Intriguing Histidine/Large Neutral Amino Acid Transporter and Its Relevance to Human Health. Front. Chem. 2018, 6, 243. [Google Scholar] [CrossRef] [PubMed]
- Bhutia, Y.D.; Babu, E.; Ramachandran, S.; Ganapathy, V. Amino acid transporters in cancer and their relevance to “glutamine addiction”: Novel targets for the design of a new class of anticancer drugs. Cancer Res. 2015, 75, 1782–1788. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, N.; Ecker, G.F. Insights into the structure, function, and ligand discovery of the large neutral amino acid transporter 1, LAT1. Int. J. Mol. Sci. 2018, 19, 1278. [Google Scholar] [CrossRef] [Green Version]
- Yan, R.; Zhao, X.; Lei, J.; Zhou, Q. Structure of the human LAT1–4F2hc heteromeric amino acid transporter complex. Nature 2019, 568, 127–130. [Google Scholar] [CrossRef]
- Furuya, M.; Horiguchi, J.; Nakajima, H.; Kanai, Y.; Oyama, T. Correlation of L-type amino acid transporter 1 and CD98 expression with triple negative breast cancer prognosis. Cancer Sci. 2012, 103, 382–389. [Google Scholar] [CrossRef] [PubMed]
- Haining, Z.; Kawai, N.; Miyake, K.; Okada, M.; Okubo, S.; Zhang, X.; Fei, Z.; Tamiya, T. Relation of LAT1/4F2hc expression with pathological grade, proliferation and angiogenesis in human gliomas. BMC Clin. Pathol. 2012, 12, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Higuchi, K.; Sakamoto, S.; Ando, K.; Maimaiti, M.; Takeshita, N.; Okunushi, K.; Reien, Y.; Imamura, Y.; Sazuka, T.; Nakamura, K. Characterization of the expression of LAT1 as a prognostic indicator and a therapeutic target in renal cell carcinoma. Sci. Rep. 2019, 9, 1–10. [Google Scholar] [CrossRef]
- Honjo, H.; Kaira, K.; Miyazaki, T.; Yokobori, T.; Kanai, Y.; Nagamori, S.; Oyama, T.; Asao, T.; Kuwano, H. Clinicopathological significance of LAT1 and ASCT2 in patients with surgically resected esophageal squamous cell carcinoma. J. Surg. Oncol. 2016, 113, 381–389. [Google Scholar] [CrossRef] [PubMed]
- Ishihara, S.; Horiguchi, A.; Miyakawa, S.; Endo, I.; Miyazaki, M.; Takada, T. Biliary tract cancer registry in Japan from 2008 to 2013. J. Hepatobiliary Pancreat. Sci. 2016, 23, 149–157. [Google Scholar] [CrossRef]
- Kaira, K.; Nakamura, K.; Hirakawa, T.; Imai, H.; Tominaga, H.; Oriuchi, N.; Nagamori, S.; Kanai, Y.; Tsukamoto, N.; Oyama, T.; et al. Prognostic significance of L-type amino acid transporter 1 (LAT1) expression in patients with ovarian tumors. Am. J. Transl. Res. 2015, 7, 1161–1171. [Google Scholar]
- Kaira, K.; Oriuchi, N.; Imai, H.; Shimizu, K.; Yanagitani, N.; Sunaga, N.; Hisada, T.; Ishizuka, T.; Kanai, Y.; Nakajima, T. Prognostic significance of L-type amino acid transporter 1 (LAT1) and 4F2 heavy chain (CD98) expression in stage I pulmonary adenocarcinoma. Lung Cancer 2009, 66, 120–126. [Google Scholar] [CrossRef]
- Kaira, K.; Sunose, Y.; Arakawa, K.; Ogawa, T.; Sunaga, N.; Shimizu, K.; Tominaga, H.; Oriuchi, N.; Itoh, H.; Nagamori, S.; et al. Prognostic significance of L-type amino-acid transporter 1 expression in surgically resected pancreatic cancer. Br. J. Cancer 2012, 107, 632–638. [Google Scholar] [CrossRef]
- Kaira, K.; Sunose, Y.; Ohshima, Y.; Ishioka, N.S.; Arakawa, K.; Ogawa, T.; Sunaga, N.; Shimizu, K.; Tominaga, H.; Oriuchi, N. Clinical significance of L-type amino acid transporter 1 expression as a prognostic marker and potential of new targeting therapy in biliary tract cancer. BMC Cancer 2013, 13, 482. [Google Scholar] [CrossRef] [Green Version]
- Namikawa, M.; Kakizaki, S.; Kaira, K.; Tojima, H.; Yamazaki, Y.; Horiguchi, N.; Sato, K.; Oriuchi, N.; Tominaga, H.; Sunose, Y. Expression of amino acid transporters (LAT1, ASCT2 and xCT) as clinical significance in hepatocellular carcinoma. Hepatol. Res. 2015, 45, 1014–1022. [Google Scholar] [CrossRef] [PubMed]
- Nikkuni, O.; Kaira, K.; Toyoda, M.; Shino, M.; Sakakura, K.; Takahashi, K.; Tominaga, H.; Oriuchi, N.; Suzuki, M.; Iijima, M.; et al. Expression of Amino Acid Transporters (LAT1 and ASCT2) in Patients with Stage III/IV Laryngeal Squamous Cell Carcinoma. Pathol. Oncol. Res. 2015, 21, 1175–1181. [Google Scholar] [CrossRef] [PubMed]
- Toyoda, M.; Kaira, K.; Ohshima, Y.; Ishioka, N.S.; Shino, M.; Sakakura, K.; Takayasu, Y.; Takahashi, K.; Tominaga, H.; Oriuchi, N.; et al. Prognostic significance of amino-acid transporter expression (LAT1, ASCT2, and xCT) in surgically resected tongue cancer. Br. J. Cancer 2014, 110, 2506–2513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ichinoe, M.; Mikami, T.; Yoshida, T.; Igawa, I.; Tsuruta, T.; Nakada, N.; Anzai, N.; Suzuki, Y.; Endou, H.; Okayasu, I. High expression of L-type amino-acid transporter 1 (LAT1) in gastric carcinomas: Comparison with non-cancerous lesions. Pathol. Int. 2011, 61, 281–289. [Google Scholar] [CrossRef] [PubMed]
- Achmad, A.; Bhattarai, A.; Yudistiro, R.; Heryanto, Y.D.; Higuchi, T.; Tsushima, Y. The diagnostic performance of 18F-FAMT PET and 18F-FDG PET for malignancy detection: A meta-analysis. BMC Med. Imaging 2017, 17, 66. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.; Achmad, A.; Higuchi, T.; Arisaka, Y.; Yokoo, H.; Yokoo, S.; Tsushima, Y. Effects of intratumoral inflammatory process on 18F-FDG uptake: Pathologic and comparative study with 18F-fluoro-alpha-methyltyrosine PET/CT in oral squamous cell carcinoma. J. Nucl. Med. 2015, 56, 16–21. [Google Scholar] [CrossRef] [Green Version]
- Wei, L.; Tominaga, H.; Ohgaki, R.; Wiriyasermkul, P.; Hagiwara, K.; Okuda, S.; Kaira, K.; Oriuchi, N.; Nagamori, S.; Kanai, Y. Specific transport of 3-fluoro-l-alpha-methyl-tyrosine by LAT1 explains its specificity to malignant tumors in imaging. Cancer Sci. 2016, 107, 347–352. [Google Scholar] [CrossRef] [PubMed]
- Wempe, M.F.; Rice, P.J.; Lightner, J.W.; Jutabha, P.; Hayashi, M.; Anzai, N.; Wakui, S.; Kusuhara, H.; Sugiyama, Y.; Endou, H. Metabolism and pharmacokinetic studies of JPH203, an L-amino acid transporter 1 (LAT1) selective compound. Drug Metab. Pharmacokinet. 2012, 27, 155–161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Endou, H.; Kanai, Y.; Tsujihara, K.; Saito, K. Aromatic Amino Acid Derivatives and Medicinal Compositions. U.S. Patent 7345086B2, 3 March 2008. [Google Scholar]
- Kongpracha, P.; Nagamori, S.; Wiriyasermkul, P.; Tanaka, Y.; Kaneda, K.; Okuda, S.; Ohgaki, R.; Kanai, Y. Structure-activity relationship of a novel series of inhibitors for cancer type transporter L-type amino acid transporter 1 (LAT1). J. Pharmacol. Sci. 2017, 133, 96–102. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.; Villoutreix, B.O.; Ecker, G.F. Rigorous sampling of docking poses unveils binding hypothesis for the halogenated ligands of L-type Amino acid Transporter 1 (LAT1). Sci. Rep. 2019, 9, 15061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uchino, H.; Kanai, Y.; Kim, D.K.; Wempe, M.F.; Chairoungdua, A.; Morimoto, E.; Anders, M.; Endou, H. Transport of amino acid-related compounds mediated by L-type amino acid transporter 1 (LAT1): Insights into the mechanisms of substrate recognition. Mol. Pharmacol. 2002, 61, 729–737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wempe, M.F.; Jutabha, P.; Kumar, V.; Fisher, J.A.; Waers, K.; Holt, M.D.; Dodson, A.M.; Bautista, J.; Gehr, D.T.; Backos, D.S. Developing selective L-Amino Acid Transport 1 (LAT1) inhibitors: A structure-activity relationship overview. Med. Res. Arch. 2019, 7. [Google Scholar] [CrossRef]
- Oda, K.; Hosoda, N.; Endo, H.; Saito, K.; Tsujihara, K.; Yamamura, M.; Sakata, T.; Anzai, N.; Wempe, M.F.; Kanai, Y.; et al. L-type amino acid transporter 1 inhibitors inhibit tumor cell growth. Cancer Sci. 2010, 101, 173–179. [Google Scholar] [CrossRef]
- Okanishi, H.; Ohgaki, R.; Okuda, S.; Endou, H.; Kanai, Y. Proteomics and phosphoproteomics reveal key regulators associated with cytostatic effect of amino acid transporter LAT1 inhibitor. Cancer Sci. 2021, 112, 871–883. [Google Scholar] [CrossRef]
- Singh, N.; Scalise, M.; Galluccio, M.; Wieder, M.; Seidel, T.; Langer, T.; Indiveri, C.; Ecker, G.F. Discovery of potent inhibitors for the large neutral amino acid transporter 1 (LAT1) by structure-based methods. Int. J. Mol. Sci. 2019, 20, 27. [Google Scholar] [CrossRef] [Green Version]
- Yun, D.-W.; Lee, S.A.; Park, M.-G.; Kim, J.-S.; Yu, S.-K.; Park, M.-R.; Kim, S.-G.; Oh, J.-S.; Kim, C.S.; Kim, H.-J. JPH203, an L-type amino acid transporter 1–selective compound, induces apoptosis of YD-38 human Oral Cancer cells. J. Pharmacol. Sci. 2014, 13154FP. [Google Scholar] [CrossRef] [Green Version]
- Enomoto, K.; Sato, F.; Tamagawa, S.; Gunduz, M.; Onoda, N.; Uchino, S.; Muragaki, Y.; Hotomi, M. A novel therapeutic approach for anaplastic thyroid cancer through inhibition of LAT1. Sci. Rep. 2019, 9, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Hafliger, P.; Graff, J.; Rubin, M.; Stooss, A.; Dettmer, M.S.; Altmann, K.H.; Gertsch, J.; Charles, R.P. The LAT1 inhibitor JPH203 reduces growth of thyroid carcinoma in a fully immunocompetent mouse model. J. Exp. Clin. Cancer Res. 2018, 37, 234. [Google Scholar] [CrossRef]
- Cormerais, Y.; Pagnuzzi-Boncompagni, M.; Schrotter, S.; Giuliano, S.; Tambutte, E.; Endou, H.; Wempe, M.F.; Pages, G.; Pouyssegur, J.; Picco, V. Inhibition of the amino-acid transporter LAT1 demonstrates anti-neoplastic activity in medulloblastoma. J. Cell Mol. Med. 2019, 23, 2711–2718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muto, Y.; Furihata, T.; Kaneko, M.; Higuchi, K.; Okunushi, K.; Morio, H.; Reien, Y.; Uesato, M.; Matsubara, H.; Anzai, N. Different Response Profiles of Gastrointestinal Cancer Cells to an L-Type Amino Acid Transporter Inhibitor, JPH203. Anticancer Res. 2019, 39, 159–165. [Google Scholar] [CrossRef] [PubMed]
- Choi, D.W.; Kim, D.K.; Kanai, Y.; Wempe, M.F.; Endou, H.; Kim, J.K. JPH203, a selective L-type amino acid transporter 1 inhibitor, induces mitochondria-dependent apoptosis in Saos2 human osteosarcoma cells. Korean J. Physiol. Pharmacol. 2017, 21, 599–607. [Google Scholar] [CrossRef] [Green Version]
- Yothaisong, S.; Dokduang, H.; Anzai, N.; Hayashi, K.; Namwat, N.; Yongvanit, P.; Sangkhamanon, S.; Jutabha, P.; Endou, H.; Loilome, W. Inhibition of l-type amino acid transporter 1 activity as a new therapeutic target for cholangiocarcinoma treatment. Tumor Biol. 2017, 39, 1010428317694545. [Google Scholar] [CrossRef] [Green Version]
- Maimaiti, M.; Sakamoto, S.; Yamada, Y.; Sugiura, M.; Rii, J.; Takeuchi, N.; Imamura, Y.; Furihata, T.; Ando, K.; Higuchi, K. Expression of L-type amino acid transporter 1 as a molecular target for prognostic and therapeutic indicators in bladder carcinoma. Sci. Rep. 2020, 10, 1–14. [Google Scholar] [CrossRef]
- Yothaisong, S.; Namwat, N.; Yongvanit, P.; Khuntikeo, N.; Puapairoj, A.; Jutabha, P.; Anzai, N.; Tassaneeyakul, W.; Tangsucharit, P.; Loilome, W. Increase in L-type amino acid transporter 1 expression during cholangiocarcinogenesis caused by liver fluke infection and its prognostic significance. Parasitol. Int. 2017, 66, 471–478. [Google Scholar] [CrossRef]
- Yanagisawa, N.; Hana, K.; Nakada, N.; Ichinoe, M.; Koizumi, W.; Endou, H.; Okayasu, I.; Murakumo, Y. High expression of L-type amino acid transporter 1 as a prognostic marker in bile duct adenocarcinomas. Cancer Med. 2014, 3, 1246–1255. [Google Scholar] [CrossRef] [PubMed]
- Sakata, T.; Ferdous, G.; Tsuruta, T.; Satoh, T.; Baba, S.; Muto, T.; Ueno, A.; Kanai, Y.; Endou, H.; Okayasu, I. L-type amino-acid transporter 1 as a novel biomarker for high-grade malignancy in prostate cancer. Pathol. Int. 2009, 59, 7–18. [Google Scholar] [CrossRef]
- Janpipatkul, K.; Suksen, K.; Borwornpinyo, S.; Jearawiriyapaisarn, N.; Hongeng, S.; Piyachaturawat, P.; Chairoungdua, A. Downregulation of LAT1 expression suppresses cholangiocarcinoma cell invasion and migration. Cell Signal. 2014, 26, 1668–1679. [Google Scholar] [CrossRef] [PubMed]
- Mikulová, M.B.; Mikuš, P. Advances in Development of Radiometal Labeled Amino Acid-Based Compounds for Cancer Imaging and Diagnostics. Pharmaceuticals 2021, 14, 167. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Achmad, A.; Lestari, S.; Holik, H.A.; Rahayu, D.; Bashari, M.H.; Faried, A.; Kartamihardja, A.H.S. Highly Specific L-Type Amino Acid Transporter 1 Inhibition by JPH203 as a Potential Pan-Cancer Treatment. Processes 2021, 9, 1170. https://doi.org/10.3390/pr9071170
Achmad A, Lestari S, Holik HA, Rahayu D, Bashari MH, Faried A, Kartamihardja AHS. Highly Specific L-Type Amino Acid Transporter 1 Inhibition by JPH203 as a Potential Pan-Cancer Treatment. Processes. 2021; 9(7):1170. https://doi.org/10.3390/pr9071170
Chicago/Turabian StyleAchmad, Arifudin, Shinta Lestari, Holis Abdul Holik, Driyanti Rahayu, Muhammad Hasan Bashari, Ahmad Faried, and Achmad Hussein Sundawa Kartamihardja. 2021. "Highly Specific L-Type Amino Acid Transporter 1 Inhibition by JPH203 as a Potential Pan-Cancer Treatment" Processes 9, no. 7: 1170. https://doi.org/10.3390/pr9071170
APA StyleAchmad, A., Lestari, S., Holik, H. A., Rahayu, D., Bashari, M. H., Faried, A., & Kartamihardja, A. H. S. (2021). Highly Specific L-Type Amino Acid Transporter 1 Inhibition by JPH203 as a Potential Pan-Cancer Treatment. Processes, 9(7), 1170. https://doi.org/10.3390/pr9071170