Predicting Host Immune Cell Dynamics and Key Disease-Associated Genes Using Tissue Transcriptional Profiles



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. Microarray Analysis of Mouse Lung Tissue
2.3. Flow Cytometry
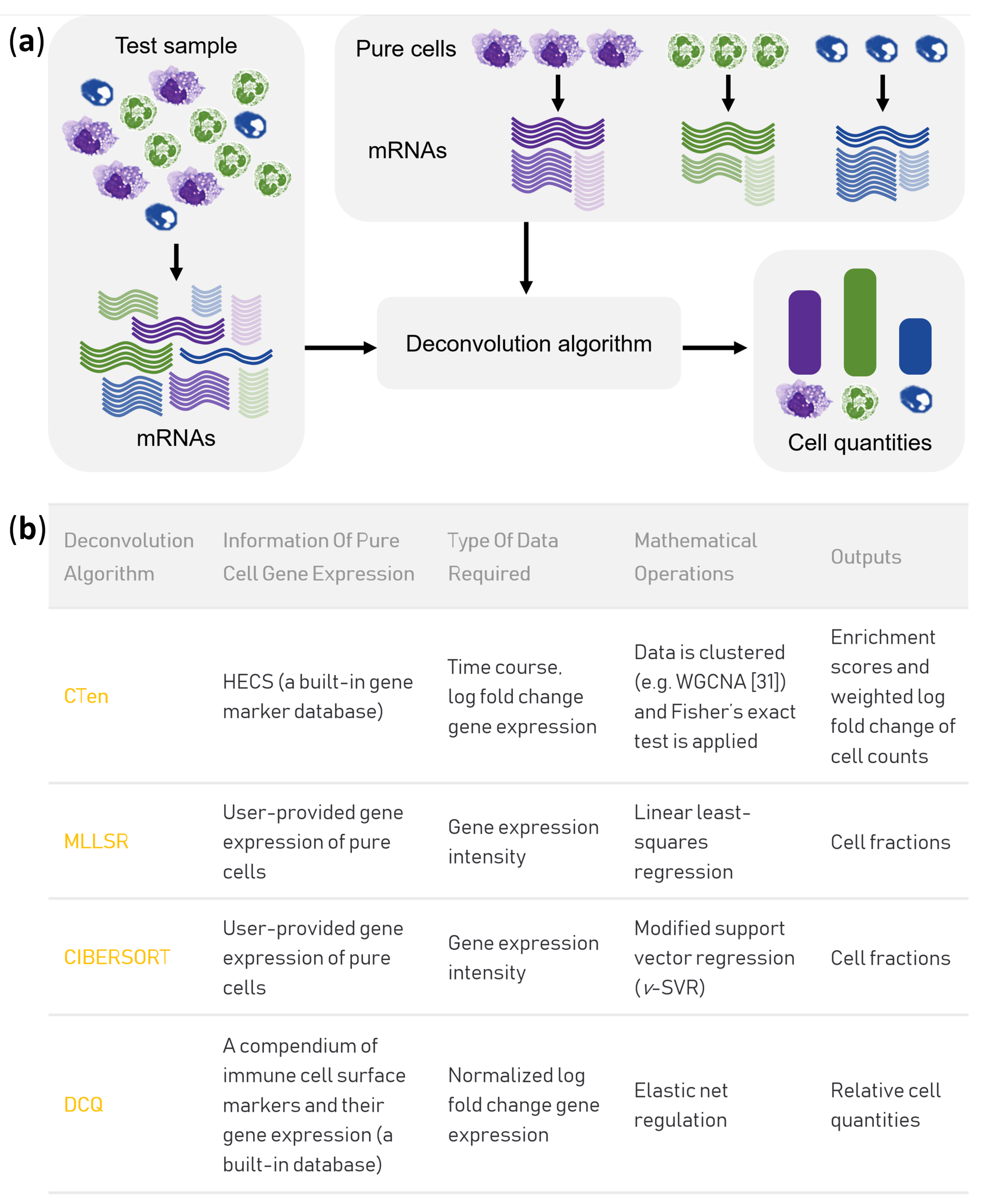
2.4. Prediction of Cell Fractions by MLLSR, CIBERSORT and DCQ
2.5. Gene Co-Expression Analysis by Wgcna and Cell Enrichment Analysis by Cten
2.6. Comparison between Estimated Cell Quantities and Flow Cytometry Data
2.7. Computation of Adjusted Gene Expression and Identification of Significant Genes
3. Results
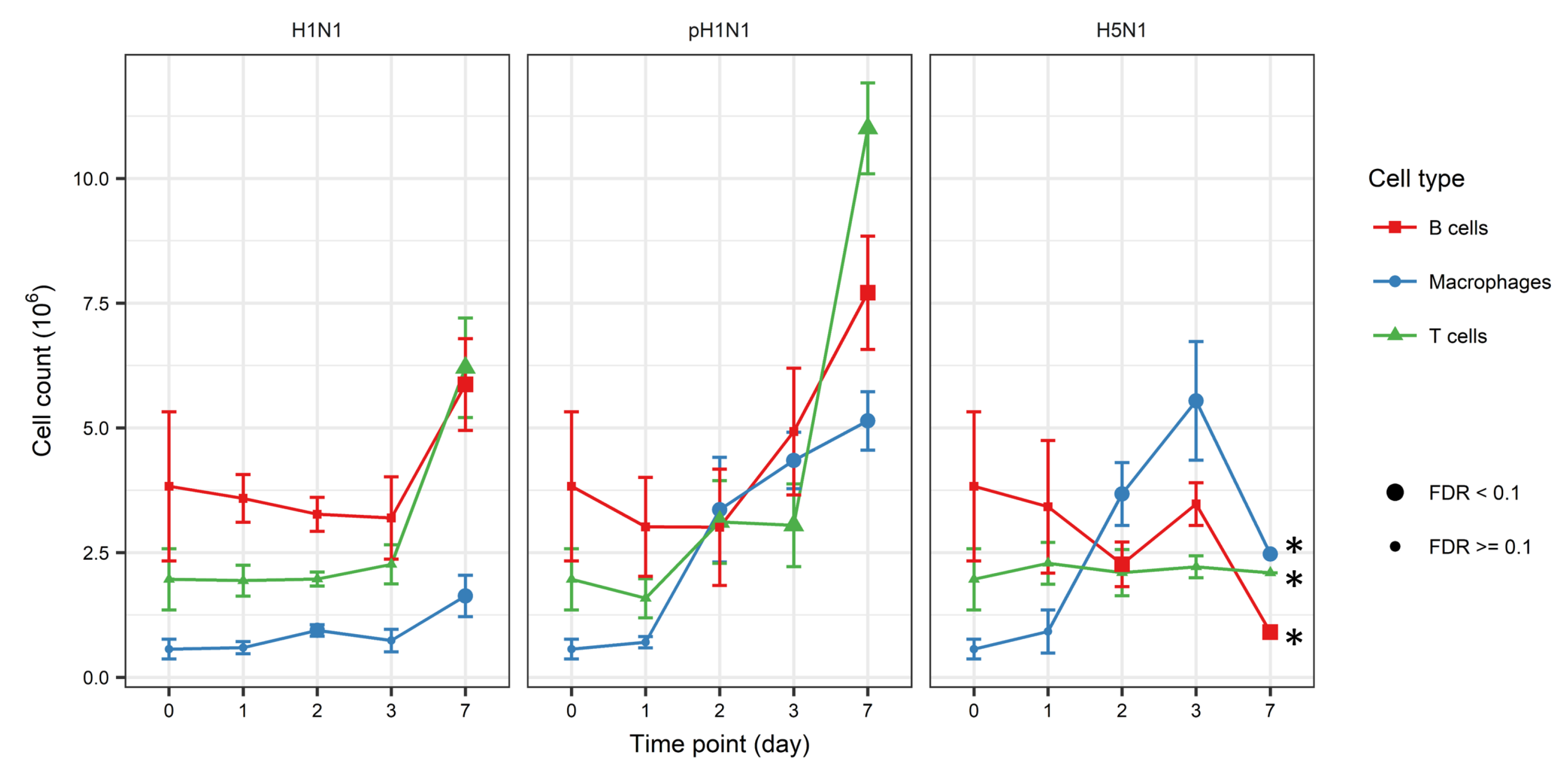
3.1. Dynamic Change of Immune Cell Quantities Induced by Influenza Infection
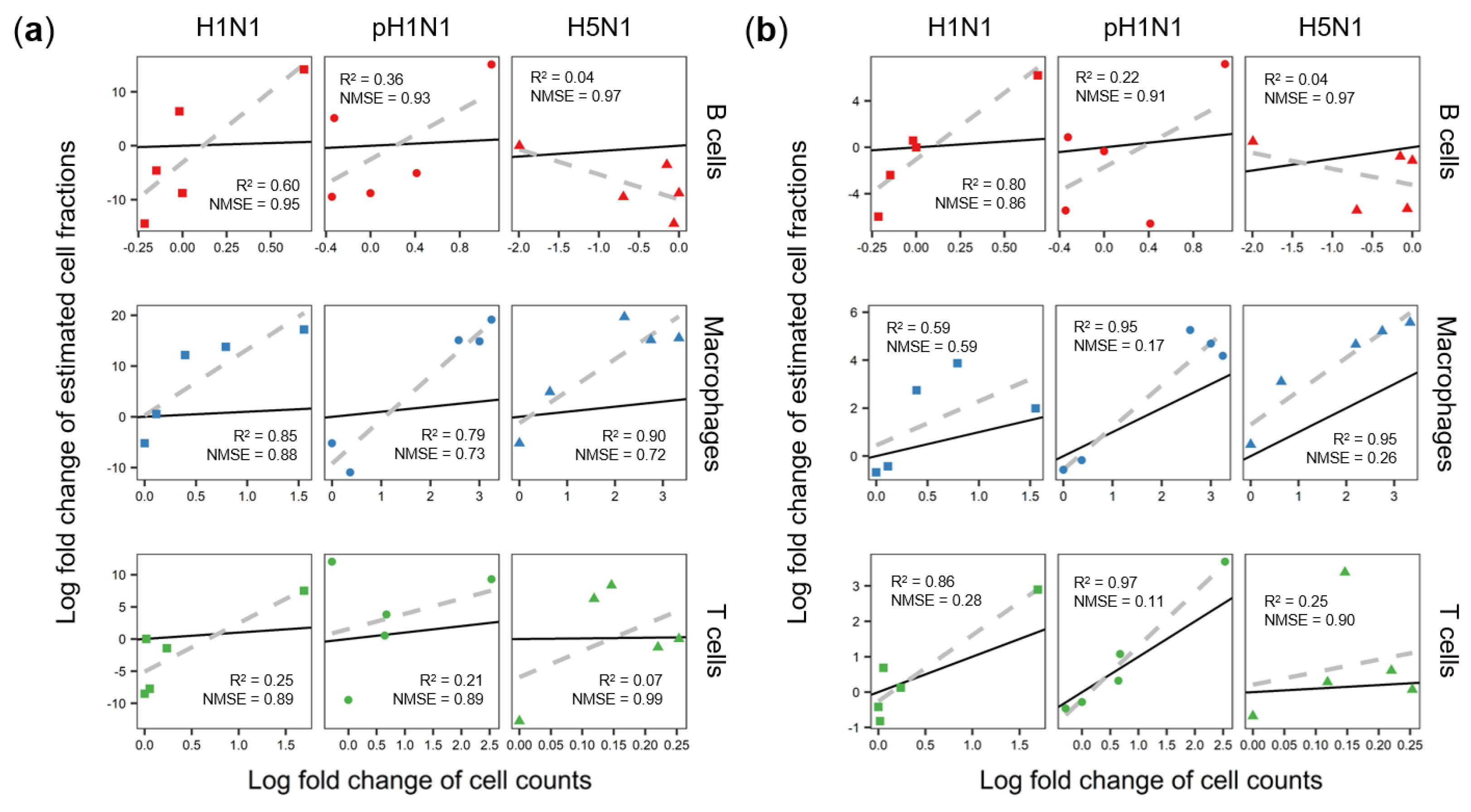
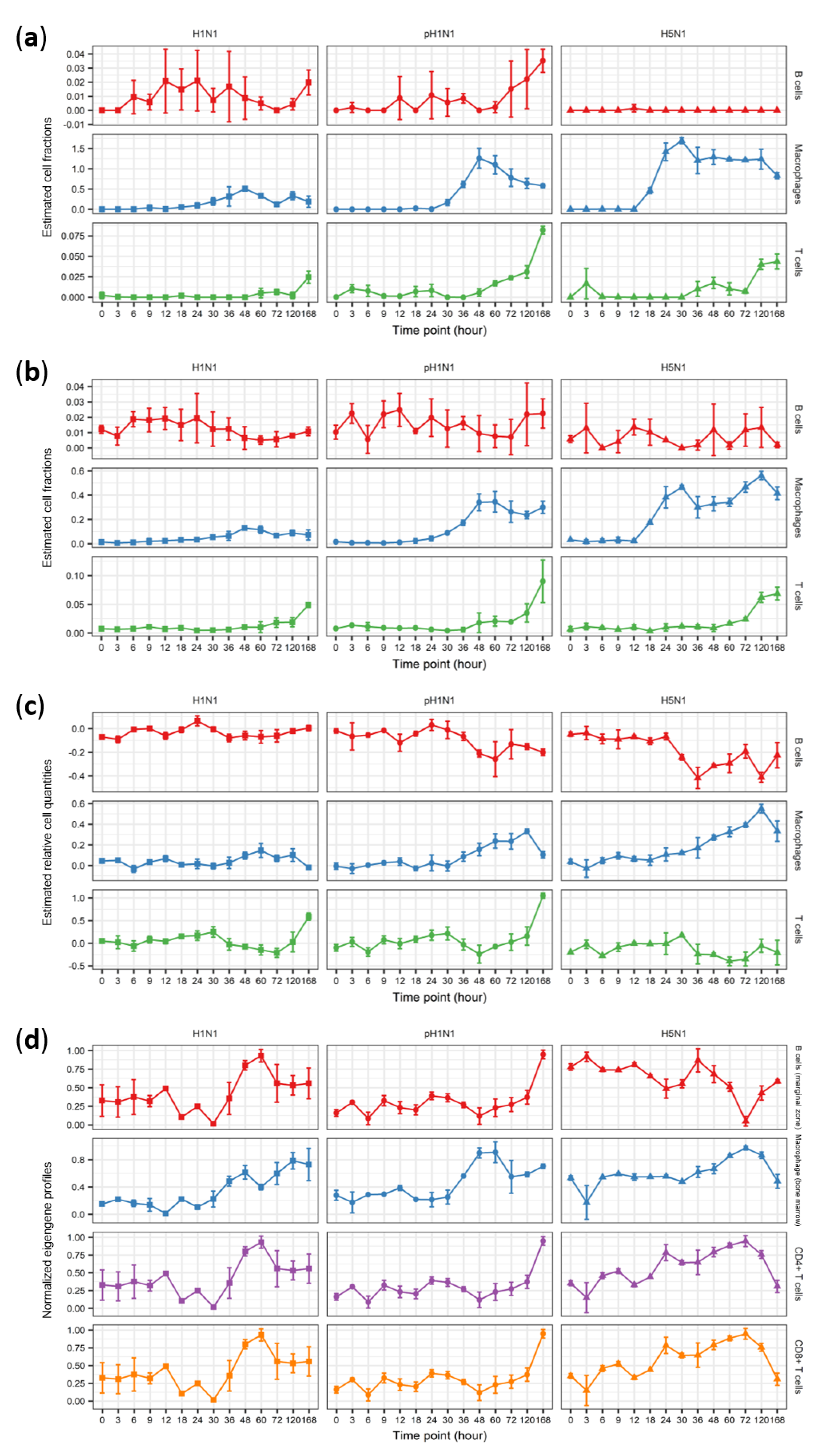
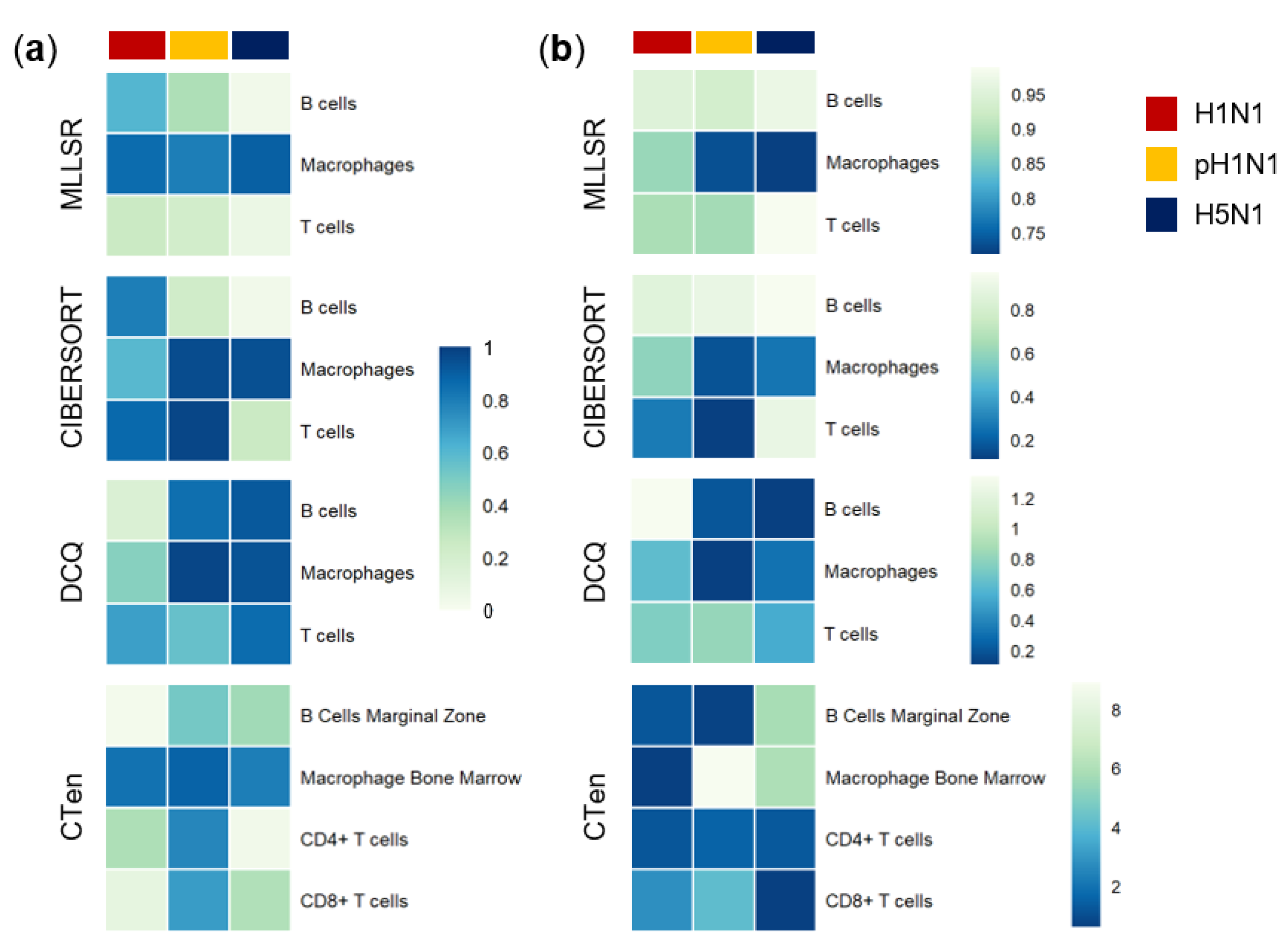
3.2. Cibersort More Accurately Predicts Quantity Changes of T Cells and Macrophages than MLLSR
3.3. DCQ Correctly Predicts Relative Cell Quantities of B Cells and Macrophages for the pH1N1 and H5N1 Cohorts
3.4. CTen Shows High Accuracy When Predicting Dynamic Changes in Macrophages and Neutrophils for All Sample Cohorts
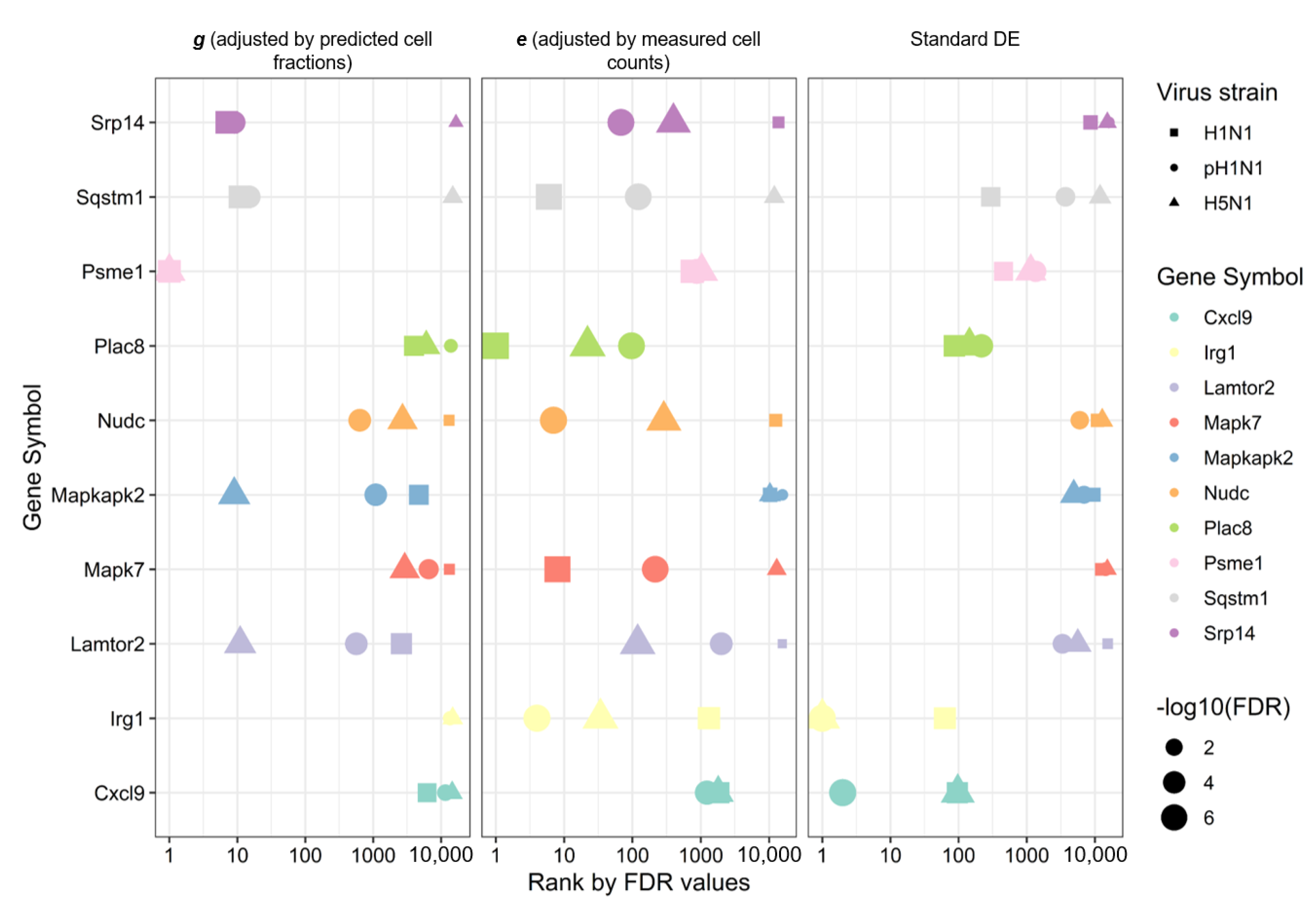
3.5. Improved Disease-Associated Gene Identification by Adjusting for Cellular Composition
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Mackroth, M.S.; Abel, A.; Steeg, C.; Schulze Zur Wiesch, J.; Jacobs, T. Acute Malaria Induces PD1+CTLA4+ Effector T Cells with Cell-Extrinsic Suppressor Function. PLoS Pathog. 2016, 12, e1005909. [Google Scholar] [CrossRef] [PubMed]
- Ostroumov, D.; Fekete-Drimusz, N.; Saborowski, M.; Kuhnel, F.; Woller, N. CD4 and CD8 T lymphocyte interplay in controlling tumor growth. Cell. Mol. Life Sci. 2018, 75, 689–713. [Google Scholar] [CrossRef]
- Dendrou, C.A.; Fugger, L.; Friese, M.A. Immunopathology of multiple sclerosis. Nat. Rev. Immunol. 2015, 15, 545–558. [Google Scholar] [CrossRef] [PubMed]
- Josset, L.; Belser, J.A.; Pantin-Jackwood, M.J.; Chang, J.H.; Chang, S.T.; Belisle, S.E.; Tumpey, T.M.; Katze, M.G. Implication of inflammatory macrophages, nuclear receptors, and interferon regulatory factors in increased virulence of pandemic 2009 H1N1 influenza A virus after host adaptation. J. Virol. 2012, 86, 7192–7206. [Google Scholar] [CrossRef] [PubMed]
- Warrington, R.; Watson, W.; Kim, H.L.; Antonetti, F.R. An introduction to immunology and immunopathology. Allergy Asthma Clin. Immunol. 2011, 7 (Suppl. 1), S1. [Google Scholar] [CrossRef] [PubMed]
- Nakaya, H.I.; Wrammert, J.; Lee, E.K.; Racioppi, L.; Marie-Kunze, S.; Haining, W.N.; Means, A.R.; Kasturi, S.P.; Khan, N.; Li, G.M.; et al. Systems biology of vaccination for seasonal influenza in humans. Nat. Immunol. 2011, 12, 786–795. [Google Scholar] [CrossRef] [PubMed]
- Obermoser, G.; Presnell, S.; Domico, K.; Xu, H.; Wang, Y.; Anguiano, E.; Thompson-Snipes, L.; Ranganathan, R.; Zeitner, B.; Bjork, A.; et al. Systems scale interactive exploration reveals quantitative and qualitative differences in response to influenza and pneumococcal vaccines. Immunity 2013, 38, 831–844. [Google Scholar] [CrossRef] [PubMed]
- Kaech, S.M.; Ahmed, R. Memory CD8+ T cell differentiation: Initial antigen encounter triggers a developmental program in naive cells. Nat. Immunol. 2001, 2, 415–422. [Google Scholar] [CrossRef]
- Luster, A.D.; Alon, R.; von Andrian, U.H. Immune cell migration in inflammation: Present and future therapeutic targets. Nat. Immunol. 2005, 6, 1182–1190. [Google Scholar] [CrossRef]
- Lang, P.A.; Lang, K.S.; Xu, H.C.; Grusdat, M.; Parish, I.A.; Recher, M.; Elford, A.R.; Dhanji, S.; Shaabani, N.; Tran, C.W.; et al. Natural killer cell activation enhances immune pathology and promotes chronic infection by limiting CD8+ T-cell immunity. Proc. Natl. Acad. Sci. USA 2012, 109, 1210–1215. [Google Scholar] [CrossRef]
- Lam, V.C.; Lanier, L.L. NK cells in host responses to viral infections. Curr. Opin. Immunol. 2017, 44, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Morrison, J.; Josset, L.; Tchitchek, N.; Chang, J.; Belser, J.A.; Swayne, D.E.; Pantin-Jackwood, M.J.; Tumpey, T.M.; Katze, M.G. H7N9 and other pathogenic avian influenza viruses elicit a three-pronged transcriptomic signature that is reminiscent of 1918 influenza virus and is associated with lethal outcome in mice. J. Virol. 2014, 88, 10556–10568. [Google Scholar] [CrossRef] [PubMed]
- Peiris, J.S.; Cheung, C.Y.; Leung, C.Y.; Nicholls, J.M. Innate immune responses to influenza A H5N1: Friend or foe? Trends Immunol. 2009, 30, 574–584. [Google Scholar] [CrossRef]
- Cilloniz, C.; Shinya, K.; Peng, X.; Korth, M.J.; Proll, S.C.; Aicher, L.D.; Carter, V.S.; Chang, J.H.; Kobasa, D.; Feldmann, F.; et al. Lethal influenza virus infection in macaques is associated with early dysregulation of inflammatory related genes. PLoS Pathog. 2009, 5, e1000604. [Google Scholar] [CrossRef]
- Brandes, M.; Klauschen, F.; Kuchen, S.; Germain, R.N. A systems analysis identifies a feedforward inflammatory circuit leading to lethal influenza infection. Cell 2013, 154, 197–212. [Google Scholar] [CrossRef]
- Carter, M.J. A rationale for using steroids in the treatment of severe cases of H5N1 avian influenza. J. Med. Microbiol. 2007, 56, 875–883. [Google Scholar] [CrossRef]
- Shinya, K.; Ito, M.; Makino, A.; Tanaka, M.; Miyake, K.; Eisfeld, A.J.; Kawaoka, Y. The TLR4-TRIF pathway protects against H5N1 influenza virus infection. J. Virol. 2012, 86, 19–24. [Google Scholar] [CrossRef]
- Tanaka, A.; Nakamura, S.; Seki, M.; Fukudome, K.; Iwanaga, N.; Imamura, Y.; Miyazaki, T.; Izumikawa, K.; Kakeya, H.; Yanagihara, K.; et al. Toll-like receptor 4 agonistic antibody promotes innate immunity against severe pneumonia induced by coinfection with influenza virus and Streptococcus pneumoniae. Clin. Vaccine Immunol. 2013, 20, 977–985. [Google Scholar] [CrossRef]
- Ibrahim, S.F.; van den Engh, G. Flow cytometry and cell sorting. Adv. Biochem. Eng. Biotechnol. 2007, 106, 19–39. [Google Scholar] [CrossRef]
- Bhattacharya, S.; Dunn, P.; Thomas, C.G.; Smith, B.; Schaefer, H.; Chen, J.; Hu, Z.; Zalocusky, K.A.; Shankar, R.D.; Shen-Orr, S.S.; et al. ImmPort, toward repurposing of open access immunological assay data for translational and clinical research. Sci. Data 2018, 5, 180015. [Google Scholar] [CrossRef]
- Edgar, R.; Domrachev, M.; Lash, A.E. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002, 30, 207–210. [Google Scholar] [CrossRef]
- Shoemaker, J.E.; Lopes, T.J.; Ghosh, S.; Matsuoka, Y.; Kawaoka, Y.; Kitano, H. CTen: A web-based platform for identifying enriched cell types from heterogeneous microarray data. BMC Genom. 2012, 13, 460. [Google Scholar] [CrossRef][Green Version]
- Shoemaker, J.E.; Fukuyama, S.; Eisfeld, A.J.; Zhao, D.; Kawakami, E.; Sakabe, S.; Maemura, T.; Gorai, T.; Katsura, H.; Muramoto, Y.; et al. An Ultrasensitive Mechanism Regulates Influenza Virus-Induced Inflammation. PLoS Pathog. 2015, 11, e1004856. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Master, S.R.; Chodosh, L.A. Computational expression deconvolution in a complex mammalian organ. BMC Bioinform. 2006, 7, 328. [Google Scholar] [CrossRef][Green Version]
- Zhong, Y.; Wan, Y.W.; Pang, K.; Chow, L.M.; Liu, Z. Digital sorting of complex tissues for cell type-specific gene expression profiles. BMC Bioinform. 2013, 14, 89. [Google Scholar] [CrossRef]
- Abbas, A.R.; Wolslegel, K.; Seshasayee, D.; Modrusan, Z.; Clark, H.F. Deconvolution of blood microarray data identifies cellular activation patterns in systemic lupus erythematosus. PLoS ONE 2009, 4, e6098. [Google Scholar] [CrossRef]
- Lu, P.; Nakorchevskiy, A.; Marcotte, E.M. Expression deconvolution: A reinterpretation of DNA microarray data reveals dynamic changes in cell populations. Proc. Natl. Acad. Sci. USA 2003, 100, 10370–10375. [Google Scholar] [CrossRef]
- Altboum, Z.; Steuerman, Y.; David, E.; Barnett-Itzhaki, Z.; Valadarsky, L.; Keren-Shaul, H.; Meningher, T.; Mendelson, E.; Mandelboim, M.; Gat-Viks, I.; et al. Digital cell quantification identifies global immune cell dynamics during influenza infection. Mol. Syst. Biol. 2014, 10, 720. [Google Scholar] [CrossRef] [PubMed]
- Newman, A.M.; Liu, C.L.; Green, M.R.; Gentles, A.J.; Feng, W.; Xu, Y.; Hoang, C.D.; Diehn, M.; Alizadeh, A.A. Robust enumeration of cell subsets from tissue expression profiles. Nat. Methods 2015, 12, 453–457. [Google Scholar] [CrossRef]
- Liebner, D.A.; Huang, K.; Parvin, J.D. MMAD: Microarray microdissection with analysis of differences is a computational tool for deconvoluting cell type-specific contributions from tissue samples. Bioinformatics 2014, 30, 682–689. [Google Scholar] [CrossRef]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Escoubet-Lozach, L.; Benner, C.; Kaikkonen, M.U.; Lozach, J.; Heinz, S.; Spann, N.J.; Crotti, A.; Stender, J.; Ghisletti, S.; Reichart, D.; et al. Mechanisms establishing TLR4-responsive activation states of inflammatory response genes. PLoS Genet. 2011, 7, e1002401. [Google Scholar] [CrossRef]
- Pan, F.; Yu, H.; Dang, E.V.; Barbi, J.; Pan, X.; Grosso, J.F.; Jinasena, D.; Sharma, S.M.; McCadden, E.M.; Getnet, D.; et al. Eos mediates Foxp3-dependent gene silencing in CD4+ regulatory T cells. Science 2009, 325, 1142–1146. [Google Scholar] [CrossRef]
- DiSpirito, J.R.; Shen, H. Expression Analysis of Resting and Stimulated naïVe and MP CD8 T Cells. 2012. Available online: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE16145 (accessed on 6 January 2017).
- Liu, X.; Qu, X.; Chen, Y.; Liao, L.; Cheng, K.; Shao, C.; Zenke, M.; Keating, A.; Zhao, R.C. Mesenchymal stem/stromal cells induce the generation of novel IL-10-dependent regulatory dendritic cells by SOCS3 activation. J. Immunol. 2012, 189, 1182–1192. [Google Scholar] [CrossRef]
- Al Moussawi, K.; Ghigo, E.; Kalinke, U.; Alexopoulou, L.; Mege, J.L.; Desnues, B. Type I interferon induction is detrimental during infection with the Whipple’s disease bacterium, Tropheryma whipplei. PLoS Pathog. 2010, 6, e1000722. [Google Scholar] [CrossRef]
- Ghigo, E.; Barry, A.O.; Pretat, L.; Al Moussawi, K.; Desnues, B.; Capo, C.; Kornfeld, H.; Mege, J.L. IL-16 promotes T. whipplei replication by inhibiting phagosome conversion and modulating macrophage activation. PLoS ONE 2010, 5, e13561. [Google Scholar] [CrossRef]
- Swirski, F.K.; Nahrendorf, M.; Etzrodt, M.; Wildgruber, M.; Cortez-Retamozo, V.; Panizzi, P.; Figueiredo, J.L.; Kohler, R.H.; Chudnovskiy, A.; Waterman, P.; et al. Identification of splenic reservoir monocytes and their deployment to inflammatory sites. Science 2009, 325, 612–616. [Google Scholar] [CrossRef]
- Latorre, A.O.; Caniceiro, B.D.; Fukumasu, H.; Gardner, D.R.; Lopes, F.M.; Wysochi, H.L.J.; da Silva, T.C.; Haraguchi, M.; Bressan, F.F.; Gorniak, S.L. Ptaquiloside reduces NK cell activities by enhancing metallothionein expression, which is prevented by selenium. Toxicology 2013, 304, 100–108. [Google Scholar] [CrossRef]
- Heng, T.S.; Painter, M.W.; Immunological Genome Project Consortium. The Immunological Genome Project: Networks of gene expression in immune cells. Nat. Immunol. 2008, 9, 1091–1094. [Google Scholar] [CrossRef]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef]
- Hasegawa, K.; Sato, A.; Tanimura, K.; Uemasu, K.; Hamakawa, Y.; Fuseya, Y.; Sato, S.; Muro, S.; Hirai, T. Fraction of MHCII and EpCAM expression characterizes distal lung epithelial cells for alveolar type 2 cell isolation. Respir. Res. 2017, 18, 150. [Google Scholar] [CrossRef]
- Huang da, W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Bioinformatics enrichment tools: Paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009, 37, 1–13. [Google Scholar] [CrossRef]
- Guirimand, T.; Delmotte, S.; Navratil, V. VirHostNet 2.0: Surfing on the web of virus/host molecular interactions data. Nucleic Acids Res. 2015, 43, D583–D587. [Google Scholar] [CrossRef]
- Duran, A.; Linares, J.F.; Galvez, A.S.; Wikenheiser, K.; Flores, J.M.; Diaz-Meco, M.T.; Moscat, J. The signaling adaptor p62 is an important NF-kappaB mediator in tumorigenesis. Cancer Cell 2008, 13, 343–354. [Google Scholar] [CrossRef]
- Scheffler, J.M.; Sparber, F.; Tripp, C.H.; Herrmann, C.; Humenberger, A.; Blitz, J.; Romani, N.; Stoitzner, P.; Huber, L.A. LAMTOR2 regulates dendritic cell homeostasis through FLT3-dependent mTOR signalling. Nat. Commun. 2014, 5, 5138. [Google Scholar] [CrossRef]
- Zhang, W.; Liu, H.T. MAPK signal pathways in the regulation of cell proliferation in mammalian cells. Cell Res. 2002, 12, 9–18. [Google Scholar] [CrossRef]
- Muller, M.; Carter, S.; Hofer, M.J.; Campbell, I.L. Review: The chemokine receptor CXCR3 and its ligands CXCL9, CXCL10 and CXCL11 in neuroimmunity—A tale of conflict and conundrum. Neuropathol. Appl. Neurobiol. 2010, 36, 368–387. [Google Scholar] [CrossRef]
- Kiso, M.; Lopes, T.J.S.; Yamayoshi, S.; Ito, M.; Yamashita, M.; Nakajima, N.; Hasegawa, H.; Neumann, G.; Kawaoka, Y. Combination Therapy With Neuraminidase and Polymerase Inhibitors in Nude Mice Infected With Influenza Virus. J. Infect. Dis. 2018, 217, 887–896. [Google Scholar] [CrossRef]
- Ueki, H.; Wang, I.H.; Fukuyama, S.; Katsura, H.; da Silva Lopes, T.J.; Neumann, G.; Kawaoka, Y. In vivo imaging of the pathophysiological changes and neutrophil dynamics in influenza virus-infected mouse lungs. Proc. Natl. Acad. Sci. USA 2018, 115, E6622–E6629. [Google Scholar] [CrossRef]
- Iwatsuki-Horimoto, K.; Nakajima, N.; Ichiko, Y.; Sakai-Tagawa, Y.; Noda, T.; Hasegawa, H.; Kawaoka, Y. Syrian Hamster as an Animal Model for the Study of Human Influenza Virus Infection. J. Virol. 2018, 92. [Google Scholar] [CrossRef] [PubMed]
- Briggs, J.A.; Weinreb, C.; Wagner, D.E.; Megason, S.; Peshkin, L.; Kirschner, M.W.; Klein, A.M. The dynamics of gene expression in vertebrate embryogenesis at single-cell resolution. Science 2018, 360, eaar5780. [Google Scholar] [CrossRef] [PubMed]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, M.; Fukuyama, S.; Kawaoka, Y.; Shoemaker, J.E. Predicting Host Immune Cell Dynamics and Key Disease-Associated Genes Using Tissue Transcriptional Profiles. Processes 2019, 7, 301. https://doi.org/10.3390/pr7050301
Wang M, Fukuyama S, Kawaoka Y, Shoemaker JE. Predicting Host Immune Cell Dynamics and Key Disease-Associated Genes Using Tissue Transcriptional Profiles. Processes. 2019; 7(5):301. https://doi.org/10.3390/pr7050301
Chicago/Turabian StyleWang, Muying, Satoshi Fukuyama, Yoshihiro Kawaoka, and Jason E. Shoemaker. 2019. "Predicting Host Immune Cell Dynamics and Key Disease-Associated Genes Using Tissue Transcriptional Profiles" Processes 7, no. 5: 301. https://doi.org/10.3390/pr7050301
APA StyleWang, M., Fukuyama, S., Kawaoka, Y., & Shoemaker, J. E. (2019). Predicting Host Immune Cell Dynamics and Key Disease-Associated Genes Using Tissue Transcriptional Profiles. Processes, 7(5), 301. https://doi.org/10.3390/pr7050301