Theoretical and Experimental Approaches Aimed at Drug Design Targeting Neurodegenerative Diseases

 , , , ,
, , , ,  and
and 
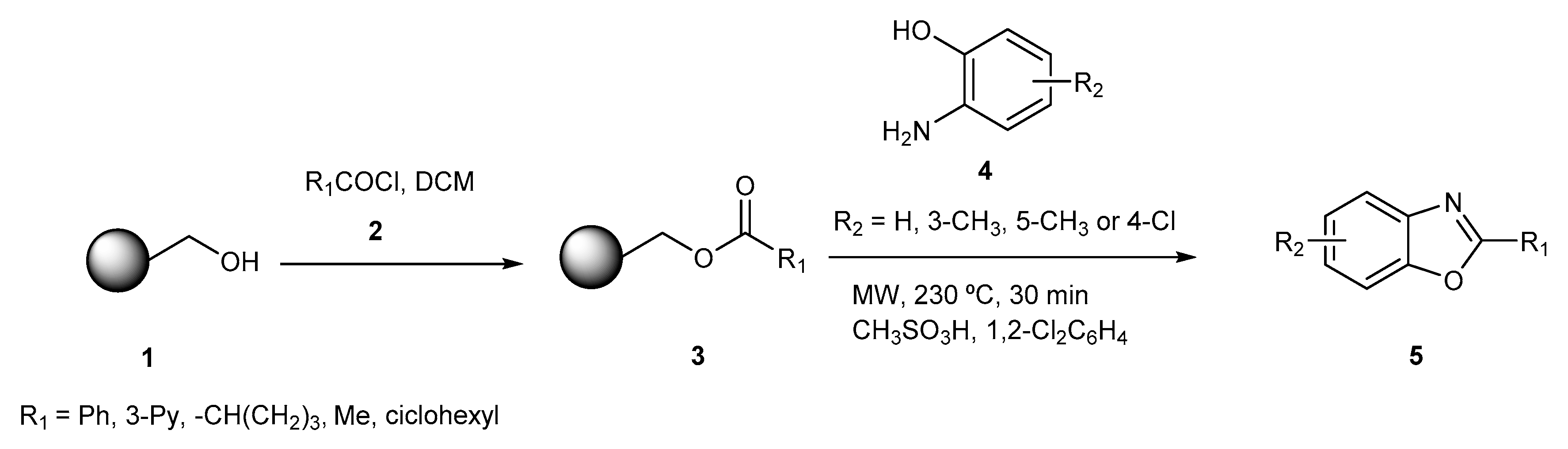{kind=link}
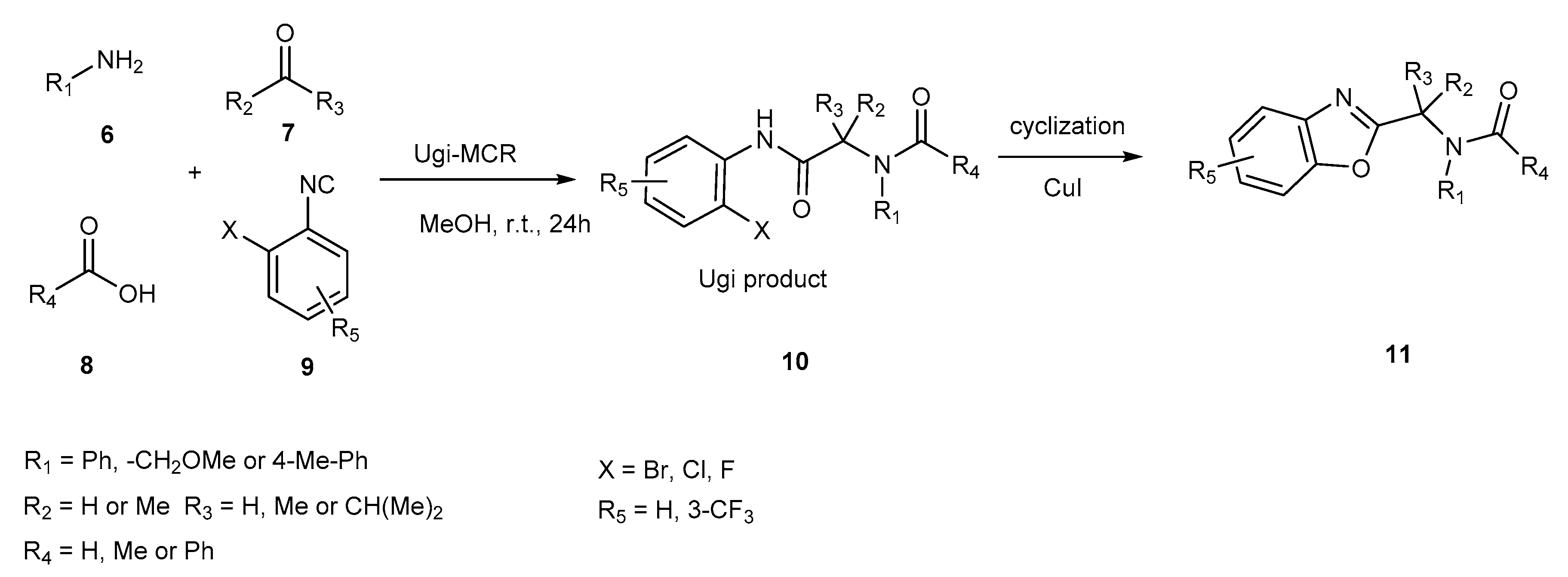{kind=link}
{kind=link}
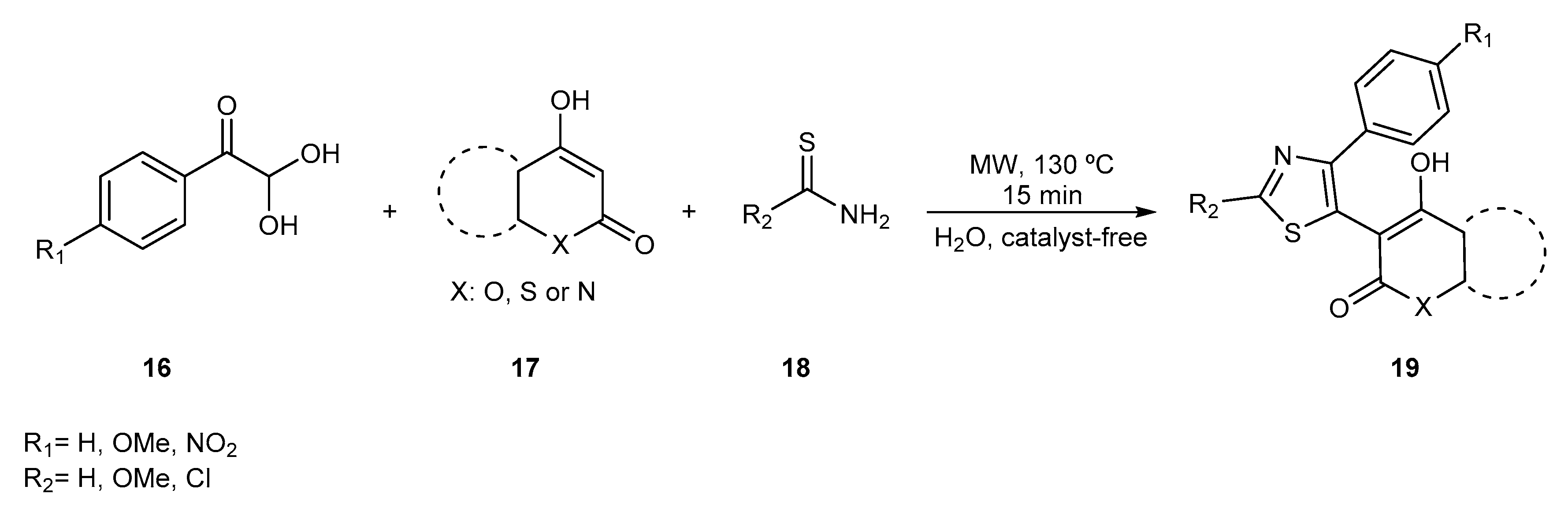{kind=link}
{kind=link}
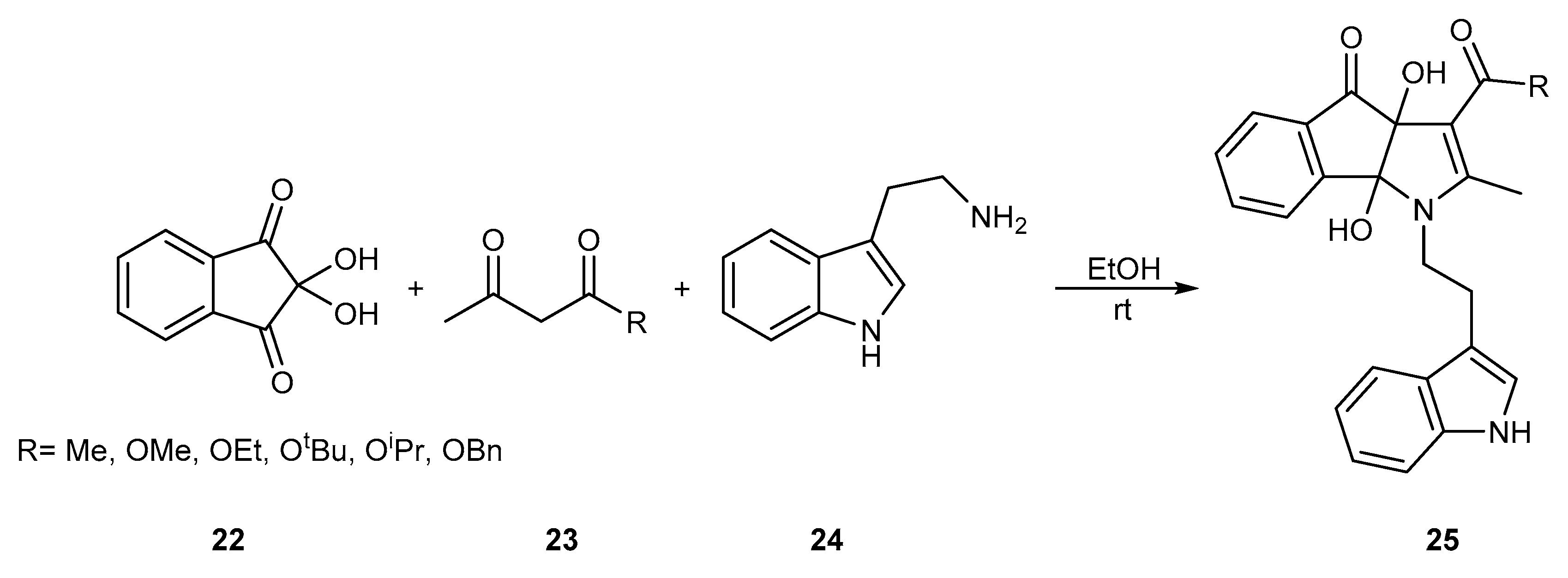{kind=link}
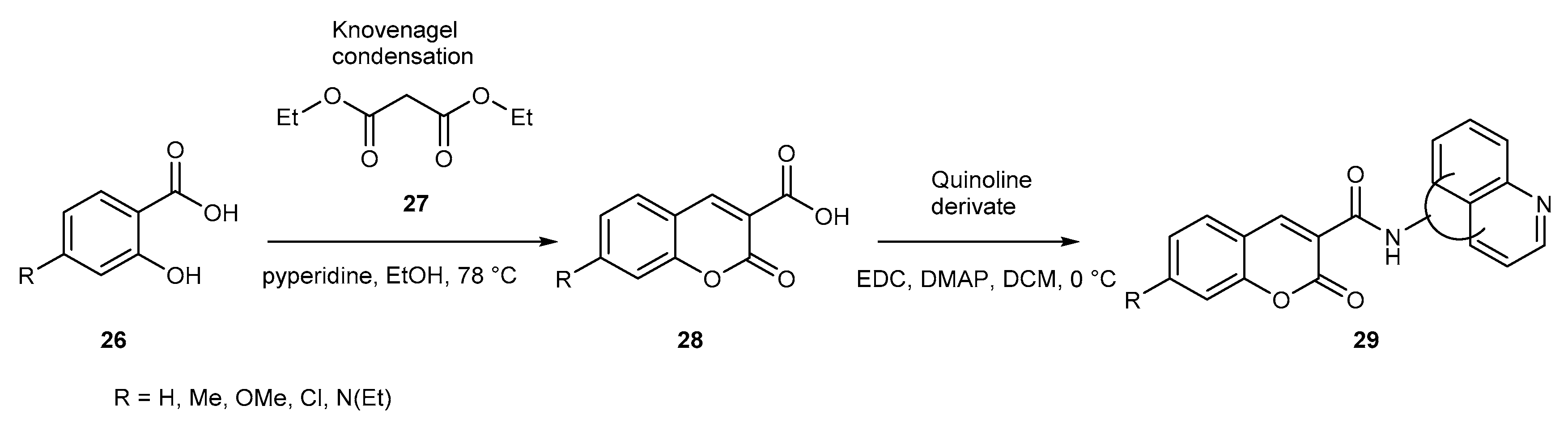{kind=link}
{kind=link}
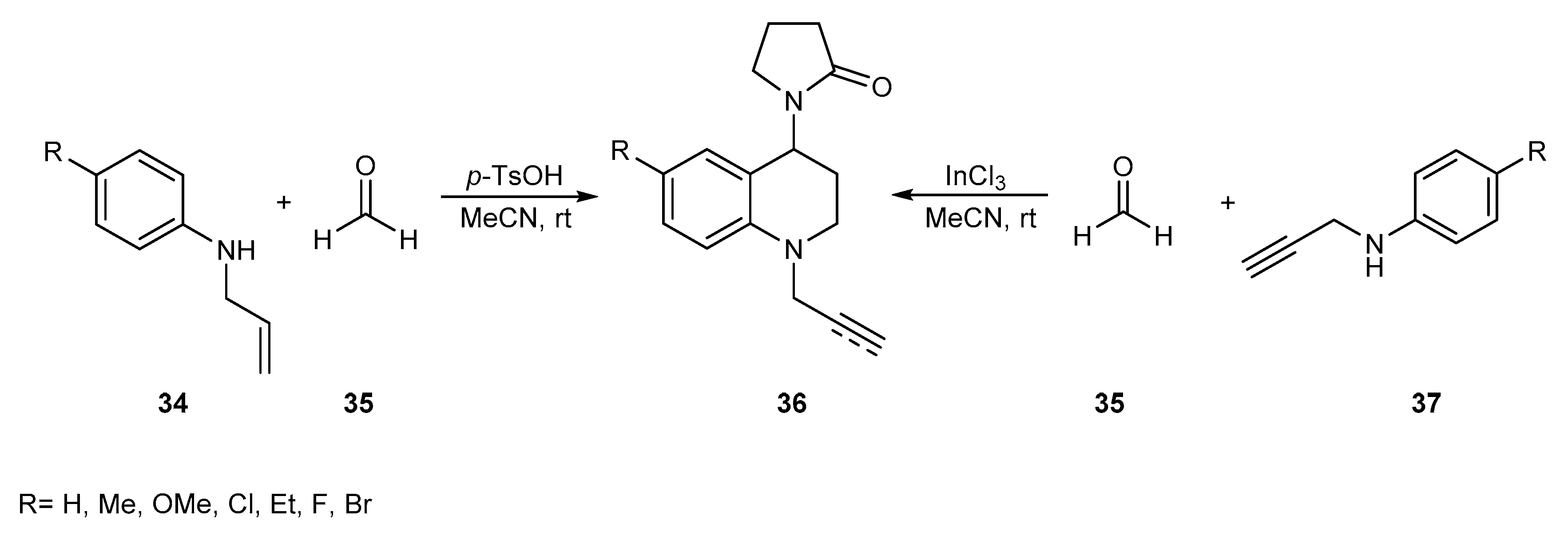{kind=link}
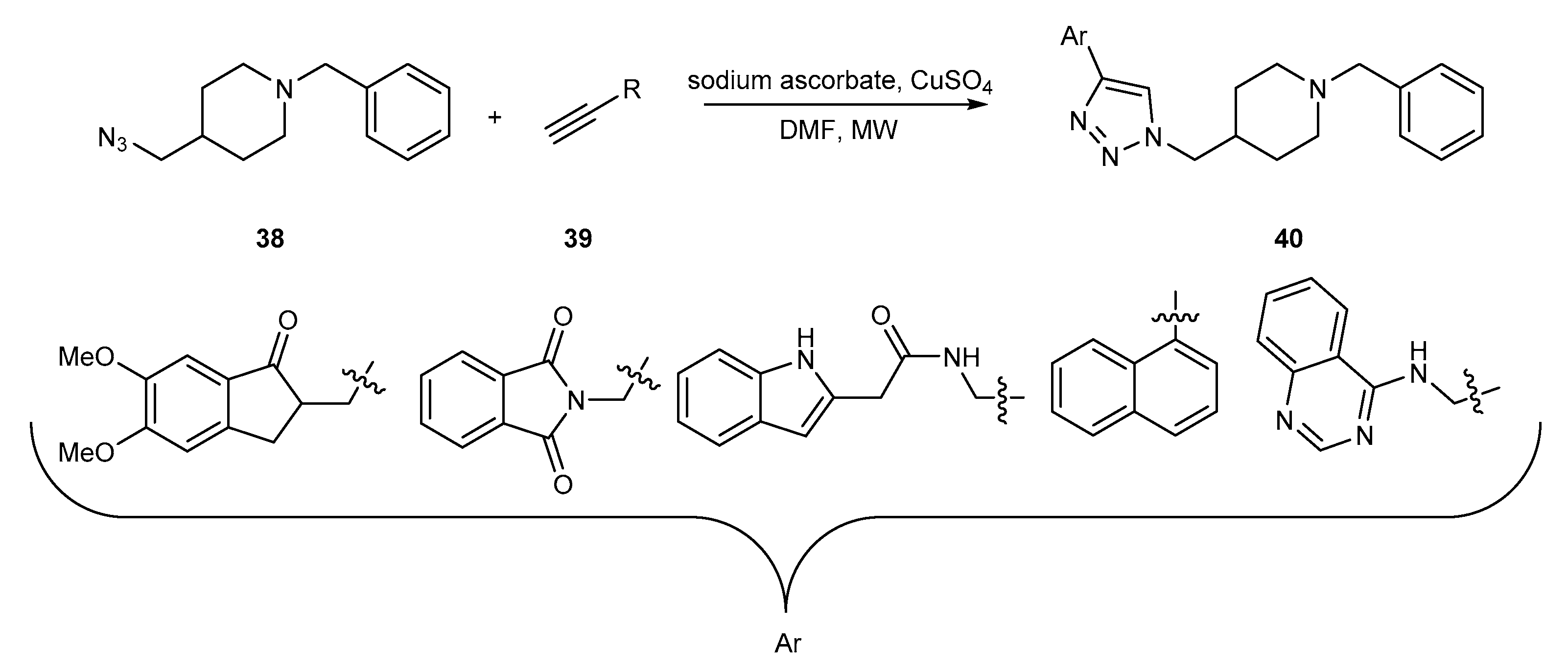{kind=link}
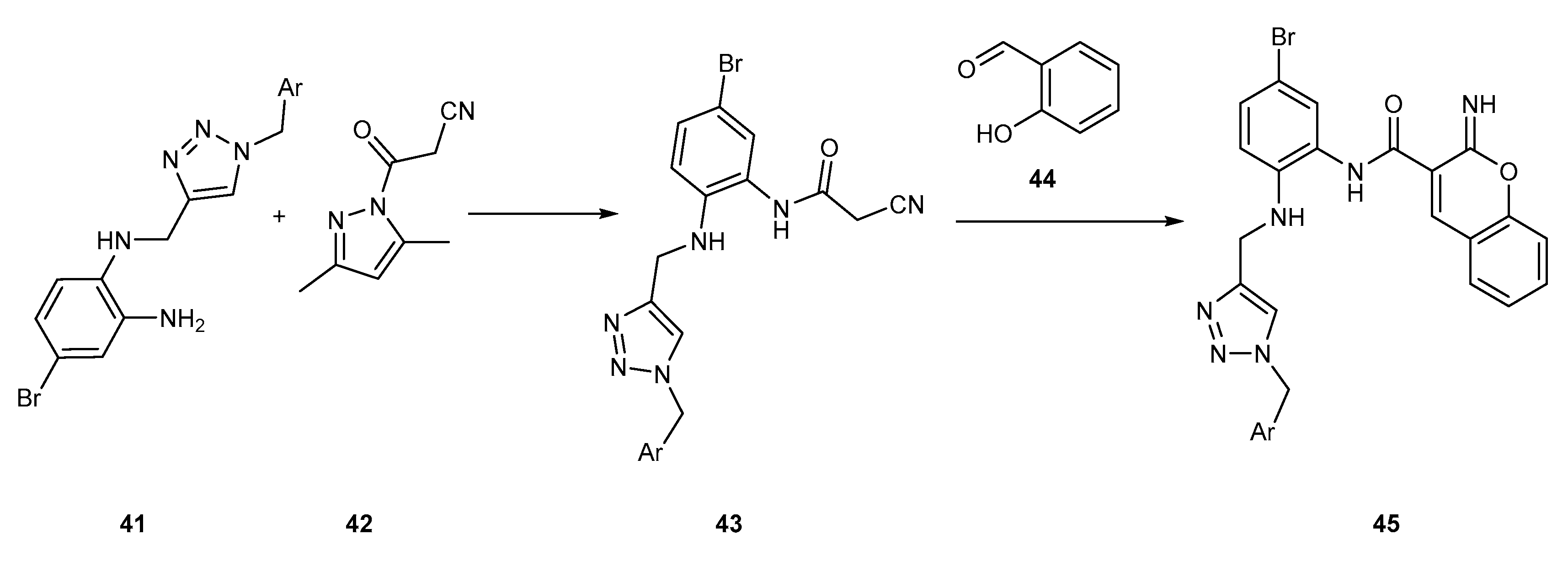{kind=link}
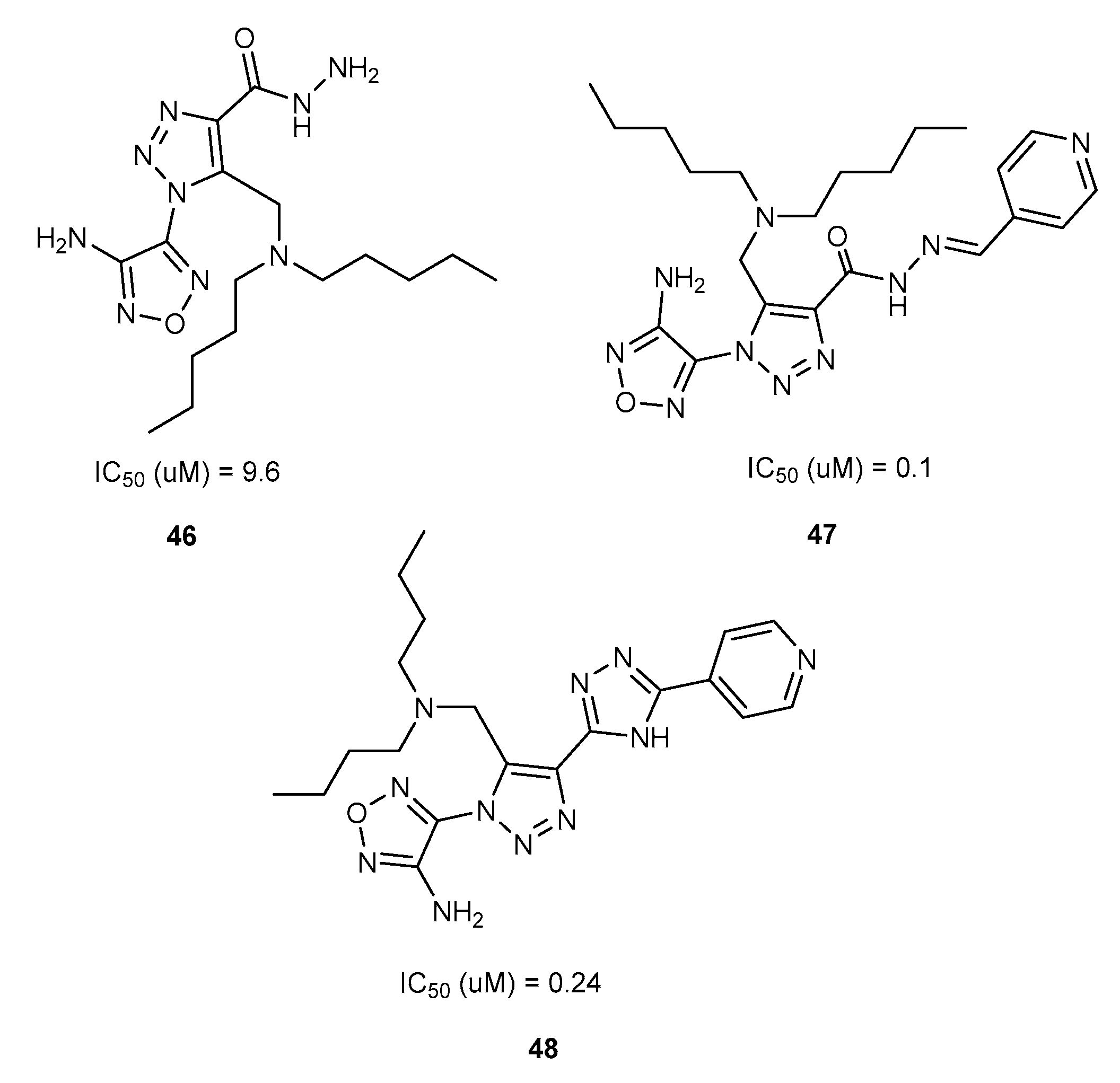{kind=link}
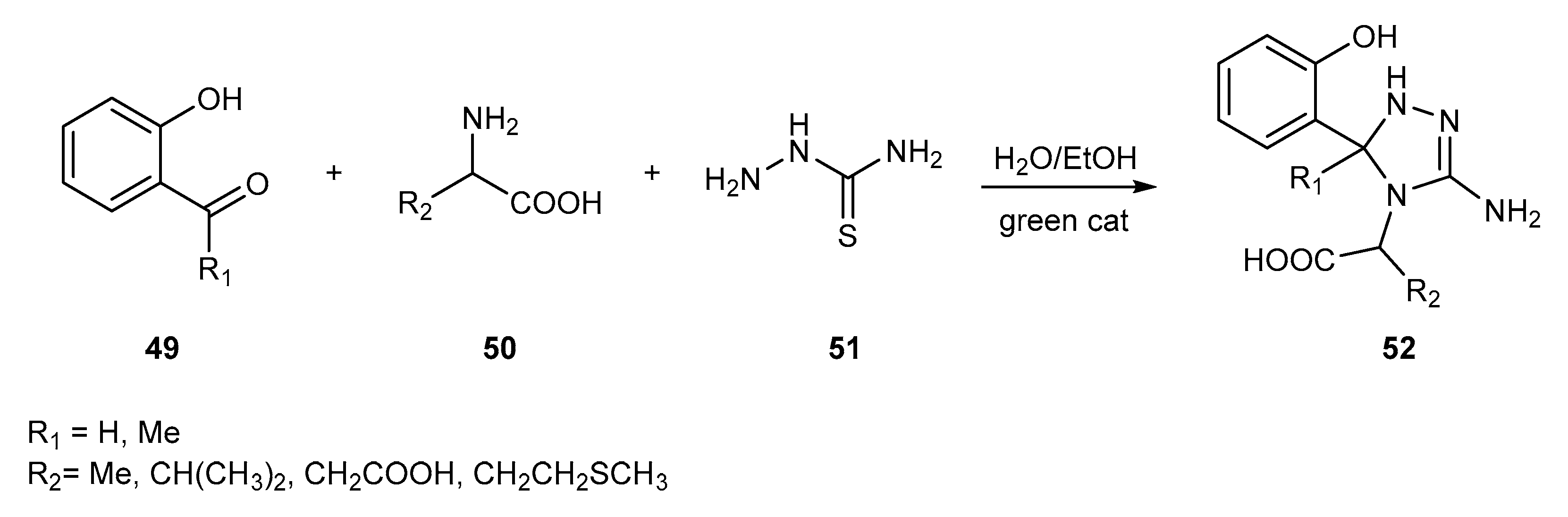{kind=link}
{kind=link}
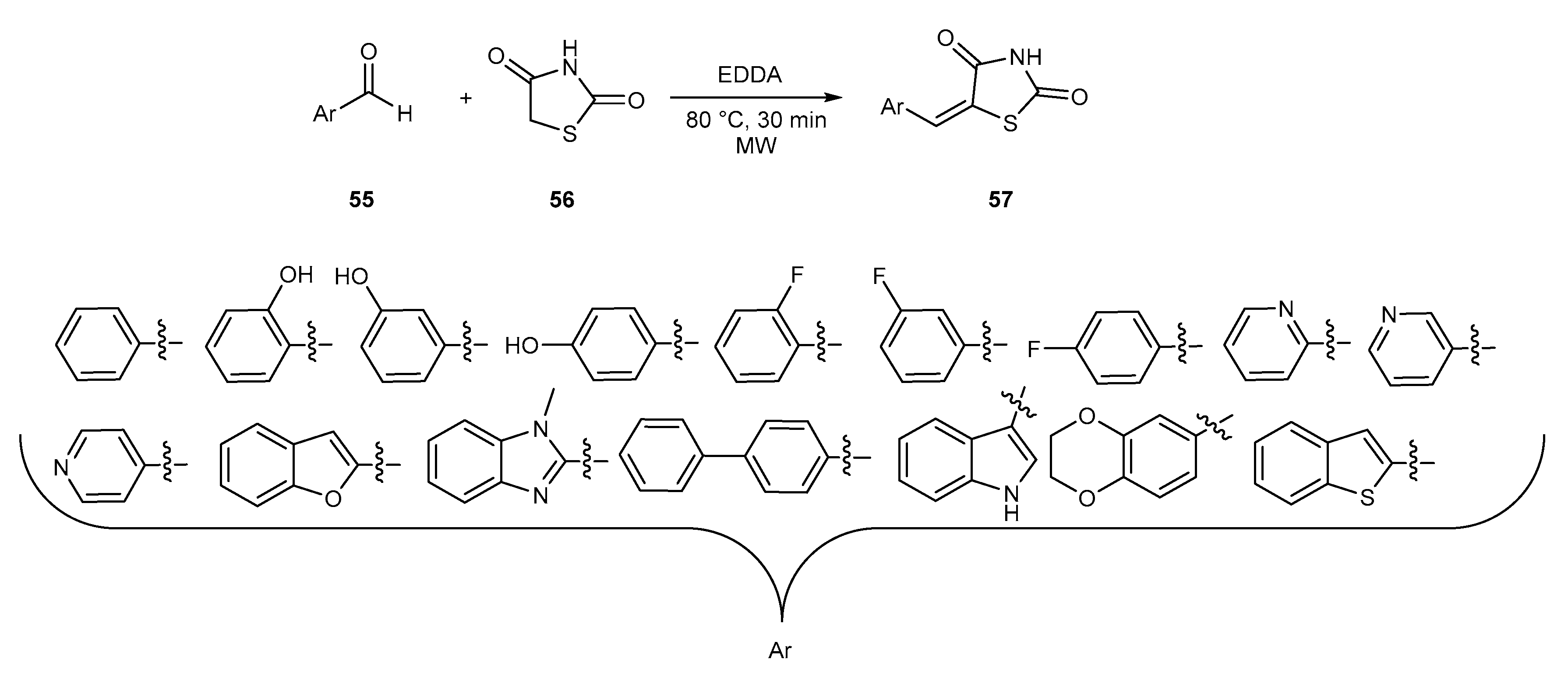{kind=link}
{kind=link}
Abstract
1. Introduction
2. A Safer Approach to NDD Drug Discovery: Green Chemistry Technology
3. Computational Methods in Drug Design and Discovery
4. Drug Design and Discovery Targeting NDD
5. Challenges and Future Prospects
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- United Nations Department of Economic and Social Affairs. World Population Ageing 2015; United Nations: New York, NY, USA, 2015; (ST/ESA/SER.A/390). [Google Scholar]
- Wyss-Coray, T. Ageing, neurodegeneration and brain rejuvenation. Nature 2016, 539, 180–186. [Google Scholar] [CrossRef] [PubMed]
- National Institute on Aging; National Institutes of Health. Global Health and Aging; World Health Organization: Geneva, Switzerland, 2011; Volume 1, pp. 1–32.
- Bhat, S.; Kamal, M.; Yarla, N.; Ashraf, G. Synopsis on Managment Strategies for Neurodegenerative Disorders: Challenges from Bench to Bedside in Successful Drug Discovery and Development. Curr. Top. Med. Chem. 2017, 17, 1371–1378. [Google Scholar] [CrossRef] [PubMed]
- Trippier, P.C.; Jansen Labby, K.; Hawker, D.D.; Mataka, J.J.; Silverman, R.B. Target- and Mechanism-Based Therapeutics for Neurodegenerative Diseases: Strength in Numbers. J. Med. Chem. 2013, 56, 3121–3147. [Google Scholar] [CrossRef] [PubMed]
- Barnham, K.J.; Masters, C.L.; Bush, A.I. Neurodegenerative diseases and oxidative stress. Nat. Rev. Drug Discov. 2004, 3, 205–214. [Google Scholar] [CrossRef] [PubMed]
- Burns, A.; Iliffe, S. Alzheimer’s disease. BMJ 2009, 338, b158. [Google Scholar] [CrossRef]
- Ponzoni, I.; Sebastián-Pérez, V.; Martínez, M.J.; Roca, C.; De la Cruz Pérez, C.; Cravero, F.; Vazquez, G.E.; Páez, J.A.; Díaz, M.F.; Campillo, N.E. QSAR Classification Models for Predicting the Activity of Inhibitors of Beta-Secretase (BACE1) Associated with Alzheimer’s Disease. Sci. Rep. 2019, 9, 9102–9114. [Google Scholar] [CrossRef]
- Hebert, L.E.; Scherr, P.A.; Bienias, J.L.; Bennett, D.A.; Evans, D.A. Alzheimer Disease in the US Population. Arch. Neurol. 2003, 60, 1119–1122. [Google Scholar] [CrossRef]
- Sebastián-Pérez, V.; Martínez, M.J.; Gil, C.; Campillo, N.E.; Martínez, A.; Ponzoni, I. QSAR modelling for drug discovery: Predicting the activity of LRRK2 inhibitors for Parkinson’s disease using cheminformatics approaches. In Advances in Intelligent Systems and Computing; Springer International Publishing: Berlin/Heidelberg, Germany, 2019; Volume 803, pp. 63–70. [Google Scholar]
- Jankovic, J. Parkinson’s disease: Clinical features and diagnosis. J. Neurol. Neurosurg. Psychiatry 2008, 79, 368–376. [Google Scholar] [CrossRef]
- Puginier, E.; Bharatiya, R.; Chagraoui, A.; Manem, J.; Cho, Y.H.; Garret, M.; De Deurwaerdère, P. Early neurochemical modifications of monoaminergic systems in the R6/1 mouse model of Huntington’s disease. Neurochem. Int. 2019, 128, 186–195. [Google Scholar] [CrossRef]
- Ribeiro, F.F.; Mendonca Junior, F.J.B.; Ghasemi, J.B.; Ishiki, H.M.; Scotti, M.T.; Scotti, L. Docking of Natural Products against Neurodegenerative Diseases: General Concepts. Comb. Chem. High Throughput Screen. 2018, 21, 152–160. [Google Scholar] [CrossRef]
- Salloway, S.; Mintzer, J.; Weiner, M.F.; Cummings, J.L. Disease-Modifying therapies in Alzheimer’s disease. Alzheimer’s Dement 2008, 4, 65–79. [Google Scholar] [CrossRef] [PubMed]
- Paul, S.M.; Mytelka, D.S.; Dunwiddie, C.T.; Persinger, C.C.; Munos, B.H.; Lindborg, S.R.; Schacht, A.L. How to improve R&D productivity: The pharmaceutical industry’s grand challenge. Nat. Rev. Drug Discov. 2010, 9, 203–214. [Google Scholar] [PubMed]
- Sheldon, R.A. The e Factor: Fifteen years on. Green Chem. 2007, 9, 1273–1283. [Google Scholar] [CrossRef]
- Rodríguez, Y.A.; Gutiérrez, M.; Ramírez, D.; Alzate-Morales, J.; Bernal, C.C.; Güiza, F.M.; Romero Bohórquez, A.R. Novel N -allyl/propargyl tetrahydroquinolines: Synthesis via Three-component Cationic Imino Diels-Alder Reaction, Binding Prediction, and Evaluation as Cholinesterase Inhibitors. Chem. Biol. Drug Des. 2016, 88, 498–510. [Google Scholar] [CrossRef] [PubMed]
- Prent-Peñaloza, L.; De la Torre, A.F.; Velázquez-Libera, J.L.; Gutiérrez, M.; Caballero, J. Synthesis of diN-Substituted Glycyl-Phenylalanine Derivatives by Using Ugi Four Component Reaction and Their Potential as Acetylcholinesterase Inhibitors. Molecules 2019, 24, 189. [Google Scholar] [CrossRef] [PubMed]
- Duarte, Y.; Fonseca, A.; Gutiérrez, M.; Adasme-Carreño, F.; Muñoz-Gutierrez, C.; Alzate-Morales, J.; Santana, L.; Uriarte, E.; Álvarez, R.; Matos, M.J. Novel Coumarin-Quinoline Hybrids: Design of Multitarget Compounds for Alzheimer’s Disease. ChemistrySelect 2019, 4, 551–558. [Google Scholar] [CrossRef]
- de Andrade, P.; Mantoani, S.P.; Gonçalves Nunes, P.S.; Magadán, C.R.; Pérez, C.; Xavier, D.J.; Hojo, E.T.S.; Campillo, N.E.; Martínez, A.; Carvalho, I. Highly potent and selective aryl-1,2,3-triazolyl benzylpiperidine inhibitors toward butyrylcholinesterase in Alzheimer’s disease. Bioorg. Med. Chem. 2019, 27, 931–943. [Google Scholar] [CrossRef]
- Chierrito, T.P.C.; Pedersoli-Mantoani, S.; Roca, C.; Sebastian-Pérez, V.; Martínez-Gonzalez, L.; Pérez, D.I.; Perez, C.; Canales, A.; Cañada, F.J.; Campillo, N.E.; et al. Chameleon-like behavior of indolylpiperidines in complex with cholinesterases targets: Potent butyrylcholinesterase inhibitors. Eur. J. Med. Chem. 2018, 145, 431–444. [Google Scholar] [CrossRef]
- Gandini, A.; Bartolini, M.; Tedesco, D.; Martinez-Gonzalez, L.; Roca, C.; Campillo, N.E.; Zaldivar-Diez, J.; Perez, C.; Zuccheri, G.; Miti, A.; et al. Tau-Centric Multitarget Approach for Alzheimer’s Disease: Development of First-in-Class Dual Glycogen Synthase Kinase 3β and Tau-Aggregation Inhibitors. J. Med. Chem. 2018, 61, 7640–7656. [Google Scholar] [CrossRef]
- Gawande, M.B.; Bonifácio, V.D.B.; Luque, R.; Branco, P.S.; Varma, R.S. Benign by design: Catalyst-free in-water, on-water green chemical methodologies in organic synthesis. Chem. Soc. Rev. 2013, 42, 5522. [Google Scholar] [CrossRef]
- Cioc, R.C.; Ruijter, E.; Orru, R.V.A. Multicomponent reactions: Advanced tools for sustainable organic synthesis. Green Chem. 2014, 16, 2958–2975. [Google Scholar] [CrossRef]
- Lim, H.J.; Myung, D.; Lee, I.Y.C.; Jung, M.H. Microwave-Assisted Synthesis of Benzimidazoles, Benzoxazoles, and Benzothiazoles from Resin-Bound Esters. J. Comb. Chem. 2008, 10, 501–503. [Google Scholar] [CrossRef] [PubMed]
- Ramírez, D. Computational Methods Applied to Rational Drug Design. Open Med. Chem. J. 2016, 10, 7–20. [Google Scholar] [CrossRef] [PubMed]
- Heck, S.; Dömling, A. A Versatile Multi-Component One-Pot Thiazole Synthesis. Synlett 2000, 2000, 424–426. [Google Scholar]
- Ugi, I.; Dömling, A.; Hörl, W. Multicomponent reactions in organic chemistry. Endeavour 1994, 18, 115–122. [Google Scholar] [CrossRef]
- LeVine, H. The challenge of inhibiting Abeta polymerization. Curr. Med. Chem. 2002, 9, 1121–1133. [Google Scholar] [CrossRef]
- Lee, S.M.; Jeon, R. Synthesis of 6-[2-(benzoxazol-2-ylmethylamino)ethoxy]-1-alkyl-1H-indole-2-carboxylic acid and inhibitory activity on beta-amyloid aggregation. Arch. Pharm. Res. 2005, 28, 1219–1223. [Google Scholar] [CrossRef]
- Spatz, J.H.; Bach, T.; Umkehrer, M.; Bardin, J.; Ross, G.; Burdack, C.; Kolb, J. Diversity oriented synthesis of benzoxazoles and benzothiazoles. Tetrahedron Lett. 2007, 48, 9030–9034. [Google Scholar] [CrossRef]
- Armstrong, R.W.; Combs, A.P.; Tempest, P.A.; Brown, S.D.; Keating, T.A. Multiple-Component Condensation Strategies for Combinatorial Library Synthesis. Acc. Chem. Res. 1996, 29, 123–131. [Google Scholar] [CrossRef]
- Jangale, A.D.; Dalal, D.S. Highly Efficient, Combinatorial and Catalyst-Free Approach for the Synthesis of 2-Benzylidenehydrazono-3-phenyl-4-thiazolidinone-5-acetates in Ethanol. ChemistrySelect 2019, 4, 1323–1329. [Google Scholar] [CrossRef]
- Karamthulla, S.; Pal, S.; Khan, M.N.; Choudhury, L.H. “On-water” synthesis of novel trisubstituted 1,3-thiazoles via microwave-assisted catalyst-free domino reactions. RSC Adv. 2014, 4, 37889–37899. [Google Scholar] [CrossRef]
- Kushwaha, P.; Fatima, S.; Upadhyay, A.; Gupta, S.; Bhagwati, S.; Baghel, T.; Siddiqi, M.I.; Nazir, A.; Sashidhara, K.V. Synthesis, biological evaluation and molecular dynamic simulations of novel Benzofuran-tetrazole derivatives as potential agents against Alzheimer’s disease. Bioorg. Med. Chem. Lett. 2019, 29, 66–72. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; Zhu, S.; Ma, Y.; Wen, R.; Cen, L.; Gong, P.; Wang, J. A Simple and Efficient Synthesis of Highly Substituted Indeno[1,2-b]pyrrole and Acenaphtho[1,2-b]pyrrole Derivatives by Tandem Three-Component Reactions. Molecules 2018, 23, 3031. [Google Scholar] [CrossRef] [PubMed]
- Scotti, L.; Scotti, M.T. Computer Aided Drug Design Studies in the Discovery of Secondary Metabolites Targeted Against Age-Related Neurodegenerative Diseases. Curr. Top. Med. Chem. 2015, 15, 2239–2252. [Google Scholar] [CrossRef]
- Ghorbani-Vaghei, R.; Toghraei-Semiromi, Z.; Karimi-Nami, R. One-pot synthesis of 4H-chromene and dihydropyrano[3, 2-c]chromene derivatives in hydroalcoholic media. J. Braz. Chem. Soc. 2011, 22, 905–909. [Google Scholar] [CrossRef]
- Mehrabi, H.; Kazemi-Mireki, M. CuO nanoparticles: An efficient and recyclable nanocatalyst for the rapid and green synthesis of 3,4-dihydropyrano[c]chromenes. Chin. Chem. Lett. 2011, 22, 1419–1422. [Google Scholar] [CrossRef]
- Heravi, M.M.; Zakeri, M.; Mohammadi, N. Morpholine Catalyzed One-pot Multicomponent Synthesis of Compounds Containing Chromene Core in Water. Chin. J. Chem. 2011, 29, 1163–1166. [Google Scholar] [CrossRef]
- Celik, F.; Unver, Y.; Barut, B.; Ozel, A.; Sancak, K. Synthesis, Characterization and Biological Activities of New Symmetric Bis-1,2,3-Triazoles with Click Chemistry. Med. Chem. 2018, 14, 230–241. [Google Scholar] [CrossRef]
- Sciú, M.L.; Sebastián-Pérez, V.; Martinez-Gonzalez, L.; Benitez, R.; Perez, D.I.; Pérez, C.; Campillo, N.E.; Martinez, A.; Moyano, E.L. Computer-aided molecular design of pyrazolotriazines targeting glycogen synthase kinase 3. J. Enzym. Inhib. Med. Chem. 2019, 34, 87–96. [Google Scholar] [CrossRef]
- Olesen, P.H.; Sørensen, A.R.; Ursø, B.; Kurtzhals, P.; Bowler, A.N.; Ehrbar, U.; Hansen, B.F. Synthesis and in Vitro Characterization of 1-(4-Aminofurazan-3-yl)-5-dialkylaminomethyl-1 H -[1,2,3]triazole-4-carboxylic Acid Derivatives. A New Class of Selective GSK-3 Inhibitors. J. Med. Chem. 2003, 46, 3333–3341. [Google Scholar] [CrossRef]
- El-Saghier, A.M.; Mohamed, M.A.; Abd-Allah, O.A.; Kadry, A.M.; Ibrahim, T.M.; Bekhit, A.A. Green synthesis, antileishmanial activity evaluation, and in silico studies of new amino acid-coupled 1,2,4-triazoles. Med. Chem. Res. 2019, 28, 169–181. [Google Scholar] [CrossRef]
- Fereidoonnezhad, M.; Mostoufi, A.; Eskandari, M.; Zali, S.; Aliyan, F. Multitarget Drug Design, Molecular Docking and PLIF Studies of Novel Tacrine-Coumarin Hybrids for the Treatment of Alzheimer’s Disease. Iran. J. Pharm. Res. 2018, 17, 1217–1228. [Google Scholar] [PubMed]
- Zhang, H.Y. One-compound-multiple-targets strategy to combat Alzheimer’s disease. FEBS Lett. 2005, 579, 5260–5264. [Google Scholar] [CrossRef] [PubMed]
- Das, M.; Prakash, S.; Nayak, C.; Thangavel, N.; Singh, S.K.; Manisankar, P.; Devi, K.P. Dihydroactinidiolide, a natural product against Aβ25–35 induced toxicity in Neuro2a cells: Synthesis, in silico and in vitro studies. Bioorg. Chem. 2018, 81, 340–349. [Google Scholar] [CrossRef]
- Porcal, W.; Hernández, P.; González, M.; Ferreira, A.; Olea-Azar, C.; Cerecetto, H.; Castro, A. Heteroarylnitrones as Drugs for Neurodegenerative Diseases: Synthesis, Neuroprotective Properties, and Free Radical Scavenger Properties. J. Med. Chem. 2008, 51, 6150–6159. [Google Scholar] [CrossRef] [PubMed]
- Chandra, N. Computational approaches for drug target identification in pathogenic diseases. Expert Opin. Drug Discov. 2011, 6, 975–979. [Google Scholar] [CrossRef]
- Malathi, K.; Ramaiah, S. Bioinformatics approaches for new drug discovery: A review. Biotechnol. Genet. Eng. Rev. 2018, 34, 243–260. [Google Scholar] [CrossRef]
- Liu, C.; Constantinides, P.P.; Li, Y. Research and development in drug innovation: Reflections from the 2013 bioeconomy conference in China, lessons learned and future perspectives. Acta Pharm. Sin. B 2014, 4, 112–119. [Google Scholar] [CrossRef]
- Sehgal, S.A. Pharmacoinformatics, Adaptive Evolution, and Elucidation of Six Novel Compounds for Schizophrenia Treatment by Targeting DAOA (G72) Isoforms. Biomed. Res. Int. 2017, 2017, 1–19. [Google Scholar] [CrossRef]
- Sehgal, S.A.; Khattak, N.A.; Mir, A. Structural, phylogenetic and docking studies of D-amino acid oxidase activator (DAOA), a candidate schizophrenia gene. Biol. Med. Model. 2013, 10, 3–16. [Google Scholar] [CrossRef]
- Yu, W.; MacKerell, A.D. Computer-Aided Drug Design Methods. In Methods in Molecular Biology; Humana Press: New York, NY, USA, 2017; Volume 1520, pp. 85–106. [Google Scholar]
- Lounnas, V.; Ritschel, T.; Kelder, J.; McGuire, R.; Bywater, R.P.; Foloppe, N. Current progress in structure-based rational drug design marks a new mindset in drug discovery. Comput. Struct. Biotechnol. J. 2013, 5, e201302011. [Google Scholar] [CrossRef] [PubMed]
- Schenone, M.; Dančík, V.; Wagner, B.K.; Clemons, P.A. Target identification and mechanism of action in chemical biology and drug discovery. Nat. Chem. Biol. 2013, 9, 232–240. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.-W.; Wang, J.-W. How cryo-electron microscopy and X-ray crystallography complement each other. Protein Sci. 2017, 26, 32–39. [Google Scholar] [CrossRef] [PubMed]
- Rankin, N.J.; Preiss, D.; Welsh, P.; Burgess, K.E.V.; Nelson, S.M.; Lawlor, D.A.; Sattar, N. The emergence of proton nuclear magnetic resonance metabolomics in the cardiovascular arena as viewed from a clinical perspective. Atherosclerosis 2014, 237, 287–300. [Google Scholar] [CrossRef] [PubMed]
- Carroni, M.; Saibil, H.R. Cryo electron microscopy to determine the structure of macromolecular complexes. Methods 2016, 95, 78–85. [Google Scholar] [CrossRef]
- Callaway, E. The revolution will not be crystallized: A new method sweeps through structural biology. Nature 2015, 525, 172–174. [Google Scholar] [CrossRef]
- Forrest, L.R.; Tang, C.L.; Honig, B. On the Accuracy of Homology Modeling and Sequence Alignment Methods Applied to Membrane Proteins. Biophys. J. 2006, 91, 508–517. [Google Scholar] [CrossRef]
- Rinné, S.; Kiper, A.K.; Vowinkel, K.S.; Ramírez, D.; Schewe, M.; Bedoya, M.; Aser, D.; Gensler, I.; Netter, M.F.; Stansfeld, P.J.; et al. The molecular basis for an allosteric inhibition of K+-flux gating in K2P channels. eLife 2019, 8, e39476. [Google Scholar] [CrossRef]
- Bedoya, M.; Rinné, S.; Kiper, A.K.; Decher, N.; González, W.; Ramírez, D. TASK Channels Pharmacology: New Challenges in Drug Design. J. Med. Chem. 2019, 62, 10044–10058. [Google Scholar] [CrossRef]
- Ramírez, D.; Concha, G.; Arévalo, B.; Prent-Peñaloza, L.; Zúñiga, L.; Kiper, A.K.; Rinné, S.; Reyes-Parada, M.; Decher, N.; González, W.; et al. Discovery of Novel TASK-3 Channel Blockers Using a Pharmacophore-Based Virtual Screening. Int. J. Mol. Sci. 2019, 20, 4014. [Google Scholar] [CrossRef]
- Pérot, S.; Sperandio, O.; Miteva, M.A.; Camproux, A.-C.; Villoutreix, B.O. Druggable pockets and binding site centric chemical space: A paradigm shift in drug discovery. Drug Discov. Today 2010, 15, 656–667. [Google Scholar] [CrossRef] [PubMed]
- Le Guilloux, V.; Schmidtke, P.; Tuffery, P. Fpocket: An open source platform for ligand pocket detection. BMC Bioinform. 2009, 10, 168–179. [Google Scholar] [CrossRef] [PubMed]
- Durrant, J.D.; McCammon, J.A. Molecular dynamics simulations and drug discovery. BMC Biol. 2011, 9, 71–80. [Google Scholar] [CrossRef] [PubMed]
- Talwar, P.; Sinha, J.; Grover, S.; Rawat, C.; Kushwaha, S.; Agarwal, R.; Taneja, V.; Kukreti, R. Dissecting Complex and Multifactorial Nature of Alzheimer’s Disease Pathogenesis: A Clinical, Genomic, and Systems Biology Perspective. Mol. Neurobiol. 2016, 53, 4833–4864. [Google Scholar] [CrossRef] [PubMed]
- Govoni, S.; Mura, E.; Preda, S.; Racchi, M.; Lanni, C.; Grilli, M.; Zappettini, S.; Salamone, A.; Olivero, G.; Pittaluga, A.; et al. Dangerous Liaisons between Beta-Amyloid and Cholinergic Neurotransmission. Curr. Pharm. Des. 2014, 20, 2525–2538. [Google Scholar] [CrossRef] [PubMed]
- Hardy, J. The Amyloid Hypothesis of Alzheimer’s Disease: Progress and Problems on the Road to Therapeutics. Science 2002, 297, 353–356. [Google Scholar] [CrossRef]
- Butterfield, D.A.; Drake, J.; Pocernich, C.; Castegna, A. Evidence of oxidative damage in Alzheimer’s disease brain: Central role for amyloid β-peptide. Trends Mol. Med. 2001, 7, 548–554. [Google Scholar] [CrossRef]
- Butterfield, D.A.; Boyd-Kimball, D. The critical role of methionine 35 in Alzheimer’s amyloid β-peptide (1–42)-induced oxidative stress and neurotoxicity. Biochim. Biophys. Acta 2005, 1703, 149–156. [Google Scholar] [CrossRef]
- Chaudhary, A.; Maurya, P.K.; Yadav, B.S.; Singh, S.; Mani, A. Current Therapeutic Targets for Alzheimer’s Disease. J. Biomed. 2018, 3, 74–84. [Google Scholar] [CrossRef]
- Danysz, W.; Parsons, C.G. The NMDA receptor antagonist memantine as a symptomatological and neuroprotective treatment for Alzheimer’s disease: Preclinical evidence. Int. J. Geriatr. Psychiatry 2003, 18, S23–S32. [Google Scholar] [CrossRef]
- Fukumoto, H.; Cheung, B.S.; Hyman, B.T.; Irizarry, M.C. β-Secretase Protein and Activity Are Increased in the Neocortex in Alzheimer Disease. Arch. Neurol. 2002, 59, 1381–1389. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.-B.; Lindholm, K.; Yan, R.; Citron, M.; Xia, W.; Yang, X.-L.; Beach, T.; Sue, L.; Wong, P.; Price, D.; et al. Elevated β-secretase expression and enzymatic activity detected in sporadic Alzheimer disease. Nat. Med. 2003, 9, 3–4. [Google Scholar] [CrossRef] [PubMed]
- Perry, E.; Perry, R.; Blessed, G.; Tomlinson, B. Necropsy evidence of central cholinergic deficits in senile dementia. Lancet 1977, 309, 189. [Google Scholar] [CrossRef]
- Park, P.; Sanderson, T.M.; Amici, M.; Choi, S.-L.; Bortolotto, Z.A.; Zhuo, M.; Kaang, B.-K.; Collingridge, G.L. Calcium-Permeable AMPA Receptors Mediate the Induction of the Protein Kinase A-Dependent Component of Long-Term Potentiation in the Hippocampus. J. Neurosci. 2016, 36, 622–631. [Google Scholar] [CrossRef] [PubMed]
- Halliwell, J.V. Physiological mechanisms of cholinergic action in the hippocampus. Prog. Brain Res. 1990, 84, 255–272. [Google Scholar] [PubMed]
- Blalock, E.M.; Geddes, J.W.; Chen, K.C.; Porter, N.M.; Markesbery, W.R.; Landfield, P.W. Incipient Alzheimer’s disease: Microarray correlation analyses reveal major transcriptional and tumor suppressor responses. Proc. Natl. Acad. Sci. USA 2004, 101, 2173–2178. [Google Scholar] [CrossRef] [PubMed]
- Castillo, P.E.; Chiu, C.Q.; Carroll, R.C. Long-term plasticity at inhibitory synapses. Curr. Opin. Neurobiol. 2011, 21, 328–338. [Google Scholar] [CrossRef]
- Garcia-Alloza, M.; Hirst, W.D.; Chen, C.P.L.-H.; Lasheras, B.; Francis, P.T.; Ramírez, M.J. Differential Involvement of 5-HT1B/1D and 5-HT6 Receptors in Cognitive and Non-cognitive Symptoms in Alzheimer’s Disease. Neuropsychopharmacology 2004, 29, 410–416. [Google Scholar] [CrossRef]
- Roca, C.; Requena, C.; Sebastián-Pérez, V.; Malhotra, S.; Radoux, C.; Pérez, C.; Martinez, A.; Antonio Páez, J.; Blundell, T.L.; Campillo, N.E. Identification of new allosteric sites and modulators of AChE through computational and experimental tools. J. Enzym. Inhib. Med. Chem. 2018, 33, 1034–1047. [Google Scholar] [CrossRef]
- Sebastián-Pérez, V.; Roca, C.; Awale, M.; Reymond, J.-L.; Martinez, A.; Gil, C.; Campillo, N.E. Medicinal and Biological Chemistry (MBC) Library: An Efficient Source of New Hits. J. Chem. Inf. Model. 2017, 57, 2143–2151. [Google Scholar] [CrossRef]
- Camps, P.; Cusack, B.; Mallender, W.D.; El Achab, R.E.; Morral, J.; Muñoz-Torrero, D.; Rosenberry, T.L. Huprine X is a novel high-affinity inhibitor of acetylcholinesterase that is of interest for treatment of Alzheimer’s disease. Mol. Pharm. 2000, 57, 409–417. [Google Scholar]
- Marco-Contelles, J.; Unzeta, M.; Bolea, I.; Esteban, G.; Ramsay, R.R.; Romero, A.; Martínez-Murillo, R.; Carreiras, M.C.; Ismaili, L. ASS234, as a New Multi-Target Directed Propargylamine for Alzheimer’s Disease Therapy. Front. Neurosci. 2016, 10, 294–301. [Google Scholar] [CrossRef] [PubMed]
- Green, K.; Fosso, M.; Garneau-Tsodikova, S. Multifunctional Donepezil Analogues as Cholinesterase and BACE1 Inhibitors. Molecules 2018, 23, 3252. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, R.J.; Wong, P.C. Amyloid Precursor Protein Processing and Alzheimer’s Disease. Annu. Rev. Neurosci. 2011, 34, 185–204. [Google Scholar] [CrossRef] [PubMed]
- Tapia-Rojas, C.; Inestrosa, N. Loss of canonical Wnt signaling is involved in the pathogenesis of Alzheimer’s disease. Neural Regen. Res. 2018, 13, 1705–1710. [Google Scholar] [PubMed]
- Atkinson, B.N.; Steadman, D.; Zhao, Y.; Sipthorp, J.; Vecchia, L.; Ruza, R.R.; Jeganathan, F.; Lines, G.; Frew, S.; Monaghan, A.; et al. Discovery of 2-phenoxyacetamides as Inhibitors of the Wnt-depalmitoleating enzyme NOTUM from an X-ray Fragment Screen. Medchemcomm 2019, 10, 1361–1369. [Google Scholar] [CrossRef]
- Gameiro, I.; Michalska, P.; Tenti, G.; Cores, Á.; Buendia, I.; Rojo, A.I.; Georgakopoulos, N.D.; Hernández-Guijo, J.M.; Teresa Ramos, M.; Wells, G.; et al. Discovery of the first dual GSK3β inhibitor/Nrf2 inducer. A new multitarget therapeutic strategy for Alzheimer’s disease. Sci. Rep. 2017, 7, 45701. [Google Scholar] [CrossRef]
- Itoh, K.; Chiba, T.; Takahashi, S.; Ishii, T.; Igarashi, K.; Katoh, Y.; Oyake, T.; Hayashi, N.; Satoh, K.; Hatayama, I.; et al. An Nrf2/Small Maf Heterodimer Mediates the Induction of Phase II Detoxifying Enzyme Genes through Antioxidant Response Elements. Biochem. Biophys. Res. Commun. 1997, 236, 313–322. [Google Scholar] [CrossRef]
- Ross, C.A.; Pickart, C.M. The ubiquitin–proteasome pathway in Parkinson’s disease and other neurodegenerative diseases. Trends Cell Biol. 2004, 14, 703–711. [Google Scholar] [CrossRef]
- Polymeropoulos, M.H.; Lavedan, C.; Leroy, E.; Ide, S.E.; Dehejia, A.; Dutra, A.; Pike, B.; Root, H.; Rubenstein, J.; Boyer, R.; et al. Mutation in the -Synuclein Gene Identified in Families with Parkinson’s Disease. Sci. Dly. 1997, 276, 2045–2047. [Google Scholar] [CrossRef]
- Leroy, E.; Boyer, R.; Auburger, G.; Leube, B.; Ulm, G.; Mezey, E.; Harta, G.; Brownstein, M.J.; Jonnalagada, S.; Chernova, T.; et al. The ubiquitin pathway in Parkinson’s disease. Nature 1998, 395, 451–452. [Google Scholar] [CrossRef] [PubMed]
- Martin, J.B. Molecular Basis of the Neurodegenerative Disorders. N. Engl. J. Med. 1999, 340, 1970–1980. [Google Scholar] [CrossRef] [PubMed]
- Mhyre, T.R.; Boyd, J.T.; Hamill, R.W.; Maguire-Zeiss, K.A. Parkinson’s Disease. Subcell. Biochem. 2012, 65, 389–455. [Google Scholar] [PubMed]
- Goldenberg, M.M. Medical management of Parkinson’s disease. PT 2008, 33, 590–606. [Google Scholar]
- Learmonth, D.A.; Vieira-Coelho, M.A.; Benes, J.; Alves, P.C.; Borges, N.; Freitas, A.P.; Soares-da-Silva, P. Synthesis of 1-(3,4-Dihydroxy-5-nitrophenyl)-2-phenyl-ethanone and Derivatives as Potent and Long-Acting Peripheral Inhibitors of Catechol- O -methyltransferase. J. Med. Chem. 2002, 45, 685–695. [Google Scholar] [CrossRef]
- Learmonth, D.A.; Palma, P.N.; Vieira-Coelho, M.A.; Soares-da-Silva, P. Synthesis, Biological Evaluation, and Molecular Modeling Studies of a Novel, Peripherally Selective Inhibitor of Catechol- O -methyltransferase. J. Med. Chem. 2004, 47, 6207–6217. [Google Scholar] [CrossRef]
- Schwede, T. Protein modeling: What happened to the “protein structure gap”? Structure 2013, 21, 1531–1540. [Google Scholar] [CrossRef]
- Vyas, V.K.; Ukawala, R.D.; Ghate, M.; Chintha, C. Homology modeling a fast tool for drug discovery: Current perspectives. Indian J. Pharm. Sci. 2012, 74, 1–17. [Google Scholar] [CrossRef]
- Chothia, C.; Lesk, A.M. The relation between the divergence of sequence and structure in proteins. EMBO J. 1986, 5, 823–826. [Google Scholar] [CrossRef]
- Makhouri, F.R.; Ghasemi, J.B. In Silico Studies in Drug Research Against Neurodegenerative Diseases. Curr. Neuropharmacol. 2018, 16, 664–725. [Google Scholar] [CrossRef]
- Cavasotto, C.N. Homology models in docking and high-throughput docking. Curr. Top. Med. Chem. 2011, 11, 1528–1534. [Google Scholar] [CrossRef] [PubMed]
- Eberini, I.; Daniele, S.; Parravicini, C.; Sensi, C.; Trincavelli, M.L.; Martini, C.; Abbracchio, M.P. In silico identification of new ligands for GPR17: A promising therapeutic target for neurodegenerative diseases. J. Comput. Aided. Mol. Des. 2011, 25, 743–752. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.-Y.; Kim, Y.-M. Comparative Homology Modeling and Ligand Docking Study of Human Catechol-O-Methyltransferase for Antiparkinson Drug Design. Bull. Korean Chem. Soc. 2005, 26, 1695–1700. [Google Scholar]
- Dhanavade, M.J.; Jalkute, C.B.; Barage, S.H.; Sonawane, K.D. Homology modeling, molecular docking and MD simulation studies to investigate role of cysteine protease from Xanthomonas campestris in degradation of Aβ peptide. Comput. Biol. Med. 2013, 43, 2063–2070. [Google Scholar] [CrossRef] [PubMed]
- Mueller-Steiner, S.; Zhou, Y.; Arai, H.; Roberson, E.D.; Sun, B.; Chen, J.; Wang, X.; Yu, G.; Esposito, L.; Mucke, L.; et al. Antiamyloidogenic and neuroprotective functions of cathepsin B: Implications for Alzheimer’s disease. Neuron 2006, 51, 703–714. [Google Scholar] [CrossRef]
- Roos, R.A. Huntington’s disease: A clinical review. Orphanet J. Rare Dis. 2010, 5, 40–48. [Google Scholar] [CrossRef]
- Walker, F.O. Huntington’s disease. Lancet 2007, 369, 218–228. [Google Scholar] [CrossRef]
- Andrew, S.E.; Paul Goldberg, Y.; Kremer, B.; Telenius, H.; Theilmann, J.; Adam, S.; Starr, E.; Squitieri, F.; Lin, B.; Kalchman, M.A.; et al. The relationship between trinucleotide (CAG) repeat length and clinical features of Huntington’s disease. Nat. Genet. 1993, 4, 398–403. [Google Scholar] [CrossRef]
- Bashir, H. Emerging therapies in Huntington’s disease. Expert Rev. Neurother. 2019, 19, 983–995. [Google Scholar] [CrossRef]
- Mestre, T.A. Recent advances in the therapeutic development for Huntington disease. Parkinsonism Relat. Disord. 2019, 59, 125–130. [Google Scholar] [CrossRef]
- Cattaneo, E.; Zuccato, C.; Tartari, M. Normal huntingtin function: An alternative approach to Huntington’s disease. Nat. Rev. Neurosci. 2005, 6, 919–930. [Google Scholar] [CrossRef] [PubMed]
- Zuccato, C.; Valenza, M.; Cattaneo, E. Molecular Mechanisms and Potential Therapeutical Targets in Huntington’s Disease. Physiol. Rev. 2010, 90, 905–981. [Google Scholar] [CrossRef] [PubMed]
- Saudou, F.; Humbert, S. The Biology of Huntingtin. Neuron 2016, 89, 910–926. [Google Scholar] [CrossRef] [PubMed]
- Arrasate, M.; Finkbeiner, S. Protein aggregates in Huntington’s disease. Exp. Neurol. 2012, 238, 1–11. [Google Scholar] [CrossRef]
- Neueder, A.; Landles, C.; Ghosh, R.; Howland, D.; Myers, R.H.; Faull, R.L.M.; Tabrizi, S.J.; Bates, G.P. The pathogenic exon 1 HTT protein is produced by incomplete splicing in Huntington’s disease patients. Sci. Rep. 2017, 7, 1307. [Google Scholar] [CrossRef]
- Richard, A.; Frank, S. Deutetrabenazine in the treatment of Huntington’s disease. Neurodegener. Dis. Manag. 2019, 9, 31–37. [Google Scholar] [CrossRef]
- Conforti, P.; Zuccato, C.; Gaudenzi, G.; Ieraci, A.; Camnasio, S.; Buckley, N.J.; Mutti, C.; Cotelli, F.; Contini, A.; Cattaneo, E. Binding of the repressor complex REST-mSIN3b by small molecules restores neuronal gene transcription in Huntington’s disease models. J. Neurochem. 2013, 127, 22–35. [Google Scholar] [CrossRef]
- Nyamoya, S.; Leopold, P.; Becker, B.; Beyer, C.; Hustadt, F.; Schmitz, C.; Michel, A.; Kipp, M. G-Protein-Coupled Receptor Gpr17 Expression in Two Multiple Sclerosis Remyelination Models. Mol. Neurobiol. 2019, 56, 1109–1123. [Google Scholar] [CrossRef]
- Mogha, A.; D’Rozario, M.; Monk, K.R. G Protein-Coupled Receptors in Myelinating Glia. Trends Pharm. Sci. 2016, 37, 977–987. [Google Scholar] [CrossRef]
- Lu, C.; Dong, L.; Zhou, H.; Li, Q.; Huang, G.; Bai, S.J.; Liao, L. G-Protein-Coupled Receptor Gpr17 Regulates Oligodendrocyte Differentiation in Response to Lysolecithin-Induced Demyelination. Sci. Rep. 2018, 8, 4502. [Google Scholar] [CrossRef]
- Kessler, R.C.; Berglund, P.; Demler, O.; Jin, R.; Koretz, D.; Merikangas, K.R.; Rush, A.J.; Walters, E.E.; Wang, P.S. The Epidemiology of Major Depressive Disorder. JAMA 2003, 289, 3095–3105. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, A.J.; Charlson, F.J.; Norman, R.E.; Patten, S.B.; Freedman, G.; Murray, C.J.L.; Vos, T.; Whiteford, H.A. Burden of Depressive Disorders by Country, Sex, Age, and Year: Findings from the Global Burden of Disease Study 2010. PLoS Med. 2013, 10, e1001547. [Google Scholar] [CrossRef] [PubMed]
- Artigas, F. Serotonin receptors involved in antidepressant effects. Pharmacol. Ther. 2013, 137, 119–131. [Google Scholar] [CrossRef] [PubMed]
- Nautiyal, K.M.; Hen, R. Serotonin receptors in depression: From A to B. F1000Research 2017, 6, 123–135. [Google Scholar] [CrossRef] [PubMed]
- Gupta, D.; Radhakrishnan, M.; Kurhe, Y. Ondansetron, a 5HT3 receptor antagonist reverses depression and anxiety-like behavior in streptozotocin-induced diabetic mice: Possible implication of serotonergic system. Eur. J. Pharm. 2014, 744, 59–66. [Google Scholar] [CrossRef]
- Bétry, C.; Etiévant, A.; Oosterhof, C.; Ebert, B.; Sanchez, C.; Haddjeri, N. Role of 5-HT3 Receptors in the Antidepressant Response. Pharmaceuticals 2011, 4, 603–629. [Google Scholar] [CrossRef]
- Rajkumar, R.; Mahesh, R. Review: The auspicious role of the 5-HT3 receptor in depression: A probable neuronal target? J. Psychopharmacol. 2010, 24, 455–469. [Google Scholar] [CrossRef]
- Reeves, D.C.; Sayed, M.F.R.; Chau, P.-L.; Price, K.L.; Lummis, S.C.R. Prediction of 5-HT3 Receptor Agonist-Binding Residues Using Homology Modeling. Biophys. J. 2003, 84, 2338–2344. [Google Scholar] [CrossRef]
- Price, K.L.; Lillestol, R.K.; Ulens, C.; Lummis, S.C.R. Palonosetron–5-HT 3 Receptor Interactions As Shown by a Binding Protein Cocrystal Structure. ACS Chem. Neurosci. 2016, 7, 1641–1646. [Google Scholar] [CrossRef]
















© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Morales-Navarro, S.; Prent-Peñaloza, L.; Rodríguez Núñez, Y.A.; Sánchez-Aros, L.; Forero-Doria, O.; González, W.; Campilllo, N.E.; Reyes-Parada, M.; Martínez, A.; Ramírez, D. Theoretical and Experimental Approaches Aimed at Drug Design Targeting Neurodegenerative Diseases. Processes 2019, 7, 940. https://doi.org/10.3390/pr7120940
Morales-Navarro S, Prent-Peñaloza L, Rodríguez Núñez YA, Sánchez-Aros L, Forero-Doria O, González W, Campilllo NE, Reyes-Parada M, Martínez A, Ramírez D. Theoretical and Experimental Approaches Aimed at Drug Design Targeting Neurodegenerative Diseases. Processes. 2019; 7(12):940. https://doi.org/10.3390/pr7120940
Chicago/Turabian StyleMorales-Navarro, Samuel, Luis Prent-Peñaloza, Yeray A. Rodríguez Núñez, Laura Sánchez-Aros, Oscar Forero-Doria, Wendy González, Nuria E. Campilllo, Miguel Reyes-Parada, Ana Martínez, and David Ramírez. 2019. "Theoretical and Experimental Approaches Aimed at Drug Design Targeting Neurodegenerative Diseases" Processes 7, no. 12: 940. https://doi.org/10.3390/pr7120940
APA StyleMorales-Navarro, S., Prent-Peñaloza, L., Rodríguez Núñez, Y. A., Sánchez-Aros, L., Forero-Doria, O., González, W., Campilllo, N. E., Reyes-Parada, M., Martínez, A., & Ramírez, D. (2019). Theoretical and Experimental Approaches Aimed at Drug Design Targeting Neurodegenerative Diseases. Processes, 7(12), 940. https://doi.org/10.3390/pr7120940