Molecular Insights into Cage Occupancy of Hydrogen Hydrate: A Computational Study

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Computational Details
3. Results and Discussion
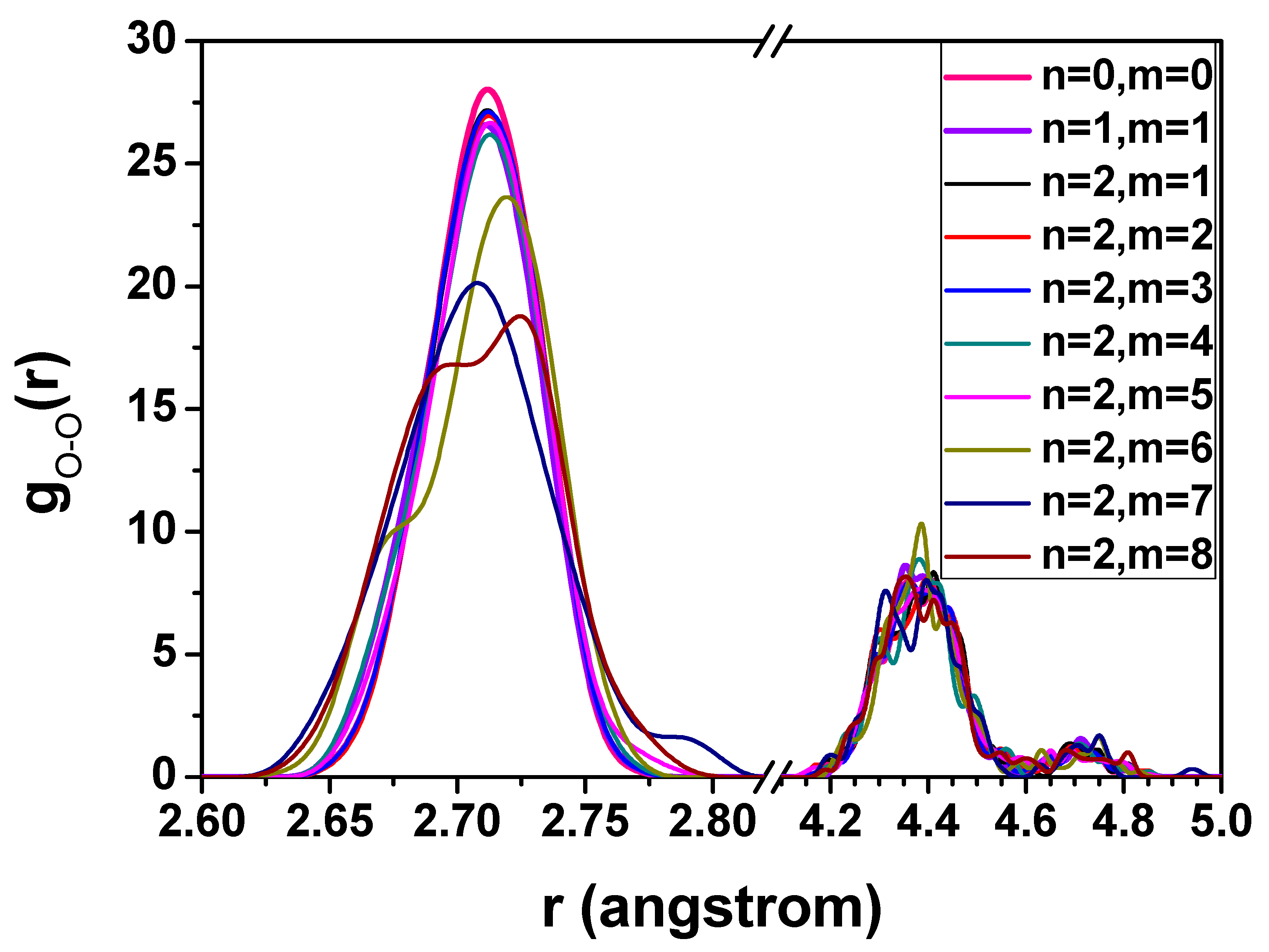
3.1. The Hydrate Stability with Respect to the Hydrogen Occupancy
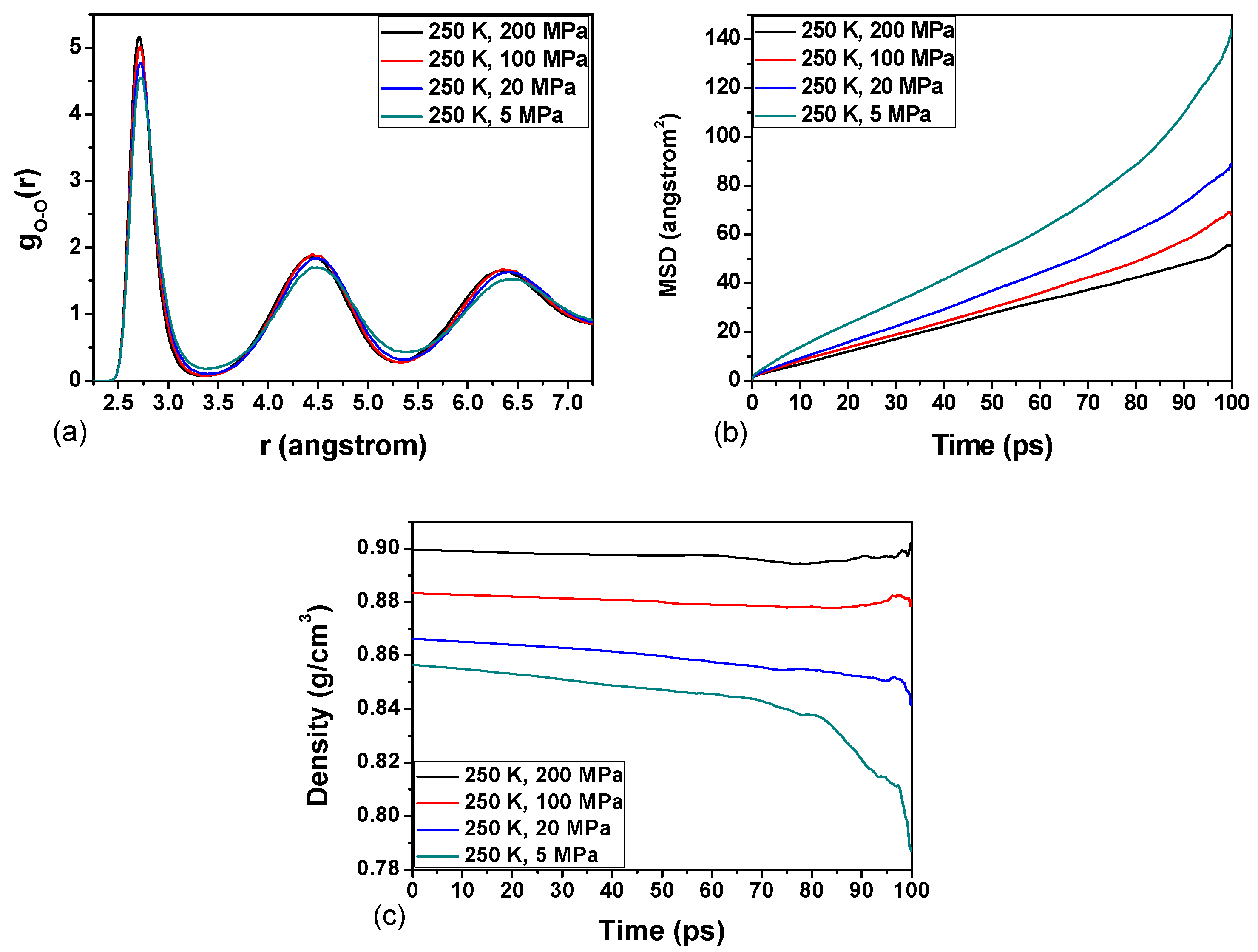
3.2. Effect of Pressure on the Hydrate Stability
3.3. Effect of Temperature on the Hydrate Stability
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Dresselhaus, M.S.; Thomas, I.L. Alternative energy technologies. Nature 2001, 414, 332–337. [Google Scholar] [CrossRef] [PubMed]
- Getman, R.B.; Bae, Y.-S.; Wilmer, C.E.; Snurr, R.Q. Review and analysis of molecular simulations of methane, hydrogen, and acetylene storage in metal–organic frameworks. Chem. Rev. 2012, 112, 703–723. [Google Scholar] [CrossRef] [PubMed]
- Schlapbach, L.; Zuttel, A. Hydrogen-storage materials for mobile applications. Nature 2001, 414, 353–358. [Google Scholar] [CrossRef] [PubMed]
- Jena, P. Materials for hydrogen storage: Past, present, and future. J. Phys. Chem. Lett. 2011, 2, 206–211. [Google Scholar] [CrossRef]
- Schüth, F. Encapsulation strategies in energy conversion materials. Chem. Mater. 2014, 26, 423–434. [Google Scholar] [CrossRef]
- Mao, W.L.; Mao, H.-K. Hydrogen storage in molecular compounds. Proc. Nalt. Acad. Sci. USA 2004, 101, 708–710. [Google Scholar] [CrossRef]
- Lee, H.; Lee, J.-W.; Kim, D.Y.; Park, J.; Seo, Y.-T.; Zeng, H.; Moudrakovski, I.L.; Ratcliffe, C.I.; Ripmeester, J.A. Tuning clathrate hydrates for hydrogen storage. Nature 2005, 434, 743–746. [Google Scholar] [CrossRef]
- Román-Pérez, G.; Moaied, M.; Soler, J.M.; Yndurain, F. Stability, adsorption, and diffusion of CH4, CO2, and H2 in clathrate hydrates. Phys. Rev. Lett. 2010, 105, 145901. [Google Scholar] [CrossRef]
- Lu, H.; Wang, J.; Liu, C.; Ratcliffe, C.I.; Becker, U.; Kumar, R.; Ripmeester, J. Multiple H2 occupancy of cages of clathrate hydrate under mild conditions. J. Am. Chem. Soc. 2012, 134, 9160–9162. [Google Scholar] [CrossRef]
- Willow, S.Y.; Xantheas, S.S. Enhancement of hydrogen storage capacity in hydrate lattices. Chem. Phys. Lett. 2012, 525, 13–18. [Google Scholar] [CrossRef]
- Sloan, E.D. Fundamental principles and applications of natural gas hydrates. Nature 2003, 426, 353–363. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Du, S.; Yu, X.; Zhang, J.; Xu, H.; Vogel, S.C.; Germann, T.C.; Francisco, J.S.; Izumi, F.; Momma, K.; et al. Encapsulation kinetics and dynamics of carbon monoxide in clathrate hydrate. Nat. Commun. 2014, 5, 4128. [Google Scholar] [CrossRef] [PubMed]
- Mao, W.L.; Mao, H.-k.; Goncharov, A.F.; Struzhkin, V.V.; Guo, Q.; Hu, J.; Shu, J.; Hemley, R.J.; Somayazulu, M.; Zhao, Y. Hydrogen clusters in clathrate hydrate. Science 2002, 297, 2247–2249. [Google Scholar] [CrossRef]
- Patchkovskii, S.; Tse, J.S. Thermodynamic stability of hydrogen clathrates. Proc. Natl. Acad. Sci. USA 2003, 100, 14645–14650. [Google Scholar] [CrossRef] [PubMed]
- Sebastianelli, F.; Xu, M.; Bačić, Z. Quantum dynamics of small H2 and D2 clusters in the large cage of structure ii clathrate hydrate: Energetics, occupancy, and vibrationally averaged cluster structures. J. Chem. Phys. 2008, 129, 244706. [Google Scholar] [CrossRef]
- Ranieri, U.; Koza, M.M.; Kuhs, W.F.; Gaal, R.; Klotz, S.; Falenty, A.; Wallacher, D.; Ollivier, J.; Gillet, P.; Bove, L.E. Quantum dynamics of H2 and D2 confined in hydrate structures as a function of pressure and temperature. J. Phys. Chem. C 2019, 123, 1888–1903. [Google Scholar] [CrossRef]
- Witt, A.; Sebastianelli, F.; Tuckerman, M.E.; Bačić, Z. Path integral molecular dynamics study of small H2 clusters in the large cage of structure II clathrate hydrate: Temperature dependence of quantum spatial distributions. J. Phys. Chem. C 2010, 114, 20775–20782. [Google Scholar] [CrossRef]
- Burnham, C.J.; Futera, Z.; English, N.J. Study of hydrogen-molecule guests in type II clathrate hydrates using a force-matched potential model parameterised from ab initio molecular dynamics. J. Chem. Phys. 2018, 148, 102323. [Google Scholar] [CrossRef]
- Wang, J.; Lu, H.; Ripmeester, J.A. Raman spectroscopy and cage occupancy of hydrogen clathrate hydrate from first-principle calculations. J. Am. Chem. Soc. 2009, 131, 14132–14133. [Google Scholar] [CrossRef]
- Gorman, P.D.; English, N.J.; MacElroy, J.M.D. Dynamical cage behaviour and hydrogen migration in hydrogen and hydrogen-tetrahydrofuran clathrate hydrates. J. Chem. Phys. 2012, 136, 044506. [Google Scholar] [CrossRef]
- Papadimitriou, N.I.; Tsimpanogiannis, I.N.; Economou, I.G.; Stubos, A.K. The effect of lattice constant on the storage capacity of hydrogen hydrates: A Monte Carlo Study. Mol. Phys. 2016, 114, 2664–2671. [Google Scholar] [CrossRef]
- Brumby, P.E.; Yuhara, D.; Hasegawa, T.; Wu, D.T.; Sum, A.K.; Yasuoka, K. Cage occupancies, lattice constants, and guest chemical potentials for structure II hydrogen clathrate hydrate from Gibbs ensemble Monte Carlo simulations. J. Chem. Phys. 2019, 150, 134503. [Google Scholar] [CrossRef] [PubMed]
- Lokshin, K.A.; Zhao, Y.; He, D.; Mao, W.L.; Mao, H.-K.; Hemley, R.J.; Lobanov, M.V.; Greenblatt, M. Structure and dynamics of hydrogen molecules in the novel clathrate hydrate by high pressure neutron diffraction. Phys. Rev. Lett. 2004, 93, 125503. [Google Scholar] [CrossRef] [PubMed]
- Inerbaev, T.M.; Belosludov, V.R.; Belosludov, R.V.; Sluiter, M.; Kawazoe, Y. Dynamics and equation of state of hydrogen clathrate hydrate as a function of cage occupation. Comput. Mater. Sci. 2006, 36, 229–233. [Google Scholar] [CrossRef]
- Del Rosso, L.; Celli, M.; Ulivi, L. Raman measurements of pure hydrogen clathrate formation from a supercooled hydrogen–water solution. J. Phys. Chem. Lett. 2015, 6, 4309–4313. [Google Scholar] [CrossRef]
- English, N.J.; MacElroy, J.M.D. Perspectives on molecular simulation of clathrate hydrates: Progress, prospects and challenges. Chem. Eng. Sci. 2015, 121, 133–156. [Google Scholar] [CrossRef]
- Rasoolzadeh, A.; Shariati, A. Hydrogen hydrate cage occupancy: A key parameter for hydrogen storage and transport. Fluid Phase Equilib. 2019, 494, 8–20. [Google Scholar] [CrossRef]
- Sugahara, T.; Haag, J.C.; Prasad, P.S.R.; Warntjes, A.A.; Sloan, E.D.; Sum, A.K.; Koh, C.A. Increasing hydrogen storage capacity using tetrahydrofuran. J. Am. Chem. Soc. 2009, 131, 14616–14617. [Google Scholar] [CrossRef]
- Chapoy, A.; Anderson, R.; Tohidi, B. Low-pressure molecular hydrogen storage in semi-clathrate hydrates of quaternary ammonium compounds. J. Am. Chem. Soc. 2007, 129, 746–747. [Google Scholar] [CrossRef]
- Cai, J.; Tao, Y.-Q.; von Solms, N.; Xu, C.-G.; Chen, Z.-Y.; Li, X.-S. Experimental studies on hydrogen hydrate with tetrahydrofuran by differential scanning calorimeter and in-situ Raman. Appl. Energ. 2019, 243, 1–9. [Google Scholar] [CrossRef]
- Liu, J.; Yan, Y.; Chen, G.; Hou, J.; Yan, Y.; Liu, H.; Li, S.; Zhang, J. Prediction of efficient promoter molecules of sh hydrogen hydrate: An ab initio study. Chem. Phys. 2019, 516, 15–21. [Google Scholar] [CrossRef]
- Kaur, S.P.; Ramachandran, C.N. Hydrogen-tetrahydrofuran mixed hydrates: A computational study. Int. J. Hydrogen Energy 2018, 43, 19559–19566. [Google Scholar] [CrossRef]
- Liu, J.; Hou, J.; Xu, J.; Liu, H.; Chen, G.; Zhang, J. Ab initio study of the molecular hydrogen occupancy in pure H2 and binary H2-THF clathrate hydrates. Int. J. Hydrogen Energy 2017, 42, 17136–17143. [Google Scholar] [CrossRef]
- Florusse, L.J.; Peters, C.J.; Schoonman, J.; Hester, K.C.; Koh, C.A.; Dec, S.F.; Marsh, K.N.; Sloan, E.D. Stable low-pressure hydrogen clusters stored in a binary clathrate hydrate. Science 2004, 306, 469–471. [Google Scholar] [CrossRef]
- Lenz, A.; Ojamäe, L. Structures of the I-, II- and H-methane clathrates and the ice−methane clathrate phase transition from quantum-chemical modeling with force-field thermal corrections. J. Phys. Chem. A 2011, 115, 6169–6176. [Google Scholar] [CrossRef]
- Becke, A.D. A multicenter numerical integration scheme for polyatomic molecules. J. Chem. Phys. 1988, 88, 2547–2553. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the colle-salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Delley, B. Ground-state enthalpies: Evaluation of electronic structure approaches with emphasis on the density functional method. J. Phys. Chem. A 2006, 110, 13632–13639. [Google Scholar] [CrossRef]
- Tkatchenko, A.; Scheffler, M. Accurate molecular van der waals interactions from ground-state electron density and free-atom reference data. Phys. Rev. Lett. 2009, 102, 073005. [Google Scholar] [CrossRef]
- Cox, S.J.; Towler, M.D.; Alfè, D.; Michaelides, A. Benchmarking the performance of density functional theory and point charge force fields in their description of sI methane hydrate against diffusion Monte Carlo. J. Chem. Phys. 2014, 140, 174703. [Google Scholar] [CrossRef]
- Parr, R.G.; Donnelly, R.A.; Levy, M.; Palke, W.E. Electronegativity: The density functional viewpoint. J. Chem. Phys. 1978, 68, 3801–3807. [Google Scholar] [CrossRef]
- Parr, R.G.; Pearson, R.G. Absolute hardness: Companion parameter to absolute electronegativity. J. Am. Chem. Soc. 1983, 105, 7512–7516. [Google Scholar] [CrossRef]
- Parr, R.G.; Szentpály, L.V.; Liu, S. Electrophilicity index. J. Am. Chem. Soc. 1999, 121, 1922–1924. [Google Scholar] [CrossRef]
- Terleczky, P.; Nyulászi, L. Dft study of possible lattice defects in methane-hydrate and their appearance in 13C NMR spectra. Chem. Phys. Lett. 2010, 488, 168–172. [Google Scholar] [CrossRef]
- Verma, C.; Lgaz, H.; Verma, D.K.; Ebenso, E.E.; Bahadur, I.; Quraishi, M.A. Molecular dynamics and Monte Carlo simulations as powerful tools for study of interfacial adsorption behavior of corrosion inhibitors in aqueous phase: A review. J. Mol. Liq. 2018, 260, 99–120. [Google Scholar] [CrossRef]
- Andersen, H.C. Molecular dynamics simulations at constant pressure and/or temperature. J. Chem. Phys. 1980, 72, 2384–2393. [Google Scholar] [CrossRef]
- Berendsen, H.J.C.; Postma, J.P.M.; van Gunsteren, W.F.; DiNola, A.; Haak, J.R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef]
- Ewald, P.P. The calculation of optical and electrostatic grid potential. Ann. Phys. 1921, 64, 253–287. [Google Scholar] [CrossRef]
- Sun, H. COMPASS: An ab initio force-field optimized for condensed-phase applications—Overview with details on alkane and benzene compounds. J. Phys. Chem. B 1998, 102, 7338–7364. [Google Scholar] [CrossRef]
- Rigby, D.; Sun, H.; Eichinger, B.E. Computer simulations of poly(ethylene oxide): Force field, pvt diagram and cyclization behaviour. Polym. Int. 1997, 44, 311–330. [Google Scholar] [CrossRef]
- Sun, H.; Ren, P.; Fried, J.R. The COMPASS force field: Parameterization and validation for phosphazenes. Comput. Theor. Polym. Sci. 1998, 8, 229–246. [Google Scholar] [CrossRef]
- Bedrov, D.; Borodin, O.; Smith, G.D. Molecular dynamics simulation of 1,2-dimethoxyethane/water solutions. 2. Dynamical properties. J. Phys. Chem. B 1998, 102, 9565–9570. [Google Scholar] [CrossRef]
- Martos-Villa, R.; Francisco-Márquez, M.; Mata, M.P.; Sainz-Díaz, C.I. Crystal structure, stability and spectroscopic properties of methane and CO2 hydrates. J. Mol. Graph. Model. 2013, 44, 253–265. [Google Scholar] [CrossRef] [PubMed]







© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ma, R.; Zhong, H.; Liu, J.; Zhong, J.; Yan, Y.; Zhang, J.; Xu, J. Molecular Insights into Cage Occupancy of Hydrogen Hydrate: A Computational Study. Processes 2019, 7, 699. https://doi.org/10.3390/pr7100699
Ma R, Zhong H, Liu J, Zhong J, Yan Y, Zhang J, Xu J. Molecular Insights into Cage Occupancy of Hydrogen Hydrate: A Computational Study. Processes. 2019; 7(10):699. https://doi.org/10.3390/pr7100699
Chicago/Turabian StyleMa, Rui, Hong Zhong, Jinxiang Liu, Jie Zhong, Youguo Yan, Jun Zhang, and Jiafang Xu. 2019. "Molecular Insights into Cage Occupancy of Hydrogen Hydrate: A Computational Study" Processes 7, no. 10: 699. https://doi.org/10.3390/pr7100699
APA StyleMa, R., Zhong, H., Liu, J., Zhong, J., Yan, Y., Zhang, J., & Xu, J. (2019). Molecular Insights into Cage Occupancy of Hydrogen Hydrate: A Computational Study. Processes, 7(10), 699. https://doi.org/10.3390/pr7100699