1. Introduction
Statins, also known as HMG-CoA reductase inhibitors, are known for their efficacy in treating primary hypercholesterolemia [
1]. Moreover, they exert antitumor effects, particularly by inhibiting proliferation and inducing apoptosis in various tumor cells [
2]. Several statins, such as atorvastatin (Lipitor), simvastatin (Zocor), and pravastatin (Pravachol), are widely used [
3,
4]. Among these, simvastatin has been adapted for use in all high-risk patients with cardiovascular disease and diabetes, regardless of the baseline LDL level [
5,
6]. Compared to other statins, simvastatin is a good lipid solvent, and from a pharmacological perspective, its ability to lower cholesterol is strong, so its dosage is relatively low. In the United Kingdom, up to 10 mg of simvastatin has been considered safe, and patients do not need a prescription within this dosage range [
6]. Thus, a huge global demand for simvastatin remains, and techniques for simvastatin synthesis are needed.
The first synthetic simvastatin was manufactured by Merck (Darmstadt, Germany) [
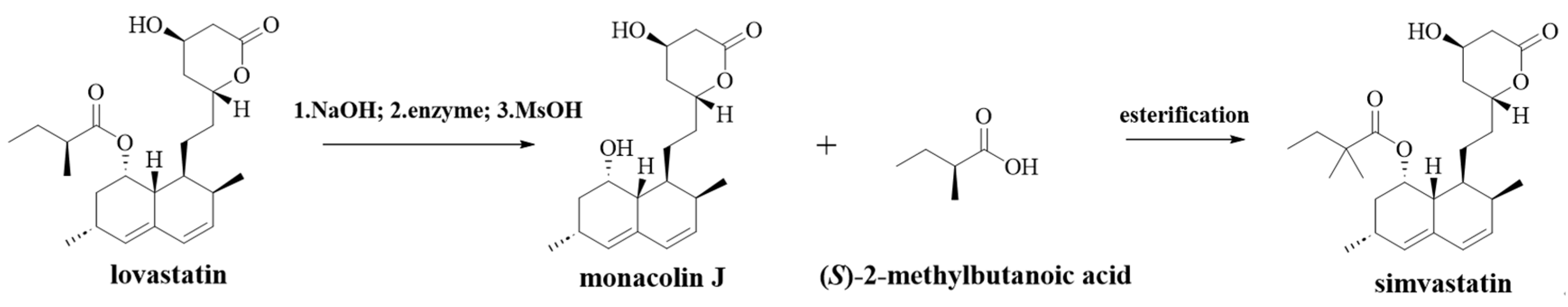
7]. In industry, simvastatin is synthesized using lovastatin as the starting material. Specifically, simvastatin is prepared using lovastatin via amidation, protection of hydroxyl groups using tert-butyl-dimethylsilyl derivatives, methylation, deprotection, alkaline hydrolysis, ammonium salt formation, and cyclization. This route has multiple steps and a low final product yield. Moreover, the reaction process uses hazardous reagents such as n-butyl lithium and hydrofluoric acid, and the reaction conditions are harsh, which is not conducive to scaling up production. Furthermore, there is a need for simple, economic, and environmentally friendly methods for synthesizing simvastatin due to its significantly increased demand. Several simple methods based on monacolin J as an intermediate and lovastatin as the raw material have been developed in previous research studies [
8]. The number of steps is significantly reduced using this approach. In addition, there is no need to use dangerous reagents. The reaction conditions are mild and friendly to the environment. In order to industrially synthesize simvastatin, it is necessary to first prepare monacolin J.
In previous research studies, monacolin J has been transformed using lovastatin through different methods [
9]. Among these approaches, chemical methods are effective regarding synthesis, but extreme conditions and contamination of the environment are typical disadvantages of this technique [
10,
11,
12]. Biological methods for monacolin J synthesis have offered a large number of advantages including mild reaction conditions and environmentally friendly synthesis, but the reaction efficiency needs be improved for it to have practical value [
13,
14,
15,
16,
17]. Lovastatin has a poor water solubility, which restricts the catalytic efficiency of lipolytic enzymes. Enhancing the water solubility of substrates is a common issue in the enzymatic production of small molecule drugs, and multiple methods have been explored for solving the problem. To address this issue, lovastatin salt has been created to enhance biocatalytic efficiency, which has proved to be a suitable method for biocatalytic synthesis. In this work, the biocatalytic process was enhanced by increasing lovastatin solubility. Lovastatin hydrolase, whose amino acid sequence was obtained from Genbank GI RHZ58343.1, was optimized to catalyze the reaction [
18,
19]. Thermostability, the ability of this enzyme to act on a high concentration substrate, and other reaction conditions needed in industry production have been utilized to assess the practical value of this enzymatic method. Furthermore, this bioprocess, which takes advantage of biocatalysis with the wild-type enzyme, exhibited tremendous potential for monacolin J synthesis at an industrial scale.
2. Results and Discussion
2.1. Optimization of Biocatalytic Activity
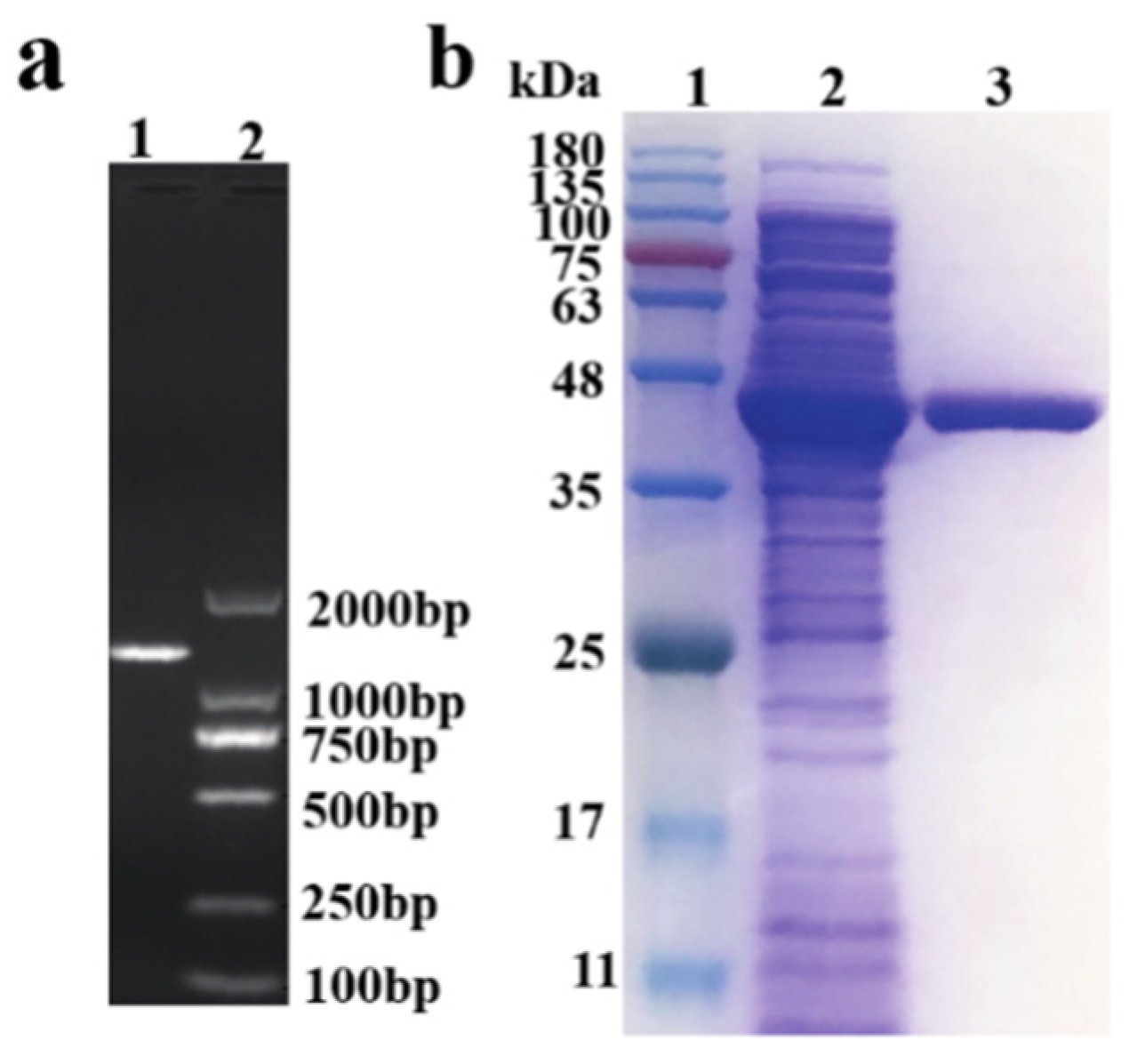
CDV55_102090, an optimized gene from
Aspergillus turcosus, was amplified and overexpressed in
E. coli BL21 (DE3). Before measuring the activity of the lovastatin hydrolase, expression analysis was performed using SDS–PAGE (
Figure 1). There was no difficulty regarding the expression of the wild-type lovastatin hydrolase. The crude enzyme extract was purified and desalted. Then, the initial activity of the enzyme was measured at 30 °C, reaching only 3.4 ± 0.2 U/mL. Low enzyme activity might increase production cost. Thus, it was necessary to employ effective methods to improve enzyme activity.
Reaction conditions such as reaction temperature, pH, and metal ion concentration can affect the activity of enzymes, which in turn affect the reaction conversion rate. Firstly, this work includes a systematic study using the above conditions. In the present study, 10 mM lovastatin was used for optimizing the biotransformation conditions.
2.1.1. Optimization of Reaction Temperature
Reaction temperature is an important parameter in enzyme-catalyzed reactions [
20]. The reaction temperature not only affects the activity of enzymes [
21] but also the degree of vibration between molecules, which is reflected in a difference in the reaction conversion rate [
22]. The activity of enzymes improved as the reaction temperature rose. By contrast, the stability of the enzymes decreased. If the reaction temperature was further increased, the enzyme was prone to lose its activity due to the instability of its structure [
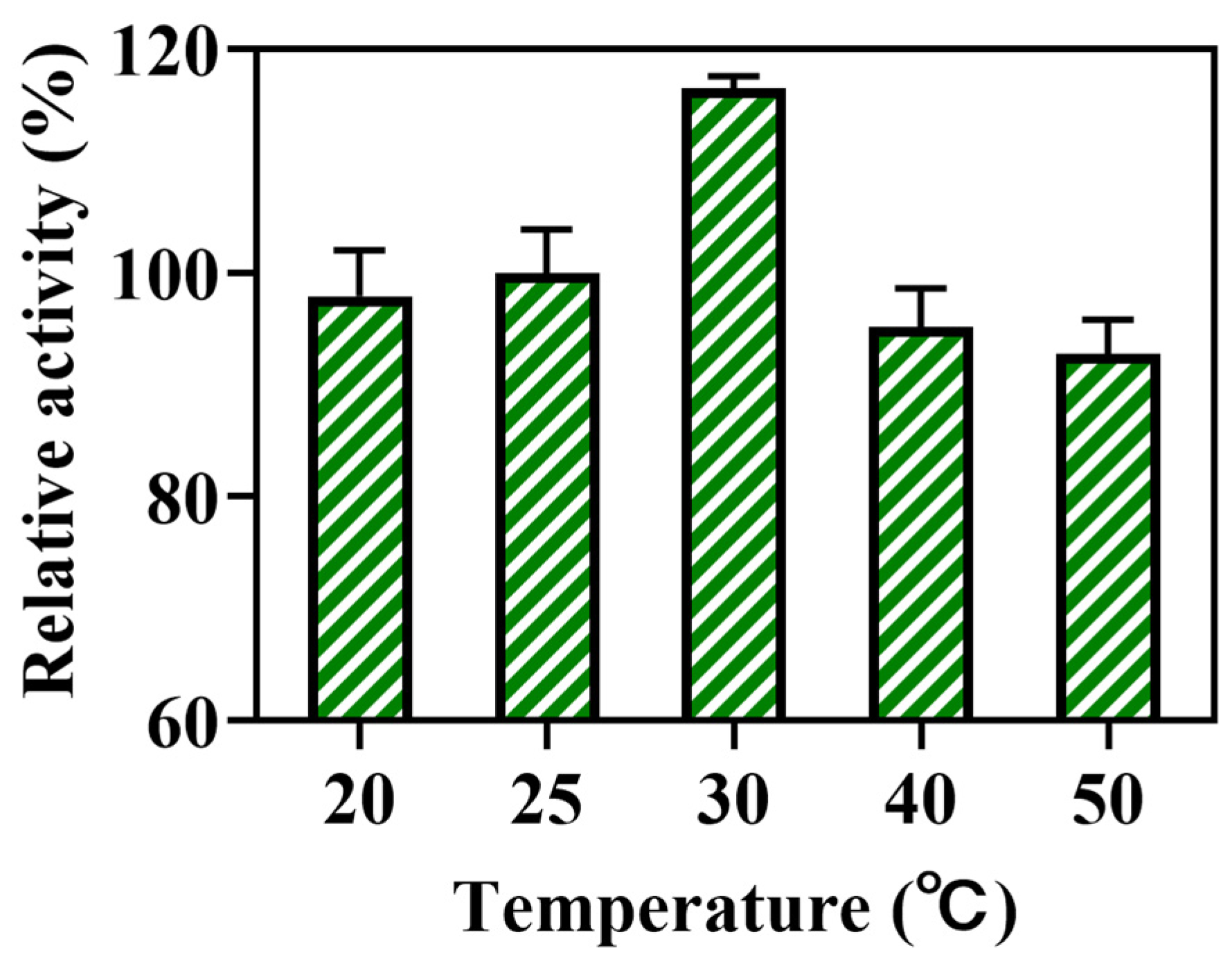
23]. In this work, the effects of different reaction temperatures (20, 25, 30, 40, and 50 °C) on biocatalytic activity were investigated, and the results are shown in
Figure 2. The biocatalytic activity increased gradually when the reaction temperature was raised to 30 °C. The activity of the lovastatin hydrolase decreased slightly when the temperature was raised to 40 °C. When the temperature was raised beyond 40 °C, no significant decrease was observed. The above results demonstrated that the lovastatin hydrolase was relatively stable at high temperatures, as it maintained more than 75% of its initial activity at 30 °C. Thus, the optimum performance temperature was 30 °C, and the wild type of the lovastatin hydrolase showed the potential stability on the higher temperature reaction.
2.1.2. Optimization of Solution pH
One reaction-related problem is a drop in pH of the system due to ester hydrolysis and the generation of byproducts such as (
S)-2-methylbutanoic acid, and a medium pH can apparently affect the biocatalytic activity [
24]. In order to better understand the resistance of the lovastatin hydrolase to acidic and alkaline conditions, we investigated the effect of solution pH (from 4.5 to 8.5) on the biocatalytic activity. The results are presented in
Figure 3. In the biocatalytic reaction, the enzymatic activity was markedly affected when the pH was raised from 4.5 to 7 at 30 °C. As the pH rose, the enzymatic activity improved significantly. When the pH was in the range of 7.5–8.0, the activity reached the highest level in the bioreaction. As the reaction pH rose to 8.5, the lovastatin hydrolase activity markedly decreased. The pH of the bioreaction medium might influence the degree of charge around the enzymes, and the addition of an acid or a base would apparently influence the biocatalytic activity of the enzyme [
25]. Thus, the appropriate pH was 7.5~8.0. Wild-type lovastatin hydrolase showed intolerance to acidic and alkaline conditions, and strict pH control was needed in the reaction.
2.1.3. Optimization of Reaction Solvent
Due to the extreme insolubility of lovastatin in water, the activity of the lovastatin hydrolase was extremely low, reaching only 3.4 ± 0.2 U/mL in the bioreaction system. In previous research studies, water/organic solvent two-phase systems were utilized in various biocatalytic reactions. Researchers discovered multiple advantages, including the protection of enzymes, the prevention of substrate decomposition, and improvements in solubilization [
26,
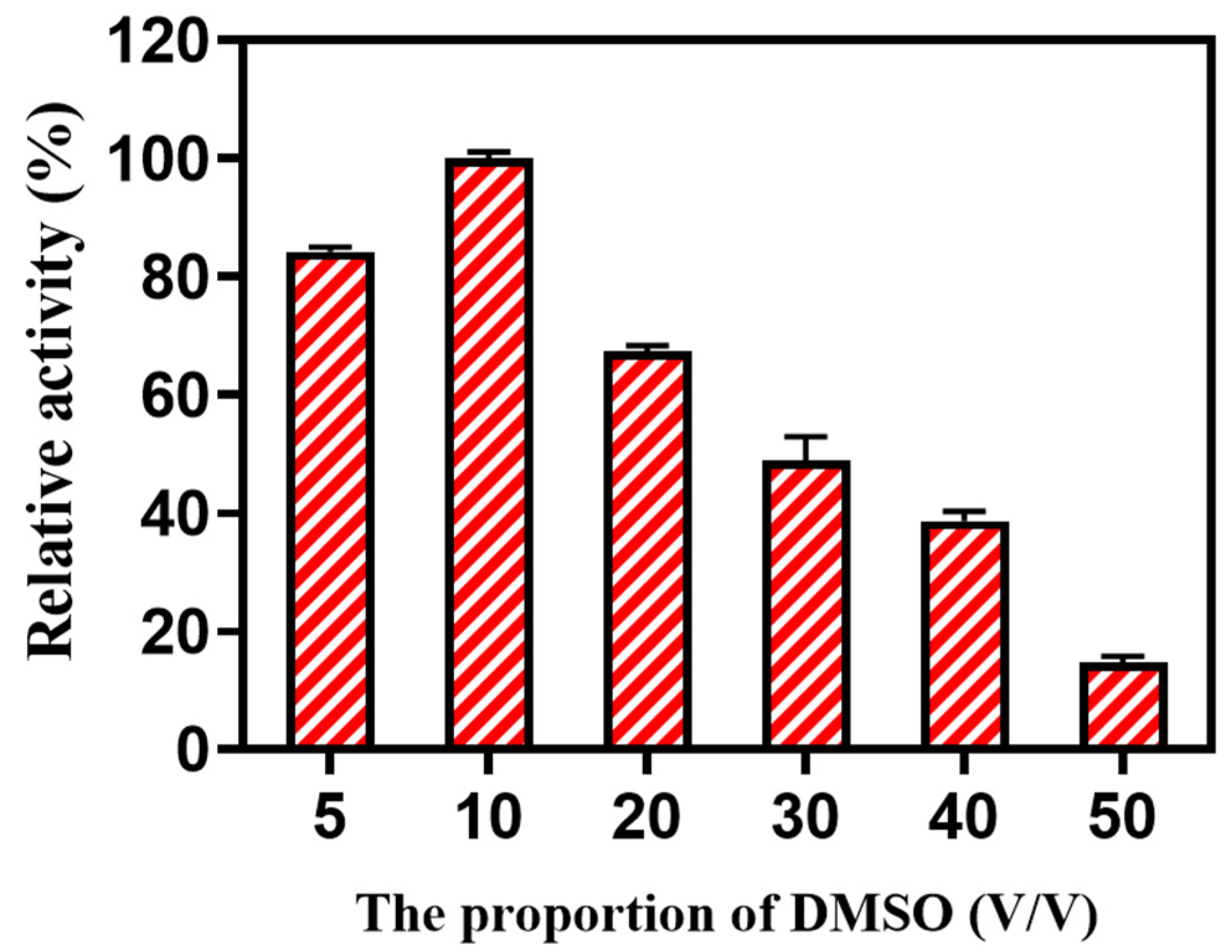
27]. In industry production, a large number of drug precursors have been produced in water/organic solvent systems, indicating that biotransformation in the organic solvent/water system is a mature and widely accepted strategy. In this work, the purpose of adding organic solvents was to increase the solubility of the reaction substrates. An enhancement of biotransformation was expected through organic solvent loading. DMSO is a universal solvent, and it was used as the organic solvent in the reaction system [
28]. In this work, when the proportion of DMSO increased to 10% (
v/
v), the enzymatic activity increased nearly 20%. As the proportion of DMSO increased to 20% (
v/
v), the enzymatic activity decreased to 67.42 ± 0.93%. As the proportion of DMSO increased to 50% (
v/
v), the enzymatic activity significantly decreased to 14.77 ± 0.96%. Clearly, the optimal level of DMSO loading was 10% (
v/
v). The addition of more DMSO failed to reverse the limitation of activity (
Figure 4).
2.1.4. Optimization of Metal Ion Concentration and Surfactants
The activities of several enzymes depend on the addition of metal ions [
29,
30,
31]. Metal ions are not only crucial cofactors for enzymatic synthesis but also have surprising functions that improve enzymatic activity [
32]. In this work, the effects of diverse metal ions (Zn
2+, Cu
2+, Ba
2+, Mg
2+, Mn
2+, Al
3+, Ca
2+, Cr
3+, Co
2+, and Fe
3+; 0.1 mM) on lovastatin hydrolase activity were evaluated at 30 °C and pH 8.0. The effects of these ions are shown in
Figure 5a; only marked inhibition of lovastatin hydrolase activity was found with various metal ions. Al
3+ decreased enzymatic activity by more than 24%. The addition of Mn
2+ reduced enzymatic activity to 80%. The addition of Cr
3+, Co
2+, and Fe
3+ lowered enzymatic activity by more than 10%. None of metal ions increased enzymatic activity. As a result, metal ion addition was not an appropriate method for the enhancement of lovastatin hydrolase activity.
Surfactants can be added to improve substrate solubility or availability and increase biocatalytic activity [
33]. In this work, diverse additives (glycerol, EDTA, CTAB, β-cyclodextrin, PEG-2000, PEG-4000, and PEG-6000; 10 mM) were added to the reaction mixture to evaluate their effect on biocatalytic activity. As illustrated in
Figure 5b, PEG-6000 strongly inhibited enzymatic activity. Only 77.76 ± 3.74% relative activity was retained after the addition of 10 mM PEG-6000. Similarly, the addition of EDTA, PEG-4000, and CTAB decreased the activity of the enzyme to less than 87% of its initial activity. Glycerol and PEG-2000 exerted no clear influence on biotransformation activity. Among all of the additives, the addition of β-cyclodextrin slightly enhanced biocatalytic activity, but this improvement was not significant. Thus, the addition of surfactants failed to significantly improve the activity of the enzyme.
Even though metal ions and surfactants have been reported to have the potential to improve enzymatic activity, no significant improvement was observed in the production of monacolin J in this work. Thus, other effective methods should be applied to solve this problem.
2.2. Substrate Treatment as an Effective Method for Enhancement of Enzymatic Activity
The suitability of the water solubility of substrates is a crucial factor to achieve optimal enzyme activity in several reactions [
34]. Here, the substrate lovastatin had a poor water solubility, but the enzyme showed better activity in aqueous solution. It is necessary to employ an appropriate method for improving the solubility of lovastatin and further promoting the enzymatic synthesis of monacolin J.
The pretreatment of substrates to improve their solubility is a potential method to address this issue. In the field of chemistry, the problem of compound solubility is often solved from the perspective of structural modification, including the introduction of polar groups, hydrogen bonding, salt formation, and other methods. Salt formation forms ionic compounds by introducing salt-forming counterions [
35,
36]. Compared with nonionic compounds, ionic compounds are more soluble in aqueous media. To solve this issue, lovastatin was pretreated in an alkaline environment to facilitate the rapid hydrolysis of the lactone ring. This pretreatment generated a highly soluble lovastatin salt that enhanced the efficiency of the ester hydrolysis reaction. The activity of the crude enzyme on the lovastatin salt substrate was dramatically improved to 74.7 ± 1.3 U/mL, which was more than 22-fold higher than that achieved with untreated lovastatin. With the pretreatment of the substrate, enzyme activity increased by an order of magnitude. Thus, the enzyme has the potential be used for industrial monacolin J production.
2.3. Lovastatin Hydrolase Stability and Scaling Up of the Biotransformation
The stability of enzymes is crucial for their use on an industrial scale. The stability of lovastatin hydrolase was assessed before using it in large-scale production. In previous studies, various mutant enzymes were prepared to improve the T50
10, the temperature at which the residual activity was half of the initial enzymatic activity after 10 min of heat treatment. Lovastatin hydrolase (PcEST) was used to synthesize monacolin J, and the mutant Q140L showed an increase in T50
10 of about 3 °C compared to the wild type (WT) [
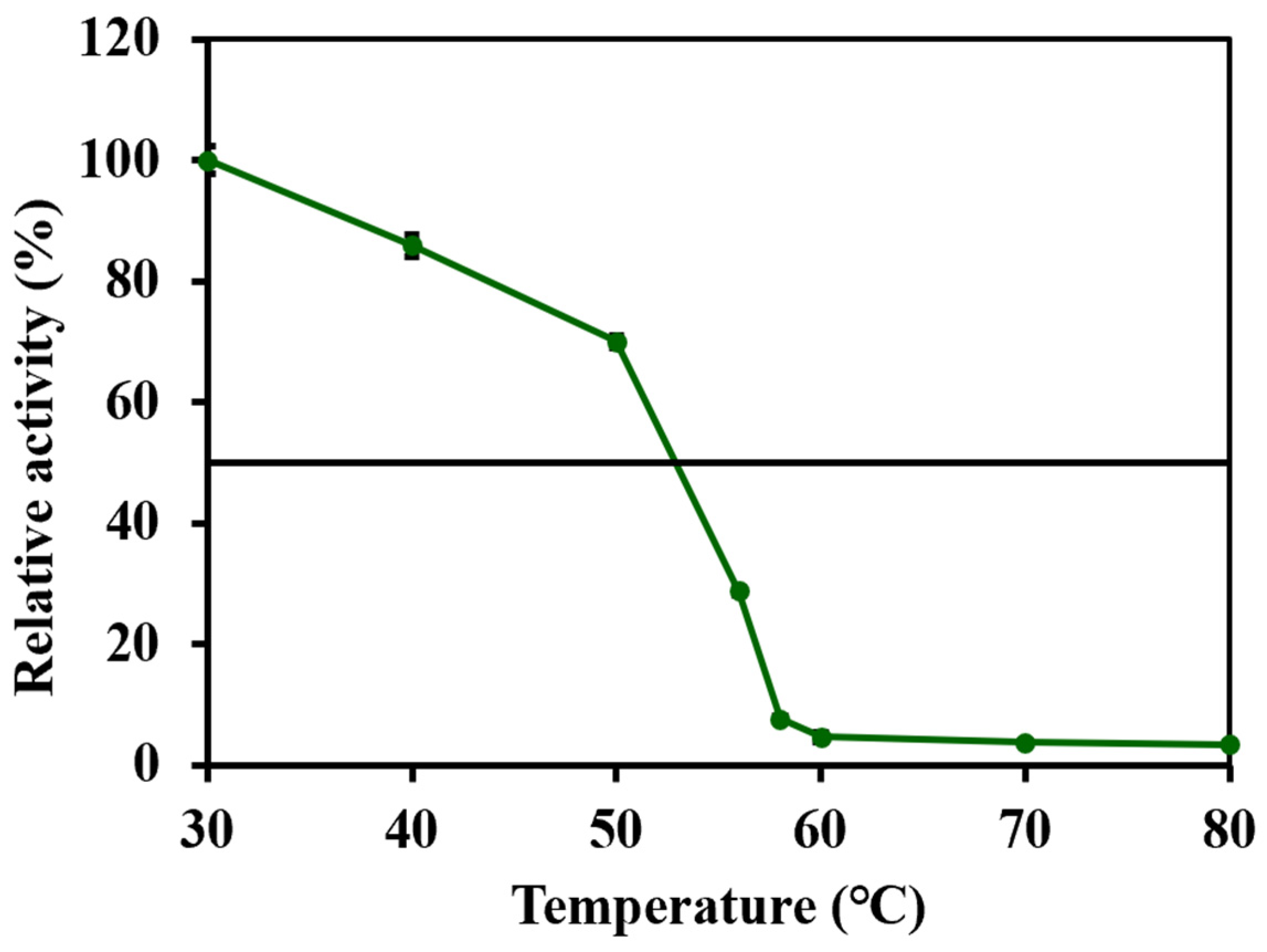
37]. Furthermore, the residual activities were measured after a 10 min incubation at a different temperature, reflecting the thermal stability of the enzyme. The enzyme exhibited a 70.0% residual activity after 10 min at 50 °C (
Figure 6), and the T50
10 was higher than 50 °C. Hence, wild-type lovastatin hydrolase encoded by the CDV55 gene showed higher thermostability for the synthesis of monacolin J on an industrial scale.
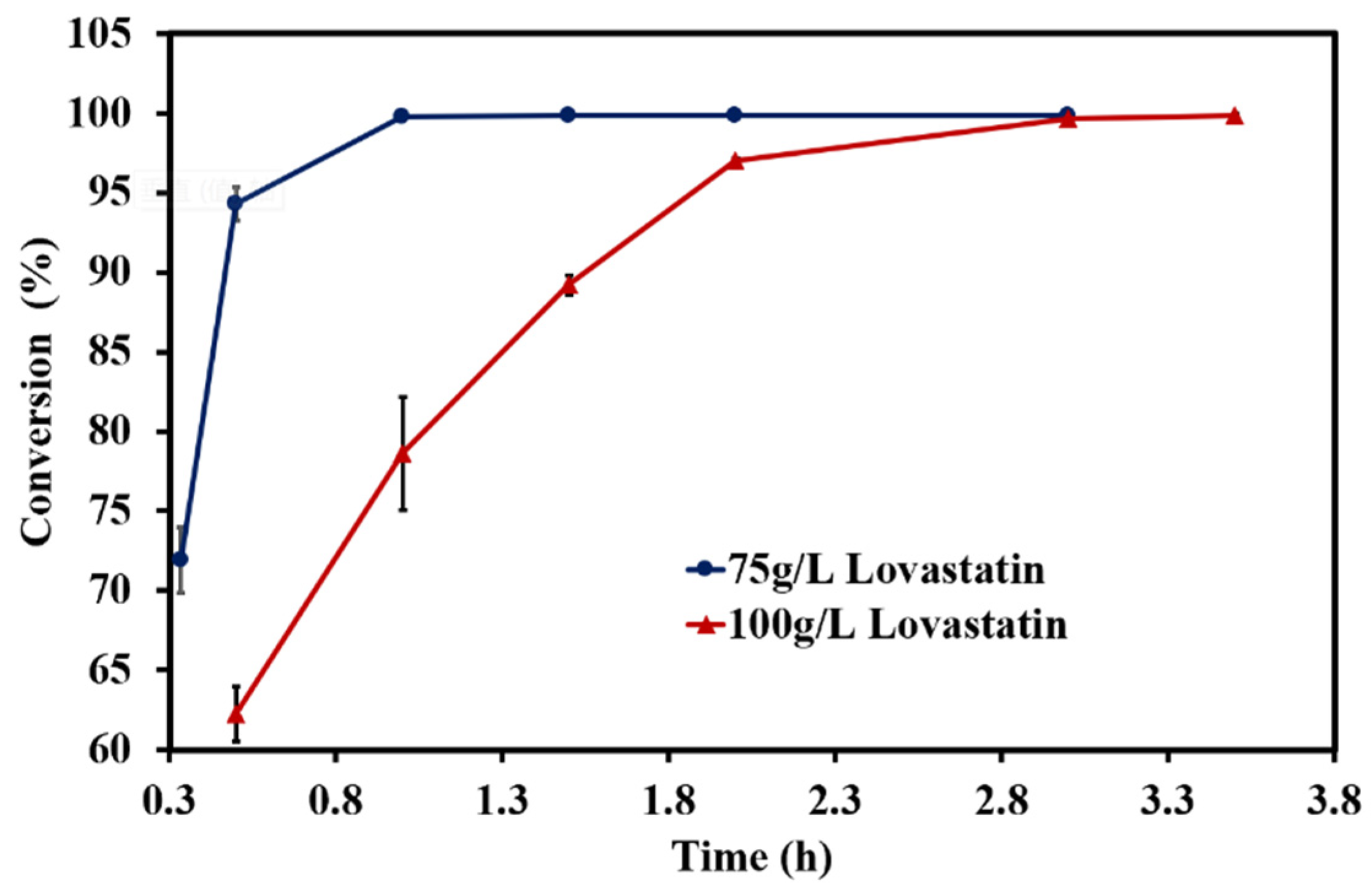
From the data shown above, the performance of lovastatin hydrolase was excellent, which demonstrated that this biocatalyst exhibited the ability to produce monacolin J on a large scale. For a further assessment of biotransformation, the pretreated lovastatin was used at concentrations of 75 g/L and 100 g/L. The results are shown in
Figure 7. When 75 g/L of substrate was incubated with 25% (
v/
v) crude enzyme at 30 °C, over70% of lovastatin was hydrolyzed to monacolin J after 0.3 hour. An hour later, the conversion of lovastatin to monacolin J approached 100%. As the concentration of substrate was further increased to 100 g/L, which is an acceptable concentration for industrial production, lovastatin was almost completely transformed into monacolin J with a conversion rate of 99.84 ± 0.08% after 3.5 h. The reaction was fast, and the suppression of biochemical reactions with high concentrations of substrates seen in earlier studies was not observed in this work.
Thus, the wild-type lovastatin hydrolase that was encoded by the CDV55 gene was an excellent biocatalyst for the conversion of lovastatin to monacolin J, which was more manageable on an industrial scale. Reaction parameters such as reaction temperature, solution pH, solvent type, metal ion concentration, and type of surfactant were systematically investigated, and the optimal reaction conditions were obtained. The pretreated lovastatin at concentrations of as high as 100 g/L could be effectively converted into monacolin J within 3.5 h at pH 8.0 and 30 °C, with a conversion rate of >99.8%.
3. Materials and Methods
3.1. Chemicals
A lovastatin substrate and a monacolin J standard were obtained from Jiangsu ALPHA Pharmaceutical Corp (Suqian, China). Isopropyl-β-D-thiogalactopyranoside (IPTG), ZnCl2, AlCl3, BaCl2, CuCl2, CoCl2·6H2O, CaCl2, CrCl3, FeCl3, MgCl2, MnCl2, and other chemical materials were bought from Aladdin Biochemical Technology Co., Ltd. (Shanghai, China).
3.2. Cloning, Expression, and Purification of Lovastatin Hydrolase
A recombination
Escherichia coli BL21 (DE3) strain harboring hydrolase from
Aspergillus turcosus was used for synthesizing monacolin J. The amino acid sequence of CDV55 was obtained from Genbank GI RHZ58343.1, and the encoding nucleotide sequence was optimized using Gene Optimizer
® expert software (GeneArt Services Dashboard (
https://www.thermofisher.cn/cn/zh/home.html)) for improved expression in
E. coli (Supplementary SEQ No. 1–2).
The CDV55 gene was amplified via PCR using the forward primer 5′-GGAATTCATGACCGACGACATCGAGACTACCTTCC-3′ (the EcoRI restriction site is underlined) and the reverse primer 5′-CCCAAGCTTCTACTGCCCCTTGAACGCATCATACTTGC-3′ (the HindIII restriction site is underlined). The PCR products were separated and purified using commonly used techniques and ligated into a pET-28a (+) plasmid that was digested with EcoRI and HindIII (the His tag in the plasmid was retained), which was then used in the transformation of
E. coli BL21 (DE3) cells (
Figure S1, in Supplementary Material).
The transformed E. coli BL21 (DE3) harboring CDV55 within pET-28a (+) were cultured in Luria–Bertani (LB) broth supplemented with 5 mg/100 mL kanamycin at 37 °C on a shaker (220 rpm). When the OD at 600 nm reached ~0.6, 0.5 mM IPTG was added to the growing culture to induce CDV55 expression over a 16 h period at 18 °C. Subsequently, the bacterial cells were harvested via centrifugation (4 °C, 10,000× g, 20 min) and washed with PBS (pH 7.5, 100 mM). Thereafter, 1 g (wet weight) of cells was resuspended in 10 mL Tris-HCl buffer (pH 8.0, 50 mM), and the cells were disrupted by sonication (260 W, 3 s pulse, 5 s pause). Cell debris was removed via centrifugation (4 °C, 10,000× g, 20 min) and the supernatant was used as the crude enzyme mixture.
The crude extract was loaded onto a Ni–NTA (nickel–nitrilotriacetic acid) agarose column (Sangon Biotech Corp., Shanghai, China), which had been equilibrated with wash buffer (Tris-HCl, 50 mM, pH 7.9–8.0) containing 0.25 M NaCl and 5 mM imidazole, and eluted with a solution containing 0.25 M of imidazole and 0.25 M NaCl. The purified protein was desalted using a HiTrap Desalting Column (GE Healthcare Corp., Piscataway, NJ, USA). Protein integrity and content were analyzed using SDS–PAGE and a BCA Protein Assay Kit (Vazyme Co., Nanjing, China), respectively, with bovine serum albumin (BSA) as a standard.
3.3. Alkaline Pretreatment of Lovastatin
Approximately 465 mL ddH2O and 35 mL methanol were added to a 1 L conical flask, followed by the addition of 10 g sodium hydroxide; finally, 100 g lovastatin was added to this solution. Lovastatin, with a solubility of approximately 200 g/L, was dissolved via continuous stirring for 12 h at 25–30 °C, and the pH was maintained at 8.0 using 6 M HCl.
3.4. Detection of Enzymatic Activity
The enzymatic activity was measured via HPLC, following a bioreaction at 30 °C and 280 rpm. Briefly, 100 µL crude enzyme was added to Tris-HCl buffer (pH 8.0, 50 mM) containing 10 mM lovastatin, and the total volume was adjusted to 1 mL. The mixture was reacted for 10 min at 30 °C. Subsequently, the bioreaction was terminated with the addition of 1 mL methanol. The resulting mixture was centrifuged at 10,000× g for 20 min, and the supernatant was filtered through 0.45 µm filters and subjected to HPLC analysis. Under the standard assay conditions, one unit (U) of enzymatic activity was defined as the amount of the enzyme required for the liberation of 1.0 mmol monacolin J per min.
3.5. Analytical Methods
The substrate (lovastatin) and the product (monacolin J) were measured using an LC–MS system consisting of an Agilent Eclipse Plus C18 column (100 × 4.6 mm, 3.5 μm, Agilent Technologies, Santa Clara, CA, USA) and UV detection at 238 nm. A mixture of acetonitrile and 0.1% H
3PO
4 (50:50
v/
v) was used as the eluent, with a flow rate of 1.5 mL/min. The retention times of lovastatin and monacolin J were 6.73 min and 1.80 min, respectively. For detection, a full scan was performed within a range of 0–1600
m/
z, and the detection analysis was carried out in positive ion mode (
Figure S2 in Supplementary Material).
3.6. Optimizing the Enzymatic Activity of Lovastatin Hydrolase
To optimize the enzymatic activity, 50 mM Tris-HCl buffer (55–90%
v/
v) and cosolvent (DMSO) (10–50%
v/
v), 0.1 M citrate (pH 4.5–7.0), 50 mM Tris-HCl buffer (pH 7.5–8.5), metal ions (ZnCl
2, AlCl
3, BaCl
2, CuCl
2, CoCl
2, CaCl
2, CrCl
3, FeCl
3, MgCl
2, and MnCl
2; 0.1 mM), other additives (glycerol, EDTA, CTAB, β-cyclodextrin, PEG-2000, PEG-4000, and PEG-6000; 10 mM), 100 µL crude enzyme, and 2.5 mM substrate were mixed in 2 mL solvent medium and incubated with shaking at 20–50 °C and 220 rpm. The enzymatic activity was assayed as described in
Section 3.4. All experiments were performed in duplicate.
3.7. Reaction Conditions for Testing the Conversion of Lovastatin
A reaction mixture containing 25 mL diluted crude enzyme and substrate solution volumes with a final concentration of substrate of 75 g/L or 100 g/L was brought up to a final volume of 100 mL with final concentration of 50 mM Tris-HCl buffer (pH 8.0) in a 500 mL conical flask and was incubated for 4 h at 30 °C with shaking at 220 rpm; the pH was adjusted to 8.0 using 0.1 M NaOH. All experiments were conducted in duplicate, and the error bars in the figures represent mean ± SD (
Scheme 1).
3.8. Intramolecular Cyclization
The reaction solution was filtered, then the filtrate was stirred under N2, and a solution of methanol (1.5 equivalents) in acetonitrile was added dropwise over a period of 15 min. After 90 min, the reaction solution was concentrated under reduced pressure to remove acetonitrile, the ethyl acetate solution was added, and the organic phase was washed with saturated sodium bicarbonate solution and water. Subsequently, n-hexane was slowly added dropwise to the ethyl acetate phase to gradually form a solid. The solid was filtered and dried to obtain the target product monacolin J.
4. Conclusions
Statins, a family of drugs known for their cholesterol-lowering properties, are also showing potential as anticancer agents. Simvastatin can be used in all high-risk patients with cardiovascular disease and diabetes. It is known that monacolin J is one of key intermediates for the production of simvastatin. In this work, a recombinant Escherichia coli BL21 (DE3) strain containing lovastatin hydrolase (encoded by CDV55_102090) from Aspergillus turcosus was constructed and used for the synthesis of monacolin J using lovastatin as a substrate. The enzyme proved to be thermostable, maintaining its activity even at relatively high temperatures. It was able to tolerate high concentrations of substrates. However, the water insolubility of lovastatin poses a challenge for this enzyme-catalyzed reaction. To address this issue, lovastatin was treated with an alkaline solution, which converted it to a more soluble salt. This treatment significantly increased the efficiency of this enzyme-catalyzed reaction. It was also found that the addition of certain solvents, metal ions, and surfactants could influence the enzymatic activity of lovastatin hydrolase. However, none of these solvents proved to be as effective as the alkaline pretreatment of the substrate. The substrate was pretreated, and the catalytic reaction was optimized by modulating various parameters; lovastatin at concentrations of as high as 100 g/L was effectively transformed to monacolin J within 3.5 h at pH 8.0 and 30 °C, with a conversion rate exceeding 99.8%. In addition, the T5010 of the enzyme was higher than 50 °C, indicating the tremendous potential of our method for the synthesis of monacolin J on an industrial scale.






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}