
Fabrication of a Charge-Conversion Polymer—Liposome for Enhancing Endosomal Escape of Drug Delivery System for α–Mangostin

 , , and
, , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Methods
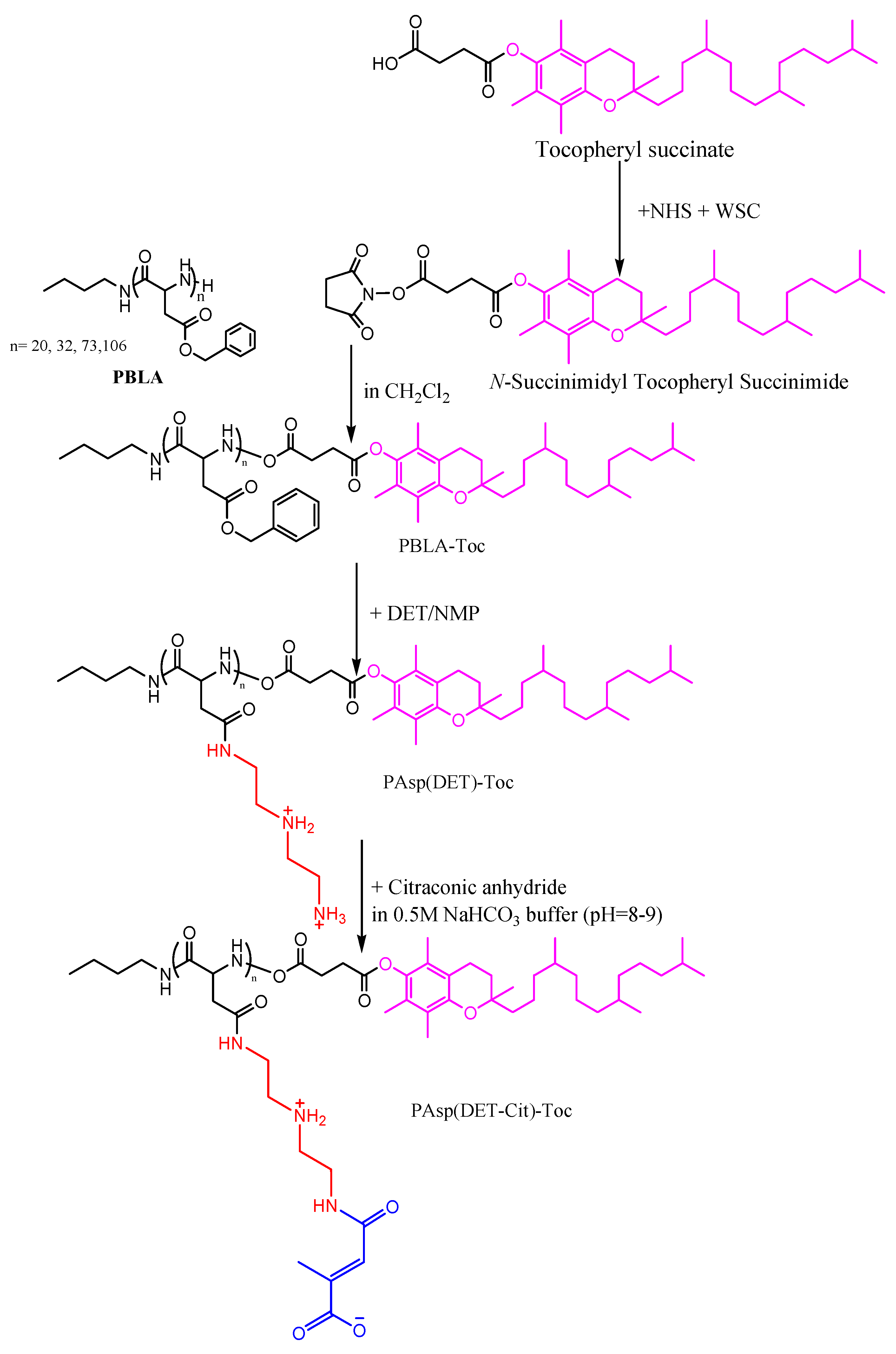
2.2.1. Synthesis of Charge–Conversion Polymer PAsp(DET-Cit)–Toc
- -
- Synthesis of N-Succinimidyl Tocopheryl Succinate
- -
- Synthesis of PBLA
- -
- Synthesis of PBLA–Toc
- -
- Synthesis of PAsp(DET)–Toc
- -
- Synthesis of PAsp(DET-Cit)–Toc
2.2.2. Preparation of Liposome
2.2.3. Measurement of Dynamic Light Scattering (DLS)
2.2.4. Drug Loading and Encapsulation Efficiency
2.2.5. Drug Release
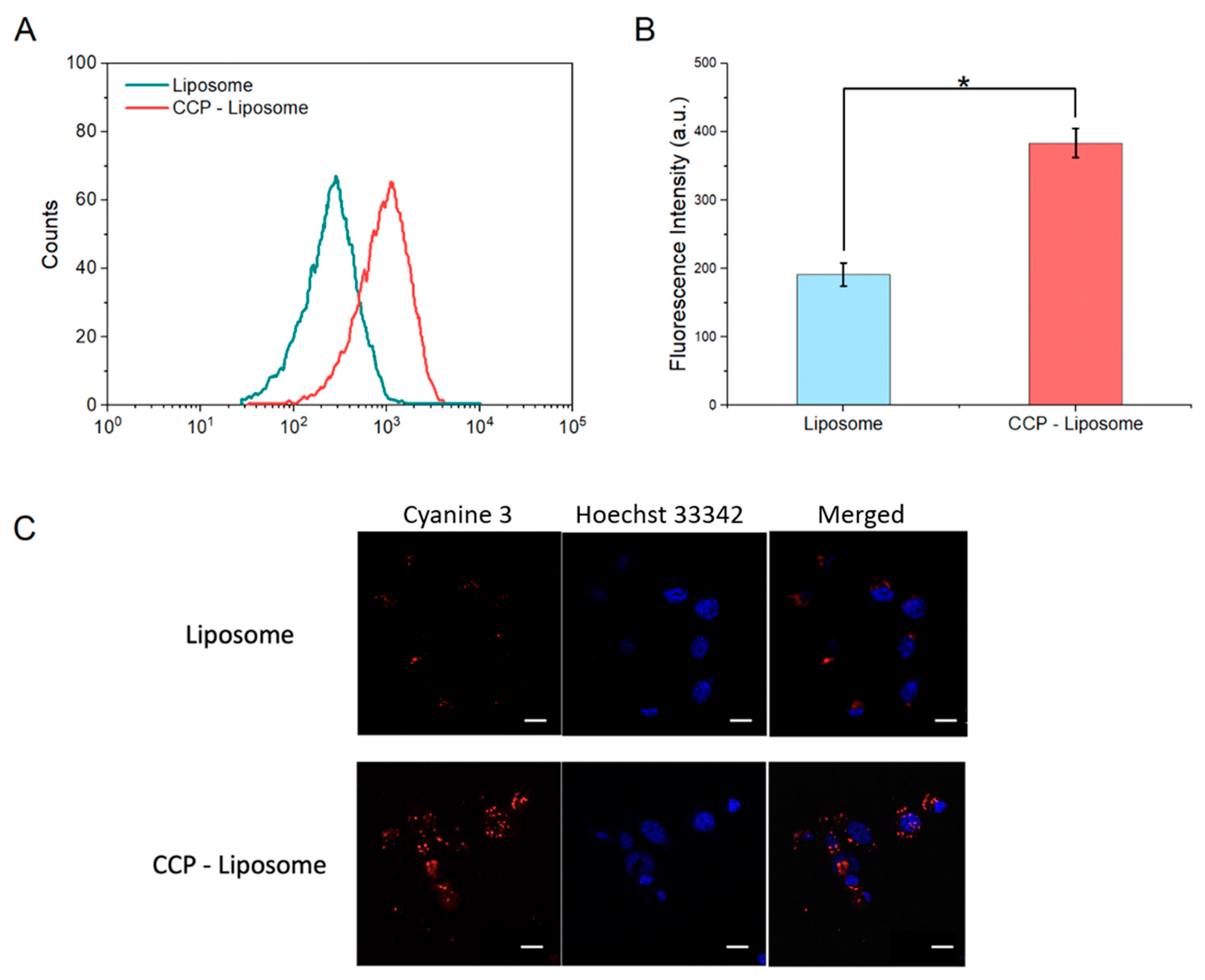
2.2.6. In-Vitro Uptake of CCP–Liposome by HepG2 Cells
2.2.7. Confocal Laser Scanning Microscopy (CLSM)
2.2.8. Cytotoxicity Assay
2.2.9. Endosomal Escape Property
2.2.10. Statistical Analysis
3. Results and Discussion
3.1. Synthesis of Charge-Conversion Polymer PAsp(DET-Cit)–Toc
3.1.1. Synthesis of N-Succinimidyl Tocopheryl Succinate
3.1.2. Synthesis of PBLA
3.1.3. Synthesis of PBLA–Toc
3.1.4. Synthesis of PAsp(DET)–Toc
3.1.5. Synthesis of PAsp(DET-Cit)–Toc
3.2. Physical Characterizations of CCP–Liposome
3.3. The Release Rate and Encapsulation Efficiency of CCP–Liposome
3.4. Endosome Escape of CCP–Liposome
3.5. In Vitro Cytotoxicity
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tsai, S.Y.; Chung, P.C.; Owaga, E.E.; Tsai, I.J.; Wang, P.Y.; Tsai, J.I.; Yeh, T.S.; Hsieh, R.H. Alpha-mangostin from mangosteen (Garcinia mangostana Linn.) pericarp extract reduces high fat-diet induced hepatic steatosis in rats by regulating mitochondria function and apoptosis. Nutr. Metab. 2016, 13, 88. [Google Scholar] [CrossRef]
- Kritsanawong, S.; Innajak, S.; Imoto, M.; Watanapokasin, R. Antiproliferative and apoptosis induction of alpha-mangostin in T47D breast cancer cells. Int. J. Oncol. 2016, 48, 2155–2165. [Google Scholar] [CrossRef]
- Ovalle-Magallanes, B.; Eugenio-Perez, D.; Pedraza-Chaverri, J. Medicinal properties of mangosteen (Garcinia mangostana L.): A comprehensive update. Food Chem. Toxicol. 2017, 109, 102–122. [Google Scholar] [CrossRef]
- Shan, T.; Ma, Q.; Guo, K.; Liu, J.; Li, W.; Wang, F.; Wu, E. Xanthones from mangosteen extracts as natural chemopreventive agents: Potential anticancer drugs. Curr. Mol. Med. 2011, 11, 666–677. [Google Scholar] [CrossRef]
- Hung, S.H.; Shen, K.H.; Wu, C.H.; Liu, C.L.; Shih, Y.W. Alpha-mangostin suppresses PC-3 human prostate carcinoma cell metastasis by inhibiting matrix metalloproteinase-2/9 and urokinase-plasminogen expression through the JNK signaling pathway. J. Agric. Food Chem. 2009, 57, 1291–1298. [Google Scholar] [CrossRef] [PubMed]
- Shih, Y.W.; Chien, S.T.; Chen, P.S.; Lee, J.H.; Wu, S.H.; Yin, L.T. Alpha-mangostin suppresses phorbol 12-myristate 13-acetate-induced MMP-2/MMP-9 expressions via alphavbeta3 integrin/FAK/ERK and NF-kappaB signaling pathway in human lung adenocarcinoma A549 cells. Cell Biochem. Biophys. 2010, 58, 31–44. [Google Scholar] [CrossRef] [PubMed]
- Phan, T.K.T.; Shahbazzadeh, F.; Pham, T.T.H.; Kihara, T. Alpha-mangostin inhibits the migration and invasion of A549 lung cancer cells. PeerJ 2018, 6, e5027. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Tian, W.; Ma, X. Alpha-mangostin inhibits intracellular fatty acid synthase and induces apoptosis in breast cancer cells. Mol. Cancer 2014, 13, 138. [Google Scholar] [CrossRef]
- Li, L.; Brunner, I.; Han, A.R.; Hamburger, M.; Kinghorn, A.D.; Frye, R.; Butterweck, V. Pharmacokinetics of alpha-mangostin in rats after intravenous and oral application. Mol. Nutr. Food Res. 2011, 55 (Suppl. S1), S67–S74. [Google Scholar] [CrossRef]
- Wathoni, N.; Rusdin, A.; Motoyama, K.; Joni, I.M.; Lesmana, R.; Muchtaridi, M. Nanoparticle Drug Delivery Systems for alpha-Mangostin. Nanotechnol. Sci. Appl. 2020, 13, 23–36. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Olenyuk, B.Z.; Okamoto, C.T.; Hamm-Alvarez, S.F. Targeting receptor-mediated endocytotic pathways with nanoparticles: Rationale and advances. Adv. Drug Deliv. Rev. 2013, 65, 121–138. [Google Scholar] [CrossRef] [PubMed]
- Cerqueira, B.B.; Lasham, A.; Shelling, A.N.; Al-Kassas, R. Nanoparticle therapeutics: Technologies and methods for overcoming cancer. Eur. J. Pharm. Biopharm. 2015, 97, 140–151. [Google Scholar] [CrossRef]
- Sercombe, L.; Veerati, T.; Moheimani, F.; Wu, S.Y.; Sood, A.K.; Hua, S. Advances and Challenges of Liposome Assisted Drug Delivery. Front. Pharmacol. 2015, 6, 286. [Google Scholar] [CrossRef] [PubMed]
- Daraee, H.; Etemadi, A.; Kouhi, M.; Alimirzalu, S.; Akbarzadeh, A. Application of liposomes in medicine and drug delivery. Artif. Cells Nanomed. Biotechnol. 2016, 44, 381–391. [Google Scholar] [CrossRef]
- Hua, S.; Wu, S.Y. The use of lipid-based nanocarriers for targeted pain therapies. Front. Pharmacol. 2013, 4, 143. [Google Scholar] [CrossRef]
- Ding, B.S.; Dziubla, T.; Shuvaev, V.V.; Muro, S.; Muzykantov, V.R. Advanced drug delivery systems that target the vascular endothelium. Mol. Interv. 2006, 6, 98–112. [Google Scholar] [CrossRef]
- Metselaar, J.M.; Storm, G. Liposomes in the treatment of inflammatory disorders. Expert Opin. Drug Deliv. 2005, 2, 465–476. [Google Scholar] [CrossRef] [PubMed]
- Das, S.S.; Hussain, A.; Verma, P.R.P.; Imam, S.S.; Altamimi, M.A.; Alshehri, S.; Singh, S.K. Recent Advances in Liposomal Drug Delivery System of Quercetin for Cancer Targeting: A Mechanistic Approach. Curr. Drug Deliv. 2020, 17, 845–860. [Google Scholar] [CrossRef]
- Li, M.; Du, C.; Guo, N.; Teng, Y.; Meng, X.; Sun, H.; Li, S.; Yu, P.; Galons, H. Composition design and medical application of liposomes. Eur. J. Med. Chem. 2019, 164, 640–653. [Google Scholar] [CrossRef]
- Trang Phan, T.K.; Tran, T.Q.; Nguyen Pham, D.T.; Nguyen, D.T. Characterization, Release Pattern, and Cytotoxicity of Liposomes Loaded With α-Mangostin Isolated From Pericarp of Mangosteen (Garcinia mangostana L.). Natural. Prod. Commun. 2020, 15, 1934578X20974559. [Google Scholar] [CrossRef]
- Rommasi, F.; Esfandiari, N. Liposomal Nanomedicine: Applications for Drug Delivery in Cancer Therapy. Nanoscale Res. Lett. 2021, 16, 95. [Google Scholar] [CrossRef] [PubMed]
- Fanciullino, R.; Ciccolini, J. Liposome-encapsulated anticancer drugs: Still waiting for the magic bullet? Curr. Med. Chem. 2009, 16, 4361–4371. [Google Scholar] [CrossRef] [PubMed]
- Torchilin, V. Tumor delivery of macromolecular drugs based on the EPR effect. Adv. Drug Deliv. Rev. 2011, 63, 131–135. [Google Scholar] [CrossRef]
- Bozzuto, G.; Molinari, A. Liposomes as nanomedical devices. Int. J. Nanomed. 2015, 10, 975–999. [Google Scholar] [CrossRef] [PubMed]
- Canton, I.; Battaglia, G. Endocytosis at the nanoscale. Chem. Soc. Rev. 2012, 41, 2718–2739. [Google Scholar] [CrossRef]
- Bae, Y.; Nishiyama, N.; Fukushima, S.; Koyama, H.; Yasuhiro, M.; Kataoka, K. Preparation and biological characterization of polymeric micelle drug carriers with intracellular pH-triggered drug release property: Tumor permeability, controlled subcellular drug distribution, and enhanced in vivo antitumor efficacy. Bioconjug. Chem. 2005, 16, 122–130. [Google Scholar] [CrossRef]
- Huang, H.; Li, Y.; Sa, Z.; Sun, Y.; Wang, Y.; Wang, J. A smart drug delivery system from charge-conversion polymer-drug conjugate for enhancing tumor therapy and tunable drug release. Macromol. Biosci. 2014, 14, 485–490. [Google Scholar] [CrossRef]
- Sun, H.; Guo, B.; Li, X.; Cheng, R.; Meng, F.; Liu, H.; Zhong, Z. Shell-sheddable micelles based on dextran-SS-poly(epsilon-caprolactone) diblock copolymer for efficient intracellular release of doxorubicin. Biomacromolecules 2010, 11, 848–854. [Google Scholar] [CrossRef]
- Gallon, E.; Matini, T.; Sasso, L.; Mantovani, G.; Arminan de Benito, A.; Sanchis, J.; Caliceti, P.; Alexander, C.; Vicent, M.J.; Salmaso, S. Triblock Copolymer Nanovesicles for pH-Responsive Targeted Delivery and Controlled Release of siRNA to Cancer Cells. Biomacromolecules 2015, 16, 1924–1937. [Google Scholar] [CrossRef]
- Selby, L.I.; Cortez-Jugo, C.M.; Such, G.K.; Johnston, A.P.R. Nanoescapology: Progress toward understanding the endosomal escape of polymeric nanoparticles. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2017, 9, e1452. [Google Scholar] [CrossRef]
- Harada, A.; Kataoka, K. Formation of Polyion Complex Micelles in an Aqueous Milieu from a Pair of Oppositely-Charged Block Copolymers with Poly(ethylene glycol) Segments. Macromolecules 1995, 25, 5294–5299. [Google Scholar] [CrossRef]
- Tang, J.; He, J.; Yang, C.; Mao, Y.; Hu, T.; Zhang, L.; Cao, H.; Tong, A.; Song, X.; He, G.; et al. Antitumor Effects of MsurvivinT34A–CaPi Complex-Embedded PLGA Nanoparticles in Combination with Doxil in Mice. J. Nanopart. Res. 2014, 16, 2682. [Google Scholar] [CrossRef]
- Lee, Y.; Fukushima, S.; Bae, Y.; Hiki, S.; Ishii, T.; Kataoka, K. A protein nanocarrier from charge-conversion polymer in response to endosomal pH. J. Am. Chem. Soc. 2007, 129, 5362–5363. [Google Scholar] [CrossRef]
- Bae, Y.; Jang, W.D.; Nishiyama, N.; Fukushima, S.; Kataoka, K. Multifunctional polymeric micelles with folate-mediated cancer cell targeting and pH-triggered drug releasing properties for active intracellular drug delivery. Mol. Biosyst. 2005, 1, 242–250. [Google Scholar] [CrossRef]
- Koide, A.; Kishimura, A.; Osada, K.; Jang, W.D.; Yamasaki, Y.; Kataoka, K. Semipermeable polymer vesicle (PICsome) self-assembled in aqueous medium from a pair of oppositely charged block copolymers: Physiologically stable micro-/nanocontainers of water-soluble macromolecules. J. Am. Chem. Soc. 2006, 128, 5988–5989. [Google Scholar] [CrossRef]
- Fan, J.; Borguet, Y.P.; Su, L.; Nguyen, T.P.; Wang, H.; He, X.; Zou, J.; Wooley, K.L. Two-Dimensional Controlled Syntheses of Polypeptide Molecular Brushes via N-Carboxyanhydride Ring-Opening Polymerization and Ring-Opening Metathesis Polymerization. ACS Macro Lett. 2017, 6, 1031–1035. [Google Scholar] [CrossRef]
- Sanjoh, M.; Hiki, S.; Lee, Y.; Oba, M.; Miyata, K.; Ishii, T.; Kataoka, K. pDNA/poly(L-lysine) Polyplexes Functionalized with a pH-Sensitive Charge-Conversional Poly(aspartamide) Derivative for Controlled Gene Delivery to Human Umbilical Vein Endothelial Cells. Macromol. Rapid Commun. 2010, 31, 1181–1186. [Google Scholar] [CrossRef]
- Dutta, K.; Das, R.; Medeiros, J.; Kanjilal, P.; Thayumanavan, S. Charge-Conversion Strategies for Nucleic Acid Delivery. Adv. Funct. Mater. 2021, 31, 2011103. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Li, L.; He, X.; Yi, Q.; He, B.; Cao, J.; Pan, W.; Gu, Z. Overcoming drug-resistant lung cancer by paclitaxel loaded dual-functional liposomes with mitochondria targeting and pH-response. Biomaterials 2015, 52, 126–139. [Google Scholar] [CrossRef]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Phan, T.T.K.; Nguyen, H.H.; Nguyen, X.T.; Van Nguyen, T.; Duong, L.A.; Nguyen, L.P.; Pham, U.T.; Le, H.N.; Tran, T.Q.; Nguyen, D.T.; et al. Fabrication of a Charge-Conversion Polymer—Liposome for Enhancing Endosomal Escape of Drug Delivery System for α–Mangostin. Processes 2023, 11, 2344. https://doi.org/10.3390/pr11082344
Phan TTK, Nguyen HH, Nguyen XT, Van Nguyen T, Duong LA, Nguyen LP, Pham UT, Le HN, Tran TQ, Nguyen DT, et al. Fabrication of a Charge-Conversion Polymer—Liposome for Enhancing Endosomal Escape of Drug Delivery System for α–Mangostin. Processes. 2023; 11(8):2344. https://doi.org/10.3390/pr11082344
Chicago/Turabian StylePhan, Trang Thi Kieu, Hoang Huy Nguyen, Xuan Thi Nguyen, Tung Van Nguyen, Linh Anh Duong, Linh Phuong Nguyen, Uyen Thu Pham, Hong Nhung Le, Toan Quoc Tran, Duong Thanh Nguyen, and et al. 2023. "Fabrication of a Charge-Conversion Polymer—Liposome for Enhancing Endosomal Escape of Drug Delivery System for α–Mangostin" Processes 11, no. 8: 2344. https://doi.org/10.3390/pr11082344
APA StylePhan, T. T. K., Nguyen, H. H., Nguyen, X. T., Van Nguyen, T., Duong, L. A., Nguyen, L. P., Pham, U. T., Le, H. N., Tran, T. Q., Nguyen, D. T., & Pham, D. T. N. (2023). Fabrication of a Charge-Conversion Polymer—Liposome for Enhancing Endosomal Escape of Drug Delivery System for α–Mangostin. Processes, 11(8), 2344. https://doi.org/10.3390/pr11082344