Abstract
Facilitating the demands of modern society, namely, smartphones, televisions, electric vehicles, and high-stability aircraft structures, requires low-cost and high-performance materials and a corresponding change in the approach needed to design them. Rare earth elements (REEs) play a significant role in achieving these objectives by adding small amounts of these elements to alloys, thereby enhancing material properties. Despite being more abundant than precious metals, the 17 REEs exhibit subtle variations in their chemical and physical characteristics. Thus, their separation is still crucial for industrial applications. There is a corresponding need to develop more effective and efficient separation methods. Adding to the separation challenge is the complexity of the sources of REEs and related materials. Thus, large-scale production of REE materials is difficult. Current REE processing techniques can be categorized into pre-treatment, beneficiation, separation, and refining. Researchers have developed various technologies encompassing chemical, physical, and biological methods, focusing on economic and environmental considerations. However, not all these approaches can be scaled up for mass production. This article focuses on feasible strategies such as precipitation and crystallization, oxidation and reduction, ion exchange, adsorption, solvent extraction, and membrane separation. Further research into these traditional and modern methods can potentially revolutionize the separation dynamics of REEs.
1. Introduction
Rare Earths Elements (REEs) are commonly associated with designing and developing sustainable technologies to meet human demands and standards of living. REEs are also recognized as critical minerals by some researchers and agencies. The REEs are the comprehensive series of elements in the IIIB group in the periodic table, with atomic numbers ranging from 57 to 71. These elements are also referred to as the lanthanide series, starting from Lanthanum to Lutetium. Further, Scandium (21 atomic number) and Yttrium (39 atomic number) are often considered to be part of the REE group [1,2]. The age-old classification of REE consists of the Ceric group (C.G.) with elements from La to Eu and the Yttric group (Y.G.) with elements from Gd to Lu including Y, the reason behind this is related to their solubility differences [3]. Additionally, Sc and Y are considered among the REEs because of their coexistence in common REE-bearing minerals and resembling properties [4]; in contrast, some authors argue not to include Sc with the rest due to ionic radii variation [5]. Later on, based on industrial applications, metallurgists classified the REEs/REMs into three groups: light/Ceric REEs from Lanthanum (La) to Neodymium (Nd), middle REEs from Samarium (Sm) to Gadolinium (Gd), and heavy REEs from Dysprosium (Dy) to Lutetium (Lu). Some of the literature uses terms like yttric REEs, which include Middle REEs, and heavy REEs, and yttrium is also considered a heavy REE because of its similar properties [6]. Figure 1 describes the chronological timeline of REE discovery.
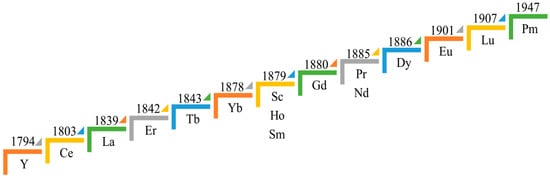
Figure 1.
A Historical Path: Mapping the Discovery of REEs.
The British Geological Survey (BGS) (www.mineralsuk.com) has reported that due to the scarcity of the original minerals from which they are extracted, the designation “rare earth elements” has become misleading because they are relatively common in the globe’s crust. REE materials are much more common in the earth’s crust than many precious metals.
Also, almost all REEs can occur in the same deposits. When viewed from different perspectives, their separation and extraction are crucial at the industrial level. For instance, given individual elemental distribution, separation becomes challenging because of similarities in physical and chemical properties. The reason behind similar chemical properties in aqueous media is attributed to the continuous increase/decrease of electrons at 4f sublevels [6], resulting in a division of REEs into LREEs/MREEs/HREEs. The difficulty in separating individual REEs is related to the similarity in electronic structure that results from a slight change in ionic radii between adjacent rare earth elements (0.01–0.03 Å for adjacent atomic number elements). Thus, individual elemental separation by precipitation methods is challenging and complicated [7].
Regarding commercial extraction, the REE’s content is often estimated and calculated based on REE oxides rather than elemental form. Jean-Claude G Bunzli stated that this is due to the electropositive nature of these ions to form oxides. Among REE series, the Oddo–Harkins rule (an odd–even strategy) is applicable where the odd atomic numbers have less availability in nature and vice versa [6].
1.1. Sources of REEs
The natural abundance (values averaged in μg/g except for Pm) of REEs in the earth’s crust was in the order of Ce > Nd > La > Y > Sc > Pr > Sm > Gd > Dy > Er > Yb > Eu > Ho > Tb > Lu > Tm [4]. As indicated in the report from the United States Geological Survey (USGS)(https://www.usgs.gov/centers/national-minerals-information-center/rare-earths-statistics-and-information (accessed on 27 March 2023)), the most widely available mineral sources for REE are monazite, bastnaesite, and xenotime. The USGS report states that the minable and extractable quantity of world REE-oxides reserves worldwide is 130,000,000 metric tons, the mined production in 2021 was 290,000 metric tons, and in 2022 it was 300,000 metric tons. As shown in Figure 2, the statistical analysis of data on REE reserves from the USGS report for the year 2022 depicts China has the maximum amount of REE reserves and the United States of America has a lesser amount of REE reserves, but not the least. The data of other countries include the cumulative reserves for Canada, Greenland, South Africa, and Tanzania. Similarly, Figure 3 represents the worldwide production of REE oxides for 2022, which informs that China has the maximum share in the REE production market, thus emerging as the global REM supply chain controller. It is worth mentioning that in Figure 3, the data of other countries include the production details of both Burma and Madagascar.
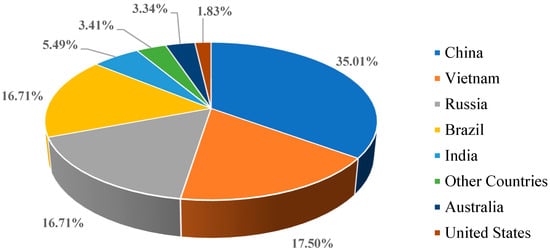
Figure 2.
Economically mineable REE reserves across the world as of 2022. Data source: USGS report 2023.
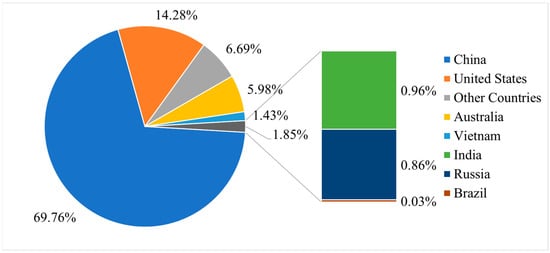
Figure 3.
Production from REE reserves across the world as of 2022. Data source: USGS report 2023.
1.1.1. Primary Sources of REEs
According to the BGS report [8] from November 2011, it was stated that there are numerous mineral resources containing rare earth elements (REEs), numbering in the hundreds. These mineral resources are characterized into three groups, and the details of the most prominent resources are here.
Cerium-dominant resources: Aeschynite, Allanite, Bastnasite, Britholite, Fergusonite, Gadolinite, Monazite, Parisite, Huanghoite, Cebaite, Florencite, and Synchysite.
Yttrium-dominant resources: Euxenite, Kainosite, and Samarskite.
Other resources: Apatite, Brannerite, Eudialyte, Loparite, Xenotime, Yttrocerite, Knopite, Gittinsite, Iimoriite, Mosandrite, Pyrochlore, Rinkite, Steenstrupine, Zircon, Carbonatites, and Ion adsorption clays [9]. Economic limitations limit commercial extraction to a small group of resources. Among the global resources, the carbonatites (associated with Bastnasite and Monazite) are the more prominent and economically extractable deposits in the USA, Australia, China, and Mongolia regions, with REE content varying from 2–10% by weight [10]. The subsequent commercial extraction is possible through placers found in the ocean belts of India, Sri Lanka, Australia, and southern USA, where the deposit is widespread but with lower REE concentration (<0.5% by weight). Ion adsorption clays are another commercial source of extraction [10]. They are preconcentrated in some REE content, and the extraction process from these deposits has low levels of radioactivity in the resulting waste compared to other sources. However, studies across the globe are continuing concerning the extraction of low REE content.
1.1.2. Secondary Sources of REEs
Many countries and industries are performing metal-life cycle assessments to counteract the harmful environmental effects of traditional exploration and extraction of resources and to meet the global metal supply demand. They are accommodating the practice of implementing recycling left-to-recycle (LTR) material such as metal scraps, electronic waste, and phosphors. This practice benefits human, technological, and environmental sustainability by creating job opportunities, developing new technologies in place of traditional and outdated methods, and reducing scrap piling at dump yards, oceans, and landfills. Some such potential sources for rare earth extraction, according to [11,12,13,14,15,16], are listed below:
- Ash is one of the significant components in Coal Combustion and contributes to REEs.
- Dross and slags from metal processing and red mud from bauxite residue are a few sources of REEs.
- Tailings generated from the gold beneficiation process can be a source of LREEs (Ce, La, and Nd).
- Neodymium–Iron–Boron permanent magnets, phosphors, fluorescent lamps, Nickel–Metal-Hydride Batteries, motors, and fluid cracking catalysts contain some REEs, and recycling is the best way of recovering them.
According to the study on the criticality of raw materials and assessment [17], the European Union (E.U.) demonstrates a 4% of HREE recovery and 3% of LREE recovery through recycling these secondary resources.
1.2. Demand for REEs in the E.U.
The increase in population across the globe has increased the demand for utility vehicles, electronic gadgets, pharmaceuticals, fuel, and energy resources. Many countries have accommodated these demands with care and concern for the environment by adopting the transformation to using clean and green energy alternatives. This emerging transition once aroused the need for rare earth supply chains and production. As cited, 75% of REEs are utilized in industries of catalysts: 10% for ceramic, glass, and polishing industries; 10% for phosphor and other optical devices; and, finally, 5% for alloy design required for wind turbines, hard disk drives, and many others [2]. According to categorization by the E.U., Sc and LREEs (referring to La–Sm) are preferred for the sectors of catalysts, metallurgy, polishing, and magnets, and HREEs (referring to Eu–Lu and Y) are used in lasers (but not only limited to) and niche applications [17]. The E.U. Criticality Assessment report for 2023 states that compared to 2020, in the E.U., there has been a 50% increase in the economic importance of La, Pr, Nd, and Tb due to their usage in magnets and catalysts, and the comparison is presented in Figure 4.
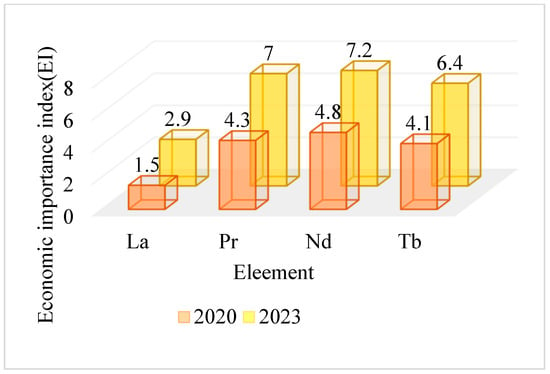
Figure 4.
Comparison of rise in Economic Importance in the year 2023 compared to 2020.
Data in Figure 5 represent the statistical analysis of the economic importance of REEs for the E.U. as of the year 2023, and it is essential to note the data described here are on a point scale, but not on a % scale. Additionally, the plot indicates that within the European Union (EU), there is a dire need for Dy, Sm, Nd, Pr, and Tb, as these metals possess economic importance values exceeding 5.0. The supporting explanation for this emerging requirement can be understandable from Figure 6, where the orange bar on the chart explains the details of the REE metal applications for different industries as of 2023, and the blue bar represents the data for 2012. This analysis is performed from the E.U. study for Critical raw materials [17,18]. Here, the rise in demand is mainly due to a jump in the utilization of REE metals in magnets, which are used in mobile phones [19], and hard disk drives [4]. Further, the increased application of REEs for catalysts such as La can increase the recovery in oil refineries, and Ce-based catalysts can reduce the pollutants and toxic gases released from cars [18], and, additionally, the application of REEs in ceramics is increased due to the application of REE oxides of Pr, Nd, Y, and Ce in manufacturing tiles and electronic components such as capacitors [20].
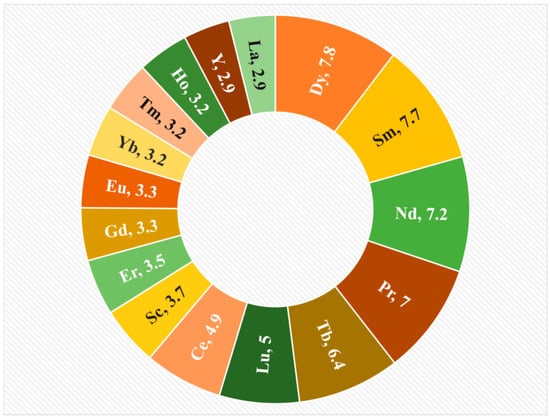
Figure 5.
Statistical analysis of the economic importance of REEs for the E.U. in 2023.
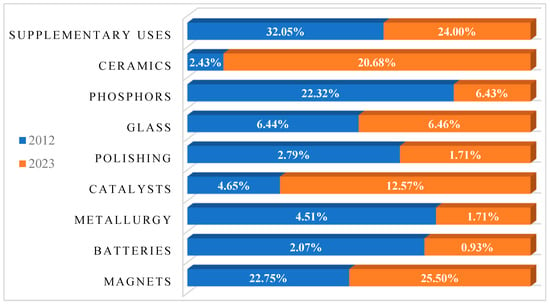
Figure 6.
Comparison between applications of REE elements in various industrial sectors for 2012 and 2023.
Furthermore, Figure 2 and Figure 3 illustrate that China holds a substantial market share in REE supply and demand. This same analysis is evident in the EU assessment report for the year 2023, which describes that among the global suppliers of REEs, China provides 100% of HREEs and 85% of LREEs. This analogy holds true for the EU as well. Thus, it may lead to the problem of a monopoly in the REE supply chain. To avoid these circumstances, the governments from almost all the countries globally had predominantly focused on demand-based REE reserve development and production. To support this, many researchers have invented different approaches for separating individual REEs, and their purpose for designing a system varies with the research interest. Most of the methods developed so far have been limited to laboratory demonstrations or research publications. Challenges such as economic and ecological concerns, time-consuming processes, and difficulties in scaling up for industrial purposes have hindered their implementation on a larger scale. Previous reviews on REE extraction and separation have often focused on promoting one or two main approaches while criticizing others, making it challenging to grasp a comprehensive understanding of the subject matter.
The critical distinction between previous reviews and this is that most of those existing reports revolve around some specific approaches for the separation of REEs. They all explained the merits, limitations, and developments that happened in a particular period. To the best of our knowledge, this article aims to bridge that gap by providing a clear and comprehensive explanation of those methods. This article will describe the mechanisms of these methods, using illustrations where necessary. By effectively comparing these approaches, this article intends to offer a comprehensive understanding and fill the void of proper explanations for each method. Thus, this article facilitates as a preliminary reference for any researcher interested in these separations.
2. Extraction of REE Minerals
Starting from the discovery of REE minerals, many scientists, industries, capitalists, and countries across the globe have invested vast amounts of knowledge, energy, time, and money in inventing and developing an Efficient-Easier-Economical-Environmentally safe (EEEE) strategy to extract these minerals for human sustainability and the circular economy [21]. Of these inventions, the most common technologies adopted by most processes from source to recycling are shown in Figure 7. As mentioned in Section 1.1.1, the concentration of REE varies by the type of deposit and, thus, the processing. In recent publications, some researchers have outlined the process of REE production from pre-concentration to refining [2]. A detailed description of each of these techniques is discussed in the subsequent sections.
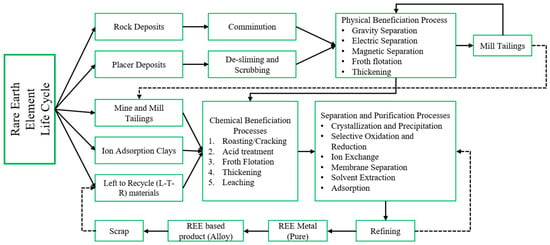
Figure 7.
A Schematic explaining the REE lifecycle from Extraction to Utilization.
2.1. Enhancement of REE Content by Physical Beneficiation Process (PBP)
As shown in Figure 7, the rock deposits mined need to be processed to remove the valuable mineral locked in the host matrix. These are subjected to subsequent crushing and grinding operations where the valuable mineral content is liberated [22,23], whereas, in the case of placer deposits (in general beach sand minerals), they do not need any size reduction because of size distribution range; instead, they need to be de-slimed and attrition scrubbed to remove the clusters of fines accumulated. The processes and their fundamental principle of separation are discussed below [10,24,25,26].
- Gravity concentration processes usually separate the heavy and lighter minerals by exploiting differences in specific gravity (S.G.). The S.G. values for REE minerals are in the order of 4–5.
- Electrical separation processes operate by the charge transfer of the conducting material onto the separator’s roll. The dominant mechanisms are charge transfer by ion bombardment, conductive induction, and triboelectric charging, usually seen in electrostatic separation. Monazite is non-conducting material, so it sticks to the roll of the separator.
- Magnetic separation uses the difference in magnetic susceptibility of the mineral particles in the presence of the applied magnetic field. In placer deposits, the difference is among the paramagnetic and ferromagnetic particles. The monazite is weakly magnetic and is separated using high-intensity magnetic separators.
- Froth Flotation is a physio-chemical separation process that operates based on the variations in the hydrophobicity differences of the minerals where the hydrophobic phase can be valuable/gangue minerals, and the hydrophobicity is induced by the addition of reagents termed as collectors and frothers. The most common collectors for REE-based mineral flotation are tall oils obtained from wood processing.
- Thickening is a dewatering process that reduces water content and increases the % of solids by weight in the slurry; thus, the product received is termed as concentrated and sent for further processing. If economically viable, the mill tailings obtained in PBS are sent for further mineral recovery.
2.2. Enhancement of REE Content by Chemical Beneficiation Process (CBP)
As shown in Figure 7, the ion adsorption clays, LTR material, and tailings (mine and mill) do not need a PBS route because they are already enriched in one or more REEs, so they are subjected to processing by chemical routes where the preliminary task is to break the bond between the coordinating elements of the REEs and waste (referred as gangue). To achieve this, the temperature of operations needs to be in the range of 573–1273 K. This process is technically termed roasting/cracking [22]; this operation produces oxides of REEs, and the purpose of this treatment is to achieve good coordination between water and mineral surface, which is needed for downstream processes [21,27]. This step is followed by acid treatment with concentrated HNO3, HCl, and H2SO4 acids to produce a slurry that is further sent for flotation and thickening processes [27]. The final cake/concentrate from the thickening stage is sent for leaching operation. Solid particles are dissolved in the leaching circuit, leaving the undissolved particles as residue (to be disposed of). The pregnant leaching solution (PLS) obtained from the leaching circuit contains all the desired REEs associated with some transition elements. Mathematical models have been tabulated to determine the leaching rate and kinetics [15]. Recycling NdFeB permanent magnets involves using pyrometallurgical principles to assist selective dissolution in hydrometallurgical operations because, in pyrometallurgy, the REE is changed from one compound to another (often from a sulfide to an oxide by roasting) [28].
2.3. Technological Advances for Individual REE Separation
Section 2.1 and Section 2.2 discussed the preliminary steps required for processing REE content from any source until it is obtained as a slurry (PLS). After PBP and CBP operations, the PLS is enriched with REE content in a solution. There can be some other multivalent ions (undesired metal ions) that need to be selectively separated to produce high-purity rare earth oxides of each element. Still, the approach from here is challenging because of the REEs’ properties, as discussed earlier. In recent years some novel methods have been invented [29]. The University of Utah researchers developed the selective electrochemical recovery of REEs based on atomic mass where the oxidation state plays a significant role [30]; further, they discovered a novel approach to REE extraction, which was designed by introducing the concept of Bio oxidation and leaching followed by a subsequent stage of precipitation with subsequent acid production, which assists the leaching process; these are two of the few economic and environmentally viable approaches for recovering REEs from secondary sources, such as coal rejects [9,14,31,32,33]. Still, most producers rely on existing methods, either solid–liquid or liquid–liquid separation processes. In most systems with REE mixtures, it is possible to separate LREEs/MREEs and HREEs, but very challenging to separate REEs with adjacent atomic numbers. The detailed description of mechanisms for processes mentioned in Figure 7 is explained in upcoming sections. Before understanding the REE separation mechanisms, it is essential to understand the solution behavior of REEs.
- i.
- Solution behavior of REEs
A few authors recently reported that coordination chemistry is crucial in segregating REEs [34]. The solid–liquid separation or liquid–liquid separation process involves the use of coordination chemistry. The larger charge densities of HREEs relative to LREEs increase electrostatic attraction, thus forming more stable complexes with ligands [34]. In the series of lanthanides, the unfilled 4f electron configuration leads to a predominant electrostatic interaction with the ligands compared to the 4d transition group of elements; the bonds formed during the interaction are relatively weaker [35]. For example, the properties of Sc are nearly equivalent to transition metals, while Y is more like lanthanides. Almost all the REEs are solely stable at +3 charge, but the same is not valid for Eu and Ce.
The polarizability of donor-atom (in other words, hard and soft acids and bases theory [36]) categorizes based on the ability of the external electronic field to cause ion distortion, where if ions are less influenced, they are hard and vice versa [34]. The hard acids (namely, the hydrated cations) interact with hard bases to form inner-sphere complexes, which form complexes into solids with low solubilities, thus assisting selectivity in precipitation.
The extent of complex formation depends on the anionic species present in the solution, and the degree to which the resulting complex can be measured by a constant of stability, the logarithm of the solubility product, logβ. The easier way of expressing this is:
REE-Cation + Ligand ⇔ REE-complex.
Also, the ligand denticity/tooth/coordination number plays a role in determining the stability of the REE complex. In aqueous chemistry, other factors that influence the logβ values are the temperature because the temperature change affects the reaction kinetics [26]; and the increase in hydrogen ion concentration increases the stability of REE complexes because of the increased tendency to deprotonate; therefore, large pKa values are often preferred for ligand design.
Additionally, it was noted that chelate stabilities also depend on the extent of the hydration layer of the REE ions in a solution [37]. Other than these factors, additional experimental variables include the strength of the ligand, ionic strength, and the hindrance caused by the steric bulk can also impact the logβ [35]. Anions of the soft bases such as nitrates, chlorides, and sulfates will only be able to produce outer-sphere complexes that are weaker in nature. Moreover, there are no such or lower logβ values of complex formation for ammonia and disulfide. Also, the interaction between these anions and a molecule is noteworthy only at high concentrations above 1 M; this is not true in the case of sulfate solutions, due to their higher logβ values; in addition, these sulfate solutions can form REE (SO4)2−, which is a negatively charged complex at higher sulfate concentrations. In the presence of hard ligand groups, creating additional bonding with softer bases, such as amine groups, is feasible. The complexing stability usually increases from La to Eu or Gd. However, for HREEs like Gd, they reach a maximum and plateau [38]. Due to their large size, REE ions can form a limited number of complexes. Ligands with high chelation capacities and the ability to attract small-sized REE cations with increased charge density can form stable and water-soluble complexes. An overview of factors influencing ligand design is presented in Figure 8.
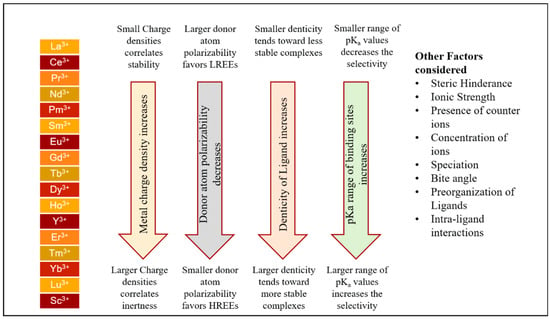
Figure 8.
Design factors considered for ligand chemistry; the image is redrawn with permission from Hovey, Dittrich, and Allen 2023 [34], copyright 2022. Published by Elsevier B.V. on behalf of the Chinese Society of Rare Earths.
The underlying fundamentals for separating individual REEs follow the variations in the basicity resulting from reduced ionic radii from La to Lu [39]. This measure of the tendency to accept hydrogen ions from water impacts the solubility limit. Further, it influences the decomposition of the water molecule and complex formation. Most separation processes are designed by utilizing this concept. Moreover, in addition to these differences, a few more possibilities that influence separation are the valency states of REEs, which can be modified by oxidation or reduction [3]. Since the discovery of REEs, there have been attempts to separate each REE. From then until 1947, the methods of crystallization and precipitation, oxidation, and reduction seemed favorable [3].
- ii.
- Separation by Crystallization and Precipitation Methods
The age-old traditional method for generating highly pure individual REEs is possible by the fractional crystallization (F.C.) approach. This method utilizes a mixture of different salts to produce REE oxides. Of these salts, nitrates based on ammonium are beneficial for LREEs (except Ce), nitrates of magnesium are helpful for MREEs (except Pm), and nitrates of manganese can separate the C.G. group of REEs, and for the Y.G. group, bromates/ethyl sulfates can be beneficial [3,40]. However, this approach is very challenging and not ideal for HREEs because of the large production cycle varying from 16 to 15,000 steps; historically, the crystallization process was performed by G. Urbain in 1907 for the separation of Yb and Lu [41], and Feit used a similar approach for the generation of Ho [3]. Moreover, salts are limited in tailoring the crystals needed for this approach [42].
Similarly, an alternative approach with extensive studies performed is the fractional precipitation (F.P.) like in F.C.; this approach generates precipitate REEs with salts of sulfates where the LREE group can form insoluble sulfates, followed by MREEs with partial solubility, and HREEs with high solubility [3]. In the case of Y.G., several precipitation techniques with hydroxides, chromates, and even nitrates were possible for preliminary Y enhancement and separation. However, the application of this method is limited to separating REEs into groups but not into individual ions [3,43].
Later on, in 2014, understanding the extensive efforts needed for F.C. separations, a size-selective crystallization (SSC) approach was tried to achieve REE separations. The duration of crystal synthesis is within a few hours, and it is environmentally friendly with a reduced use of reagents. The basis for this approach is a group of variations in a solubility product with an REE complex formation, thus resulting in the shape of a metal–organic framework (MOF) [42,44]. This approach relies on the coordination between the ionic radii and the prepared MOF to facilitate the crystallization of a specific target REE. If the ionic radii difference is significant, then a particular REE will concentrate on the specific type of crystal synthesized, which can be a suitable approach to large-scale production. However, this method fails when the difference is slight (in the range of 1–5 picometers), which is the case of adjacent REE separation leading to contamination in the range of 20–25% of undesired REEs in the final crystal [42].
A few more developments in 2017 were made to improve the separation by F.C., where crystals are formed by controlling the reaction kinetics and, later, selective separation by flotation. Still, this method did not explain the selectivity factor (S.F.) among adjacent REEs [1,45]. Later on, in 2019, a valence selective crystallization (VSC) approach was designed to selectively separate Ce from trivalent REEs because of its tetravalent nature; this work proved effective in the separation of Ce and other metal ions when there is a difference in the valence state of ion and without distinction in the valence state there is no chance of separation [1,45].
- iii.
- Separation by Selective Oxidation and Reduction Method
As discussed above, among the REEs, Ce can exhibit a tetravalent state so that the separation can be even achieved by utilizing differences in the valence between other ions and Ce. Heat treatment of cerium ore in the presence of natural atmosphere/air at elevated temperatures of 650 °C can lead to oxidation as can treatment at lower temperatures (>100 °C) using hydroxides in the presence of chlorine [3]. Oxidation of Ce from a +3 to +4 state can be achieved using ozone. Leaching the ore in concentrated acids to dissolve +3 REEs, followed by precipitation of +4 Ce can facilitate separation from other REEs. Still, some Ce is left in the system, which can be further removed.
Among other lanthanides, Pr [46] and Tb [47] exhibit the tetravalent state and thus are less stable in aqueous media. These can be separated from the system by forming precipitated and separated oxides [3].
When the oxidation methods for REEs are applicable, there can be the possibility for an approach through reduction. Sm, Eu, and Yb separations can be performed after reducing them to a +2-valence state. The reduction can be performed electrolytically or by photolysis (referred as photochemical reduction using a laser), where a concentrated beam of consistent wavelength is directed to an aqueous solution. However, in the presence of calcium, reducing REE halides is not feasible, and thus they can be separated from the slag later on, if they are pyrometallurgically processed. But, Sm can be reduced to a +2 state electrolytically among a mixture of REE chlorides. Pre-concentration or enrichment by PBP/CBP forms the basis for any reduction method because of their low crystal abundance (values averaged in μg/g) in an ore body [3].
Due to the increasing demand for the supply of rare earth metals (REM) globally, REM producers employ the most versatile and widely applicable rare earth techniques for high-quality products. Thus, years after the initial separation work in the 1950s, the REM supply market changed tremendously. Before the 1960s, the ion exchange technology (solid–liquid separation strategy) was the only possible approach to separate REE. However, post-1960s, solvent extraction technology produced high-purity REEs for specified electronics and analytical measurements [48]. Other than these techniques, there are a few more methods: membrane separation and adsorption [1].
- iv.
- Separation by Ion Exchange
The ion exchange techniques offer another approach to REE separation. The ion exchange (IX) process separates the desired ions from the solution/slurry with the help of either organic/inorganic materials [49]. Researchers have categorized IX media into natural, modified natural, and synthetic [50]. The additional classification is shown in Figure 9. As for inorganic materials, they can be clays and zeolites; the primary difference between them is layering for clays, and the porosity for zeolites [26]. Most of the ion exchange process uses resins with a porous network with embedded functional groups for enhancing selectivity. In most recent reviews, the list of commercial resins is tabulated [35].
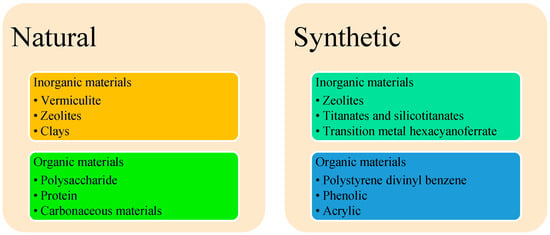
Figure 9.
Classification of IX exchangers.
A critical feature of the material used in this IX process is its ability to stoichiometrically and reversibly swap the ions from its resin-ion functional group with other ions (REE) found in the solution in which it is submerged. The equation for ion exchange reaction is expressed in equation-1,
REE ion + Resin-Ion-Functional group ⇔ Ion + Resin-REE-Functional group
The selectivity index for the reaction in Equation (1) is defined as KResin-Ion/REE ion, and this ratio is explained in Equation (2) where
The Extraction coefficient, EREE ion, for the reaction in Equation (1) is explained using Equation (3) where
In addition to these equations, the IX data are correlated and analyzed at equilibrium using several models used to design the industrial systems. Some of the most widely used and applicable models are the Langmuir, Freundlich, and Tempkin models [51]. Generally, ion diffusion in the resin phase is the rate-controlling mechanism for the IX process. In some cases of low-concentrated solutions, the rate is often controlled by diffusion through the boundary layer due to a higher dilution ratio. Numerous models of the IX process are presented in the literature, but the most often used are Freundlich, Langmuir, and Tempkin models [51]. Also, based on experimental data, the pseudo-second-order kinetic model is most applicable to IX processes [35].
The most common IX resin matrix comprises crosslinked poly (styrene-co-divinylbenzene) with desired functional groups. The ion exchange rate is attributed to the type of active group, the extent of polymerization, the size of exchangers [1], and the classification of Exchangers is dependent on the charge of the functional group; thus, they are classified as strongly acidic cation exchangers with sulfonic acid functional groups and weakly acidic cation exchangers with carboxylic acid functional groups, strongly basic anion exchangers with quaternary ammonium groups and weakly basic anion exchangers with primary, secondary, and tertiary ammonium groups. The matrix of resin can be either microporous with permanent pores or gel where diffusion of ions happens through penetration in a concentrated solution, and the resulting diffusion coefficients (measured in m2s−1) are smaller than the former.
The process of IX is cyclic, with two primary steps of loading and elution. In this, the desired ions are extracted from PLS in the loading phase and are loaded into the resin phase, and the resin is removed from the liquid. Subsequently, an elution operation is performed to exchange the loaded ions into the eluent to obtain a purer form of the target ions. This operation can be performed either in batch or continuous process. The loading step can only be effective in separating the REE from other dissolved ions (like Cd, Mg, Fe, etc.) in the system but is difficult to use to fractionate into individual REE elements. Thus, an effective IX process needs a supporting selective elution that uses a complex solvent to desorb the loaded metals, and the degree of selectivity in the elution is a factor of logβ and the type of complex solvent. The most commonly used solvent is EDTA, which typically elutes REEs in decreasing order of atomic number due to the increasing stability constants of the complexes from light to heavy REEs [51]. In recent studies, the separation of REE from sulfate solutions has been studied using conventional resins (CVRs), chelating resins (CRs), solvent-impregnated resins (SIRs), hybrid organic mineral materials (HOMSs), and ion-imprinted polymers (IIP).
CVRs with different types of functional groups showed a difference in adsorption behavior toward REEs; for instance, the strongly acidic cation type can load to higher capacities with greater affinity for LREEs than HREEs because of the small hydration layer surrounding the metal cation. But for strong basic anion types, there is a weaker affinity of REEs, other than Scandium, but further studies are needed to commercialize this type of resin. Also, for this resin type, the adsorption behavior depends on the kind of medium, like sulfate or chloride, for a given system. Strong acids can damage the resin, and weak acids can withstand the resin, thus limiting the choice of extractions. Elution operations from CVR depend more on concentrations, temperature (high temperatures are needed for REE compared to Sc [52]), and types of complexing agents like the EDTA, sodium carbonate, which are suitable for REE separation. EDTA is used for Nd separation from the bulk [35], and Sm/Eu and Eu/Tb can also be separated using EDTA, which has higher selectivity [53], but the only disadvantage is the solubility at a given pH, which needs to be addressed.
The CRs are another class of resins used to separate REEs from complex media, but these do not have good loading capacities compared to the CVRs (strongly acidic cation resins). Still, their selection depends on a trade-off between loading capacity and selectivity. As experiments with the iminodiacetic acid-based resin showed a good affinity for separating La, Sm, and Ho from the bulk or mixture of elements with REEs [35,54]. However, the CRs’ loading capability varies with variations in sulfate concentration [35].
The HOMS are IX resins that combine inorganic supports and organic ion exchange groups. These materials offer non-swelling supports and outstanding operational stability by using inorganic matrices. At the same time, incorporating organic ion exchange groups provides good selectivity and excellent binding ability to metal ions. Silica is considered the most suitable inorganic matrix for producing organo-mineral compounds because it is affordable, readily available, and allows for precise control over structural parameters like grain sizes, average porosity, and specific surface area. However, inorganic supports such as titanium and zirconium oxides can also be used. There are three techniques for modifying inorganic support surfaces to create these resins: modification of the silica surface, alteration of inorganic matrices by polymeric functional groups, and polycondensation of applicable organosilicon modifiers or their co-condensation with a non-functional composite [35]. Various functional groups, such as N-methylimidazolium, iminodiacetic acid, and phosphonic and carboxylic acids, have been used to prepare these ion exchangers for complex REE separation mixtures.
The Ion imprinting method is another effective way to enhance the selectivity of metal ion separation. This method involves synthesizing functional monomers with chelating groups into a porous solid polymer particle, incorporating metal templates during polymerization to bind to metal cations selectively [35]. Numerous studies have demonstrated the potential of IIPs in selectively removing REEs from complex media. The selection of functional monomers is critical to preparing IIPs, with vinyl groups being the most suitable polymerizable functions for chelating ligands. Studies have shown that IIPs can achieve high sorption capacities and selectivity coefficients for targeted metals using a range of functional monomers such as methacrylic acid, ethylene glycol dimethyl acrylate, 4-vinyl pyridine-acetylacetone complexes/monomers, and N-methacryloylamido folic acid. Some studies have also explored using conductive polypyrrole and iminodiacetic acid (IDA) or sulfonic groups in developing IIPs. However, despite their potential, there are still uncertainties about isolating and separating IIPs from complex matrices. The SIRs are another class of IX resins where a desired solvent is impregnated into a resin matrix, thus enhancing the selectivity. Still, their application is limited by the leakage of organic solvents into the solution, thus leading to contamination. This problem can be addressed by adding a polymer layer to resin, but the merits and demerits of this approach are not yet published [1].
- v.
- Separation by Solvent Extraction
After the development and implementation of solvent extraction (liquid–liquid separation strategy) in the 1960s, the commercial production of REMs received special attention using this method because it can handle vast volumes of solutions [22], consume less energy for operation, and increase the reaction rate by vigorous mixing for efficient separation of REEs [55]. This solvent extraction is a new approach in the field of extractive metallurgy, which is a subset of hydrometallurgy. The schematic in Figure 10 explains that the solute (target metal) is dissolved in the aqueous phase in a conventional solvent extraction. The target metal is distributed into the organic phase by vigorously mixing the organic phase with an aqueous phase [51]. The organic phase extracts the metal ion from the aqueous phase. However, the non-aqueous solvent extraction systems are less available in practical applications for rare earth systems and are primarily used for p-block and early d-block elemental separations [56]. In the traditional approach of conventional solvent extraction, the organic phase is filled with the utilization of numerous traditional organic solvents (TOS) (like kerosene, isooctane, and n-dodecane). These solvents are not eco-friendly, and the safe practices required for their handling and application can be tiresome due to their highly flammable nature, significant volatility, and potential risks to human and aquatic health [57]. Moreover, their utilization often involves saponification, which necessitates the use of acid and base [58]. A decade ago, the drafted term for this metallurgical process was ionometallurgy, in which the approach is similar to solvent extraction but with ionic solvents replacing the organic phase, which is typically made up of TOS [59]. Thus, the ionic solvents are broadly classified as ionic liquids, which consist of distinct cations and anions in the molecular structure [59]. Deep eutectic solvents are liquids that result from the eutectic mixtures of specific hydrogen bond acceptors and hydrogen bond donors [60]. The schematic classification of organic phases is explained in Figure 11.
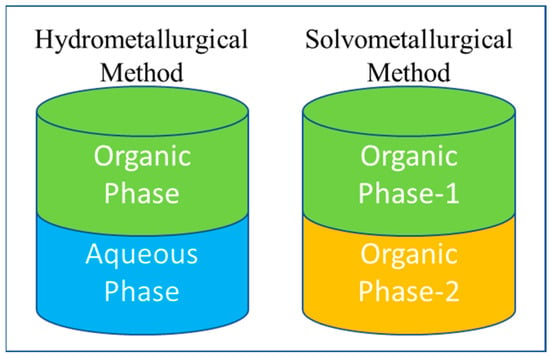
Figure 10.
Schematic of solvent extraction in Hydrometallurgy and Solvometallurgy routes image is reprinted with permission from (Binnemans and Jones 2017 [56]), Copyright © 2017, The Author(s).
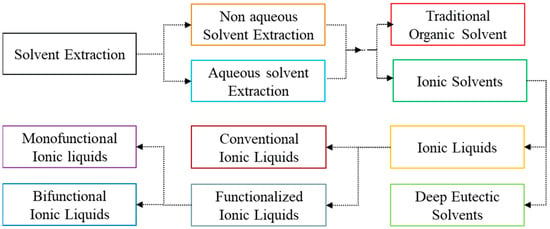
Figure 11.
Schematic explaining the classification of solvents for the solvent extraction process.
Difference between the conventional method and the ionic liquid-assisted method of solvent extraction
- Traditional solvent extraction system (Extractant based)
The separation in solvent extraction depends entirely on the efficiency of extracting a target metal from the aqueous phase to the organic phase; this transfer cannot be spontaneous due to differences in solvation energy. Adding an extractant is necessary to assist the rate of metal transfer. Therefore, a practical solvent extraction system can be expressed as
[MREE3+-Acid] aqueous phase-[Extractant-Organic phase] organic phase.
Based on the requirements for extractants, there are four categories of extractants [60,61,62,63], as discussed below:
- Acidic type (with cation as exchanger)
- ○
- Carboxylic acid
- ⮚
- Versatic acids (Versatic 10, Versatic 911);
- ⮚
- Naphthenic acids.
- ○
- Sulfonic acids (Dinonylnaphthalene sulfonic acid).
- ○
- Phosphinic acids (P229, Cyanex-272).
- ○
- Phosphonic acids (EHEHPA, HEHEHP, P507, and PC88A).
- ○
- Phosphoric acids (D2EHPA).
- ○
- Monothiophosphorous acids (Cyanex-302).
- ○
- Dithiophosphorous acids (Cyanex-301).
- A basic type (with anion as exchanger)
- ○
- Primary amines (Primene JMT, N1923).
- ○
- Secondary amines (N-Methylaniline).
- ○
- Tertiary amines (Alamines).
- ○
- Quaternary amines (Aliquat 336, Adogen 464).
- Neutral type (solvation exchanger)
- ○
- Phosphorous ester (TBP, DBBP).
- ○
- Phosphine oxides (TOPO/Cyanex 923, Cyanex921, and CMPO).
- Chelating type
- ○
- Beta diketones (LIX 54, HTTA).
Of these extractants for solvent extraction, the acidic type is most preferred, and the chelating ones are the least applicable [62,64]. However, their extraction mechanism is similar to the acidic ones. The extraction mechanism is a neutral exchange governed by the electroneutrality principle, where only neutral species are extracted. The application of this mechanism varies with the extractant type, and a detailed description is given in Table 1, and a pictorial representation of this mechanism is described in Figure 12 [60]. As in Figure 12, the red line explains that the hydrogen ion is released from the organic to the aqueous phase using an acidic extractant during the solvent extraction process. The black line indicates that the REE ion is captured from the aqueous to the organic phase. The same analogy applies to the basic extractant mechanism, where an anion is released in place of a hydrogen ion. For neutral extractants, additional cation/anion is not released into the aqueous phase [55]. In Table 1, MREE3+ denotes an REE ion, E refers to the extractant, HE is the acidic extractant, H+ is the hydrogen ion, BE refers to the basic extractant, and X refers to Halides.

Table 1.
A detailed description of mechanisms applicable to traditional solvent extraction systems.
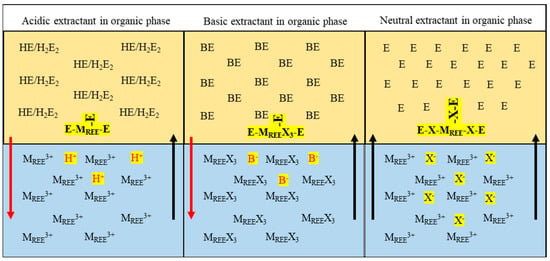
Figure 12.
Pictorial representation of mechanisms applicable to traditional solvent extraction systems.
- b.
- Modern Solvent Extraction system (Ionic Liquid Based)
- Ionic liquidsIonic liquids (ILs) are the organic salts prepared by combining different cations (organic) and anions (can be organic/inorganic), forming a chemical structure, which generates designing a specific property for an ionic liquid [65]. Some of the dominating properties that widen the application of ionic liquids over TOS are the low melting points (<less than 100 °C), and hence these are referred to as room temperature ionic liquids (RTILs); the non-volatility due to negligible vapor pressure under room temperature conditions; and that almost all ionic liquids are non-flammable. Due to the wide versatility in changing the cation and anion combinations, the available number of solvents is in the order of 1,000,000. ILs have viscosity levels that range from 20 to 97,000 mPa. The low density of ionic liquids helps in more accessible phase separation. ILs have high thermal stability and high electrical conductivity values of nearly 120 mS cm−1. An overview of promising high-potential IL utilization and potential future applications is reviewed and explained using Figure 13 [66].
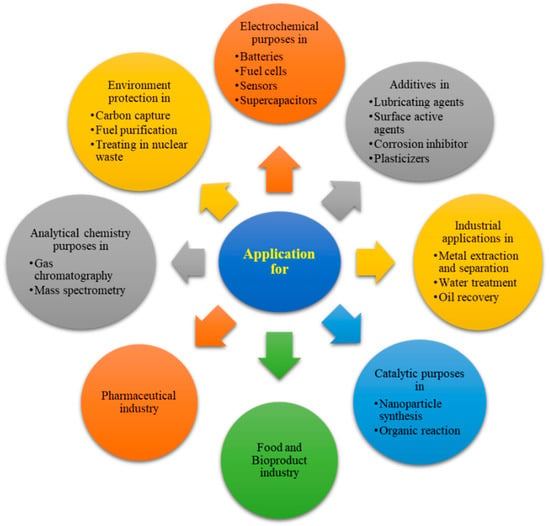 Figure 13. Application potential of Ionic liquids in diverse fields of study.
Figure 13. Application potential of Ionic liquids in diverse fields of study. - Common Cations and Anions for ionic liquids [62]
- Cations
- ○
- Ammonium, Phosphonium, Imidazolium, Pyrrolindium, Pyridinium, Piperidinium, Morpholinium, Isoquinolinium, and Pyrazinium.
- Anions
- ○
- Inorganic Anions are Halides, Nitrate, Tetrafluoroborate, and Hexafluorophosphate.
- ○
- Organic Anions are Trifluoromethanesulfonate, Bis(trifluoromethyl sulfonyl)imide, and Carboxylate.
- Significance of properties on the selection of ionic liquids
- Viscosity
- ✓
- Some general parameters like temperature, water, and organic solvents modify viscosity in any ionic liquid.
- ✓
- Other than these subtle parameters, like the chemistry of ionic liquid, which is dependent on cationic and anion structures, impact the viscosity.
- ✓
- Cationic structure modifies the viscosity due to van der Waals interactions and ionic mobility [62].
- ✓
- Anionic structure modifies viscosity due to hydrogen bonding and basicity [62].
- ✓
- For a similar cation, the viscosity can vary from low to high by changing the anion structure from weak to complex [60].
- ✓
- Viscosity and density are interrelated as these affect solute mass transport to the organic phase, impacting the extraction rate [67].
- ✓
- Due to the addition of excess water, the Coulombic force of interaction between ions tries to reduce the viscosity [62,68].
- Solubility
- ✓
- The solubility of ionic liquids in water is a concern because of the cost involved. The solubility is dependent on the chemical structure of the ionic liquid and the charged species in the solution (predominantly the anions). The charge is related to the size of anions, and this is associated with the activity of the species in solution, which determines whether a species is favorable to interact with water or not [62,69].
- ✓
- Ionic liquids made up of the anions Hexafluorophosphate and Bis (trifluoromethyl sulfonyl) imide have lower solubilities in water. Ionic liquids with halides and nitrates are readily soluble in water [62,70,71].
- HydrophobicityThe hydrophobicity of ionic liquids is dependent on the alkyl chain length geometry of the cation and anion present in the structure, but having higher chains lengths of alkyl groups results in low values of density, more hydrophobicity, and high values of viscosity; these impacts may be different for different anion combinations [60,72].
- Extraction mechanism of conventional (non-functional) ionic liquids in solvent extraction
- ⮚
- Organic phase: Conventional ionic liquidsIn conventional ionic liquids, extraction is favored because they are hydrophobic and can rapidly extract the naturally hydrophobic compounds from the aqueous phase to the organic phase, which hastens the extraction process [63,73]. The extraction of cerium using [C8mim]PF6 is one such application, but it cannot fully extract the other REEs due to the non-coordination nature of ionic liquid with water.
- ⮚
- Organic phase: Mixture of extractants and conventional ionic liquidsAs discussed previously, the mechanism of the extraction of REEs is only possible when the compounds are naturally hydrophobic, but such types of systems are not possible all the time; in such situations, extractants such as chelating agents will form a complex with metal ions of the aqueous phase, and these complexes are more soluble in ionic liquids. Other than this possibility, the extraction depends on the structure of the cations, and the anions play a significant role in leading to cation, anion, and neutral exchange mechanisms.A detailed description and pictorial representation of these mechanisms are given in Figure 14 and Table 2, respectively. As in Figure 14, the red line explains the release of ionic liquid cation (for the cationic exchange mechanism), the mixture of ionic liquid anion and hydrogen ion (for the anionic exchange mechanism), and the mixture of ionic liquid cation and hydrogen ion (for anionic exchange mechanism with more hydrophilic anions) from the organic to the aqueous phase. The black line indicates the transfer of REE ions from the aqueous to the organic phase, except in the case of the neutral exchange mechanism, where both REE ions and hydrogen ions are transferred [63]. In Table 2, MREE3+ denotes REE ion, E refers to the extractant, ILC+ is the cation of ionic liquid, HE is an acidic extractant, H+ is the hydrogen ion, and ILA- is the anion of ionic liquid.
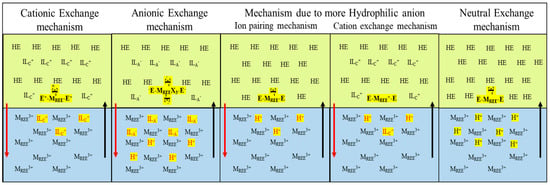 Figure 14. Pictorial representation of mechanisms applicable to conventional ionic liquid solvent extraction systems.
Table 2. Detailed description of mechanisms applicable to conventional ionic liquid solvent extraction systems.
Figure 14. Pictorial representation of mechanisms applicable to conventional ionic liquid solvent extraction systems.
Table 2. Detailed description of mechanisms applicable to conventional ionic liquid solvent extraction systems. - ⮚
- Cation exchange mechanismFrom the available literature, many investigations have been conducted to demonstrate the effect of cation variation on the cation exchange mechanism on the extraction efficiency of the lanthanide series. Still, all of them depended on the cation alkyl chain length, where increased size resulted in hydrophobicity, thus resulting in a decrease in extraction efficiency and, thus, leading to a smaller loss of cations in the aqueous phase and vice versa [60,62,63]. So, an appropriate cost–benefit analysis should be performed before utilizing this approach when processing in large volumes.
- ⮚
- Anion exchange mechanismOnly a few experiments have been conducted to demonstrate the anion behavior in the extraction of REEs where the anion of the ligand forms an anionic-ligand rare earth complex (ALREC) with the REE, and this, when it interacts with the ionic liquid cation, forms a stable structure preventing the loss of the ionic liquid cation. According to the principle of electroneutrality for balancing the charge in both aqueous and anionic phases, the anionic component of the ionic liquid goes to the aqueous phase. After this, at the stripping stage, to prevent loss of ionic liquid, one possibility is to use the ionic liquid as a stripping solution for replacing ALREC with an anionic component of ionic liquid [60,62,63]. Some researchers tried another approach for the anionic exchange mechanism, whereby they modified the anionic part of an ionic liquid by applying the strategy of hydrophobicity as conducted in the cation exchange mechanism, but, here, the hydrophilicity of anion was improved to extract more ALREC. This modification resulted in a negative impact on the solvent extraction system where water tends to dissolve, leading to an aqueous–aqueous extraction system instead of organic–aqueous extraction system, thereby resulting in a decrease in the extraction of ALREC; the reason behind this impact is related to the more hydrophilic nature of anions of ionic liquid, which paves the way for ion-pairing and cation exchange mechanisms.
- ⮚
- Neutral exchange mechanismTo prevent the loss of either of the ions as described in the above two mechanisms neutral exchange mechanism works similarly to the extraction mechanism as in TOS, but modification is performed in the organic phase where it consists of acidic extractant and ionic liquid such that the protons exchange to the aqueous phase leaving behind the cationic and anionic components of the ionic liquids, thereby limiting the loss of ionic liquid, which also enables the potential for large scale industrial applications [60,62,63].The extraction mechanism of conventional ionic liquids differs from TOS because the extraction mechanisms are more related to cation and anion interactions with the REEs; therefore, to consider ionic liquids as eco-friendly solvents, the loss of cation and anion should be prevented in the aqueous phase; at the same instance, there should not be any complications in operation as well as extraction efficiencies. Thus, the application of ionic liquids varies with the type of mechanism needed for a particular extraction process. One such approach is to modify the cation/anion structure of the ionic liquid with specialized functional groups, which have a higher tendency to attract REEs.
- Functionalized ionic liquids (FILs) (referred to as task-specific ionic liquids in some of the literature)In some solvent extractant systems, the targeted metal is associated with different types of ions, so the extraction of a specific metal can be challenging in such cases. Ionic liquids are modified with target-specific functional groups either at the cation or anion positions; these types of tailormade structures are referred to as functionalized ionic liquids [74]. The properties of ionic liquid, and the formation of the metal complex can be achieved within a functionalized ionic liquid. In most solvent extraction systems, ionic liquids with the functional group at the anionic site are utilized [75]. This approach has led to the development of different types of ionic liquids.
- ▪
- Common functionalized cations and anions for ionic liquids [62]
- ○
- Cations
- ▪
- Betainium, Trioctyl(2-ethoxy-2oxyethyl) ammonium, Tricapryl methylammonium, ammonium, Poly (ethylene glycol) functionalized imidazolium, and 1-Alkyl carboxylic acid 3-methyl imidazolium.
- ○
- Anions
- ▪
- Di-(2-ethylhexyl) phosphate [P204], Di-(2-ethylhexyl) orthophosphinate [P507], Sec-octylphenoxyl acetate, Sec-non octylphenoxyl acetate, cyanex272, Dioctyldiglycolamate (DGA), and Dihexyldiglycolamate (DHDGA).
- ▪
- Monofunctional ionic liquidsThe monofunctionalized ionic liquid structure is synthesized by placing the functional group of extractants either in the cation/anionic position of conventional ionic liquids, which results in two dominant benefits: One of them is the prevention of loss of the ionic liquid component to an aqueous phase, which correspondingly hinders the cation/anion exchange mechanism, thus supporting neutral extraction mechanism for the separation of REMs. Another benefit is that this single ionic liquid can perform the task of both diluent and extractant. Positioning the functional group at the anionic site is much easier than at the cationic site. Thus, most monofunctional ionic liquids are anionic-based. Some functional ionic liquids with carbamoylmethylphosphine oxide (CMPO) moieties showed reduced extraction due to high viscosity in such situations. It is noteworthy that a conventional ionic liquid can be used as a diluent to reduce the viscosity.
- ▪
- Bifunctional ionic liquidsBifunctional ionic liquids are designed by attaching functional groups to cation and anion parts. The extraction can be achieved in the presence or absence of TOS, and the mechanism is related to the neutral extractant mechanism but in a much more complex way due to the selection of functional groups, thereby influencing interactions between the functional group and ionic liquid, and interactions between the ionic liquid and aqueous phase.Other functionalized ionic liquids discussed in the literature are acid-functionalized ionic liquids that give a mixed advantage in the form of liquid with an appreciable contact area [76]. Based on this, acid-functionalized ionic liquids are classified into carboxyl-functionalized ionic liquids and sulphonyl-functionalized ionic liquids. The selective exchange mechanism is similar to that of the neutral extractant mechanism where the protons (H+) are released into aqueous media, thereby preventing the loss of cations and anions to the aqueous phase [76].A sulphonyl-functionalized ionic liquid was pioneered in solvent extraction for rare earth separation. According to the literature, acid–base-coupled ionic liquids are one other type of interesting ionic liquids for REE separations. Although most ionic liquids utilize an anionic-based exchange mechanism as part of the separation, this type of approach helps to combine the coordination ability and hydrophobic nature in a single ionic liquid [77]. On the whole, the ionic liquid application for solvent extraction enhances selectivity, and the type of ionic liquid application varies by the target metal required as a part of the extraction. Because these solvents are tailormade, it is easy to extract metal if the impact of the solution chemistry of the desired ions is understood and managed. The primary challenge lies in using these liquids appropriately. The selectivity enhancement of these liquids is not feasible below a certain feed concentration level. Thus, to extract them, a synergistic mixture of these liquids is required, which contributes to a factor of cost so there is a need to properly consider their application with an emphasis on not releasing some ecologic pollutant of either cations or anions.
- vi.
- Separation Using Membranes
The membrane Separation Method (MSM) is an eco-friendly process that combines extraction and stripping without thermal heating. MSMs help separate organic mixtures, gases, and metal cations, including ferrous, non-ferrous, heavy, and precious metals. Liquid Membranes (LMs) have efficiently and rapidly separated metal ions. However, due to stability issues, research has shifted toward more stable Non-Liquid Membranes (NLM). MSMs are divided into liquid membranes (LMs) and non-liquid membranes (NLMs). LMs include several types, such as bulk liquid membranes (BLMs), Emulsification liquid membranes (ELMs), supported liquid membranes (SLMs), hollow-fiber-supported liquid membranes (HFSLMs), and electrostatic-pseudo-liquid membranes (ESPLMs). NLM comprises polymer inclusion membranes (PIMs) and ion-imprinted membranes (IIMs) [1]. HREEs, or heavy rare earth elements, are often preferred in membrane separation processes due to the absence of the issues typically associated with traditional extraction methods, such as the need for large amounts of organic solvents, supersaturated extraction, and poor phase separation. The membrane transport process involves a non-equilibrium mass transfer process across various types of membrane surfaces, including BLM, ELM, SLM, and HFSLM. In PIMs and IIMs, carriers are chemically or physically bound within the membrane matrix, making these membranes more stable. There are two types of mass transport in membrane separation: co-transport, which is suitable for neutral carriers like tributyl phosphate (TBP); and counter-transport, which is ideal for acidic carriers with cation exchange mechanisms like P507. The coupled transport process can be broken down into five possible steps [78].
- Diffusion of REEs from the feed phase to the boundary layer of the feed-membrane side.
- Carriers extract REEs at the interface.
- Diffusion of REE complexes across the membrane.
- Disintegration of REE complexes near the stripping interface of the membrane.
- Diffusion of REEs from the stripping interface to the stripping phase and vice versa.
The use of BLMs was limited due to their small transport surface area.
ELMs have gained popularity for their rapid separation abilities, incorporating the benefits of solid-phase membranes and solvent extraction. The process of ELM involves carriers, surfactants, diluents, feed, and stripping solutions. The three primary stages include emulsion formation and target species permeation across the membrane, as well as emulsification of the emulsion after phase separation. Carriers, surfactants, and diluents are all crucial in the membrane separation process for rare earth ions, each with specific functions and properties. Despite their advantages, ELMs encounter instability problems such as membrane leakage and emulsion swelling that can impact transport efficiency.
Another effective method for the separation of REEs is the use of SLMs, which utilize a porous membrane that is infused with carrier-containing solvents. It separates the feed and stripping phases, has a higher interfacial area-to-volume ratio, and consumes fewer carriers (functional groups), thus providing an opportunity for the utilization of costly but effective extractants such as ionic liquids. Improved SLM strategies such as combined SLM and dispersion SLM (DSLM) have been developed to enhance stability and carrier renewal. However, challenges such as partial miscibility between the organic liquid membrane phase and aqueous phase, and blockage of carriers in the supporting membrane, must be addressed. Further advancements in supporting membrane structures and protection of the organic liquid membrane phase are required to fully realize the potential of SLMs for practical REE separation applications [78,79].
Hollow-fiber liquid membranes (HFLM) with micropores have a high surface area per volume and are promising for separating REEs. Polypropylene is commonly used in the matrix, and HFLMs can be operated in co-current or counter-current flow modes. HREEs permeate more quickly in the counter-current flow mode, and the presence of Na+ accelerates the permeation rate. The stability of the HFLM process is good, and HFSLMs integrate extraction and stripping into one module [78,79]. Future research directions include developing highly selective and permeative membrane materials and inorganic membranes with thinner thicknesses and smaller pores.
The development of BLMs faced the challenge of a low interfacial area-to-volume ratio, while ELMs and SLMs encountered instability during long-term tests. To address these issues, ESPLMs with a high contact surface area per volume, low energy and reagent consumption, and minimal leakage and swelling rate were developed. A typical ESPLM consists of a baffle plate in the middle of the reaction tank that separates the organic phase, allowing it to move freely while the aqueous phases are isolated. The bottom of the tank contains two settler areas where the raffinate and concentrated solutions can settle independently without the high-voltage electrostatic field. ESPLMs rely on an electrical field to disintegrate the organic phase into tiny droplets, and the concentration gradient drives the transport of complex and carrier. ESPLMs have demonstrated high efficiency and selectivity for preconcentrating and separating REEs from dilute solutions but require careful maintenance of the electrode devices [78,79].
Polymeric membranes have become a preferred alternative to liquid membranes due to their stability and ease of synthesis. The PIM is the most commonly used polymeric membrane for water treatment, which includes a polymer matrix, plasticizer, and carrier. While there is limited literature on separating REEs using PIMs, various polymeric membranes have been developed and tested for separation, purification, or pre-concentration. However, there are challenges with the fouling of membranes, low flux, and limited thermal and chemical stability [1,78]. More research is needed to develop new synthetic polymers designed explicitly for REE separation and to improve the economy and sustainability of the membrane technique for industrial applications.
The molecular imprinting technique, developed in 1993, is widely applied for separating molecules or ions by creating cavities in polymers that match the size and configuration of the template. Since 1994, IIM/IIP has separated REEs through functional groups from polymerization monomers. Retarded and facilitated permeation mechanisms are two ways for an imprinted membrane, but the retarded mechanism is more common. Dual-template and interconnected 3D macro-porous structure methods improve the separation efficiency of adjacent REEs. However, higher demands for membrane applications have restricted the functional monomers and crosslinking agents for imprinted systems. Another developing direction is exploring functional monomers with a specific selectivity for target template REE ions [78,79].
- vii.
- Separation by Adsorption
The adsorption-based separation for REEs is one of the most accessible and versatile methods, especially when traces of REEs are present in the source material. This method is an environmentally, gentle, and economically friendly approach. The operating principle mechanism behind adsorption is whereby the adsorbates (ions being adsorbed onto the surface) accumulate on adsorbent materials (the material on which ions are collected) with variations in surface area and porosity, thus forming a thin film on the adsorbent surface on the molecular scales [80]. The adsorption process has higher values of the selectivity index and enrichment indices. Talan et.al stated that on the surface of the adsorbent, specifically on the stern layer, the REE-complex formation occurs through covalent/ionic bonding, and the complex formation rate increases with the charge of the ion [81]. After the complex formation, the REE cation forms an outer sphere where the structure resembles a water molecule trapped between the REE cation and the adsorbent surface. There is another possibility of the REE cation remaining in the diffusion layer where charge cancellation happens with adjacent water molecules instead of complex formation, and the bonding here is purely ionic. The process is more susceptible to control parameters such as temperature, time, concentration of adsorbent, surface area, target ion concentration, etc. [7]. In extracting REEs, the adsorbent linked with the targeted ion selective functional group enhances the selectivity [1]. The process is exothermic; the continual mechanism of adsorption is the desorption process that focuses on removing adsorbed molecules from the surface. This adsorption process works in two ways: physisorption (adsorption through electrostatic forces and van der Waals interactions) and chemisorption (formation of irreversible chemical bonds, mainly the covalent bond between the adsorbent surface and the molecules) [80]. For any adsorption mechanism, we need to know the type of adsorbent, kinetics, isotherms, desorption efficiency, thermodynamics, and solution chemistry as prerequisites; the mathematical models for this are presented in [51,80,82]. In recent years, there has been widespread growth in the synthesis of these adsorbents, which are further classified and discussed in this section.
Natural and biodegradable adsorbents are cheaper, readily available, and exhibit considerable adsorption capacities. These materials also offer a typical advantage of being low cost. However, adsorption capacities in typical solutions with competitive ions are not as high as in media such as solvent extractants.
REE element production or separation using ion adsorption clays is another low-cost medium. REEs are naturally found on some clays and they can often be extracted from them due to their physisorption [7]. Thus, they can be easily separated. Some clays that are suitable for REE separation are kaolinite, halloysite, muscovite, montmorillonite, vermiculite, and illite [83,84]. The adsorption of REE on clay minerals is dependent on the surface morphology of these minerals. Natural and expanded vermiculite show good selectivity for REE extraction. Among kaolinite and halloysite, Eu adsorption was good on halloysite rather than Kaolinite due to the increased uptake capacity and surface area [85].
Zeolite is another material class that can be naturally available or synthetically manufactured. This material has good adsorption properties, especially for Nd and Gd, where a selective separation can be achieved with non-heat-treated zeolite-type adsorbent material for Nd and heat-treated adsorbent material for Gd [86]. Modifications to the surface of these materials can enhance the separation properties [1,7].
Chemical materials such as polysaccharides and carboxyl groups are found in some low-cost adsorbents, which are naturally available and biodegradable. Specifically, materials like cellulose, starch, gum arabic, chitosans, etc., function as binding sites for REE adsorption and surface modification of these adsorbents by chemical treatment can enhance the REE extraction and separation abilities. Selective separation of Dy from Nd, Pr, Tb, and Fe [87]; Gd from Dy, Tb, Pr, and Nd [88]; and Sc from Lanthanides [89] has been reported by using these kinds of materials.
Metal–organic frameworks (MOFs) are a well-known class of porous and crystalline materials with larger surface areas and adjustable pore properties. This type of material is formed by the chemical interaction of a single metal ion/cluster of ions with an organic linker [90]. As reported, a MOF-functionalized Cr-MIL-101 enhanced the separation of Gd from transition metals; also, for another compound, UiO-66, the adsorption rate of Gd increased due to the modification of structure with ionic solvents. Due to the poor mechanical properties of these types of materials, some literature used composite materials, which combine a MOF with natural adsorbents. Still, the literature did not explain the reusability of those materials. Similar to MOFs, a covalent organic framework has been found, but its application in the REE field is not yet known [1].
Using the impregnation of functional groups into adsorbents as discussed in ion-exchange technology, polymeric-based adsorbents have been developed that work on the ion-exchange mechanism [7,91]. Polymers can be either ion imprinted, natural polymers, or nanofibers. These nanostructures enhance REE adsorption due to a larger area-to-volume ratio, porosity, stability, and reduced density. In this sense, some geopolymers that help to separate HREE and LREE help to remove REE from the bulk and can facilitate subsequent separation [92].
Carbon-based adsorbents are also increasing due to the wide application of activated carbon in precious metals like gold [51]. This class of material can be graphene; the oxides of graphene with C, O, and H atoms in the structure; or carbon nanotubes, which include the single and multiwalled, activated carbon, fullerene, carbon dots, carbon nanofibers, and carbon black [18]. As reported, carbon-based nanomaterials showed good extraction values of REEs from harmful contaminants like Al, Fe, and Cu, but the selective fractionation of REE has not yet been achieved [7,18]. It is important to note that the term “harmful” indicates the negative impact these elements have on the separation process when present as contaminants.
Silica-based adsorbents are also booming for REE separation because of their increased surface area (KIT-6 silica), mechanical and thermal abilities, more readily available and easily manufactured materials, and more susceptibility for functionalization. Some functionalized silica adsorbents are selective for Nd separation from LREEs [93] and La separation from MREEs [94]. But due to the poor resistance of these materials to acidic environments and low selectivity compared to metal adsorbents, these materials are reinforced with carbon-based adsorbents to improve the acidic stability of the material [1]. Some carbon-silica-based composites showed selectivity for La and Sc separation from the rest of the REEs [7].
The magnetic-based adsorbents are developing and have improved properties compared to other adsorbents, and an easier separation from the REE stream; these adsorbents are quickly produced using co-precipitation and, similar to other adsorbents, surface modification helps to increase their selectivity for REE separation [1,7]. The principle of co-precipitation paved the way for the creation of nanocomposite adsorbents because they synthesize layered double hydroxides, but their potential application is yet to be researched [7].
The last class of adsorption materials discovered is hydrogel-based adsorbents, which show a promising application because of pore stability and functionalization; however, the penetration of REE ions and the stability of gel limits their use [1].
After these separations processes, the individual REEs are sent to an electrochemical refining process for metal recovery or recovery of REEs into oxides.
3. Holistic View of REE Separation: Perspectives and Conclusions
Information related to research and developments in REE separation is much needed because of the increased demand for the utilization of REEs in several sectors. Therefore, discovering a standard technology for base metals and other precious metals may not be suitable for REEs because of variations in sources of availability. However, for commercial operations, a solid knowledge of solution chemistry will be a guiding tool to decide which process will be technologically viable to meet business goals. Moreover, selecting a particular method depends on the operation’s economic goals accordingly to a flowsheet designed for REE recovery streams. Of the methods explained in the previous sections, the merits and demerits of each approach are summarized.
- The fractional crystallization (FC) approach has the potential for the selective separation of individual REEs, but the production cycle needed is tiresome. Additionally, the availability of salts required for this approach is limited, and the HREE products generated are less pure than any other method.
- The fractional precipitation (FP) approach can resolve some of the problems caused by the fractional crystallization approach regarding salt availability. Still, this method is only suitable for the separation of REEs into groups as individual separation is challenging.
- The size-selective crystallization (SSC) approach is the best suited when the REEs in aqueous media have a more significant ionic radii difference, but for the adjacent REEs with a smaller ionic radii difference, the contamination of unwanted REE cations into targeted REE crystals is the major setback.
- The reaction-controlled crystallization (RCC) reported to be the best practical strategy for the Nd/Dy system but is impractical with the Nd/Sm system.
- Valence selective crystallization (VSC) is a well-suited approach when dealing with systems that contain differences in valencies among metal cations, and thus it is limited to use for Ce.
- The selective oxidation (SO) approach applies to Ce, Pr, and Tb where enhancement of their oxidation states to +4 can enhance their separation. The separation post enhancement of valence state differs for Ce compared to Pr and Tb.
- The selective reduction (SR) approach can separate Sm, Eu, and Yb individually or in bulk from a mixture of REEs. The approach type varies depending on the interest of the operation needs and the desired product. However, this approach’s detailed operation cost analysis is rarely discussed in the literature.
- The ion exchange (IX) process is simple as it exchanges a counter ion for a targeted cation. This process is capable and can efficiently separate multiple elements in one go. However, the availability and development of appropriate resin material limit its use. Additionally, future research should address the issues of longer operation time, clogging of resin by unwanted metal, and loss of functional carriers to scale up this approach to industrial needs.
- The membrane separation (MS) approach delimits the few concerns of IX as this is fast, versatile, and continuous. Further, this is eco-friendly and energy-saving because of the low operational equipment needed. Still, as in ion exchange, membrane separation is also hindered due to the availability of suitable membrane material. The MS can potentially revolutionize the individual HREE separation from the bulk if the problem of suitable membrane material is resolved.
- Solvent extraction (SX) is suitable for large-scale industrial operations as bulk volumes can be processed in a short span. Additionally, selectivity can be enhanced by modifying and tailoring the organic phase. The only demerit of this method is the loss and contamination of the organic phase, which contains both harmful and costlier species, thus leading to pollution of the aqueous phase and increasing the cost of operation. Therefore, this is a significant takeaway for further research in this direction.
- Adsorption (AD) is a rapid and straightforward approach. This method needs the creation of a proper adsorbent that should meet the selectivity required for the targeted REE. In recent years researchers have synthesized many adsorbents; the hurdle in using them industrially is making adsorbents that have a high use of chemicals and more sophisticated equipment. Thus, there should be a trade-off between technology and the cost of operation. Moreover, desorption is the major challenge for natural clay-based adsorbents, and most of the available technology either uses highly concentrated acids or some less practical industrial procedures like microwave treatment or ultrasonicators. Thus, further research is needed to push these barriers for industrial application.
Of all the methods described in this study, adsorption and solvent extraction can ramp up the commercial production of REEs if few developments are performed. Additionally, it is necessary to know that other methods can be helpful and categorized as auxiliary tools to the above techniques. This hybrid application is again bound by the Efficient-Easier-Economical-Environmentally Safe (EEEE) strategy, where all these factors should be addressed commercially. However, relying entirely on auxiliary approaches for commercial REE production would require extensive research.
Author Contributions
Conceptualization, S.V.S.H.P. and P.K.S.; methodology, S.V.S.H.P. and P.K.S.; formal analysis, M.L.F. and P.K.S.; investigation, S.V.S.H.P.; resources, M.L.F. and P.K.S.; data curation, S.V.S.H.P.; writing—original draft preparation, S.V.S.H.P.; writing—review and editing, P.K.S. and M.L.F.; visualization, P.K.S. and M.L.F.; supervision, P.K.S. and M.L.F.; project administration, M.L.F. and P.K.S.; funding acquisition, M.L.F. and P.K.S. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Data Availability Statement
The data presented in this study are available on request from the authors.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Chen, Z.; Li, Z.; Chen, J.; Kallem, P.; Banat, F.; Qiu, H. Recent advances in selective separation technologies of rare earth elements: A review. J. Environ. Chem. Eng. 2022, 10, 107104. [Google Scholar] [CrossRef]
- Opare, E.O.; Struhs, E.; Mirkouei, A. A comparative state-of-technology review and future directions for rare earth element separation. Renew. Sustain. Energy Rev. 2021, 143, 110917. [Google Scholar] [CrossRef]
- Gupta, C.K.; Krishnamurthy, N. Extractive metallurgy of rare earths. Int. Mater. Rev. 1992, 37, 197–248. [Google Scholar] [CrossRef]
- Balaram, V. Rare earth elements: A review of applications, occurrence, exploration, analysis, recycling, and environmental impact. Geosci. Front. 2019, 10, 1285–1303. [Google Scholar] [CrossRef]
- Galhardi, J.A.; Luko-Sulato, K.; Yabuki, L.N.; Santos, L.M.; da Silva, Y.J. Rare Earth Elements and Radionuclides; Elsevier Inc.: Amsterdam, The Netherlands, 2022. [Google Scholar] [CrossRef]
- Bünzli, J.-C.G. Lanthanides. In Kirk-Othmer Encyclopedia of Chemical Technology; Wiley Blackwell: Hoboken, NJ, USA, 2013; pp. 1–43. Available online: https://www.researchgate.net/publication/257921324_Lanthanides (accessed on 27 March 2023).
- Asadollahzadeh, M.; Torkaman, R.; Torab-Mostaedi, M. Extraction and Separation of Rare Earth Elements by Adsorption Approaches: Current Status and Future Trends. In Separation and Purification Reviews; Taylor and Francis Inc.: Abingdon, UK, 2020; pp. 1–28. [Google Scholar] [CrossRef]
- Zhou, H.; Chen, X.; Fang, L.; Hu, C.; Wang, H. A new microwave dielectric ceramic for LTCC applications. J. Mater. Sci. Mater. Electron. 2010, 21, 849–853. Available online: http://www.mineralsuk.com/ (accessed on 6 March 2023). [CrossRef]
- Sarswat, P.; Leake, M.; Allen, L.; Free, M.; Hu, X.; Kim, D.; Noble, A.; Luttrell, G. Efficient recovery of rare earth elements from coal based resources: A bioleaching approach. Mater. Today Chem. 2020, 16, 100246. [Google Scholar] [CrossRef]
- McNulty, T.; Hazen, N.; Park, S. Processing the ores of rare-earth elements. MRS Bull. 2022, 47, 258–266. [Google Scholar] [CrossRef]
- Gaustad, G.; Williams, E.; Leader, A. Rare earth metals from secondary sources: Review of potential supply from waste and byproducts. Resour. Conserv. Recycl. 2020, 167, 105213. [Google Scholar] [CrossRef]
- Omodara, L.; Pitkäaho, S.; Turpeinen, E.-M.; Saavalainen, P.; Oravisjärvi, K.; Keiski, R.L. Recycling and substitution of light rare earth elements, cerium, lanthanum, neodymium, and praseodymium from end-of-life applications—A review. J. Clean. Prod. 2019, 236, 117573. [Google Scholar] [CrossRef]
- Dutta, T.; Kim, K.-H.; Uchimiya, M.; Kwon, E.E.; Jeon, B.-H.; Deep, A.; Yun, S.-T. Global demand for rare earth resources and strategies for green mining. Environ. Res. 2016, 150, 182–190. [Google Scholar] [CrossRef]
- Zhang, Z.; Allen, L.; Podder, P.; Free, M.L.; Sarswat, P.K. Recovery and Enhanced Upgrading of Rare Earth Elements from Coal-Based Resources: Bioleaching and Precipitation. Minerals 2021, 11, 484. [Google Scholar] [CrossRef]
- Brahim, J.A.; Hak, S.A.; Achiou, B.; Boulif, R.; Beniazza, R.; Benhida, R. Kinetics and mechanisms of leaching of rare earth elements from secondary resources. Miner. Eng. 2022, 177, 107351. [Google Scholar] [CrossRef]
- Echeverry-Vargas, L.; Ocampo-Carmona, L.M. Recovery of Rare Earth Elements from Mining Tailings: A Case Study for Generating Wealth from Waste. Minerals 2022, 12, 948. [Google Scholar] [CrossRef]
- European Commission. Study on the Critical Raw Materials for the EU. 2023. Available online: https://single-market-economy.ec.europa.eu/publications/study-critical-raw-materials-eu-2023-final-report_en (accessed on 27 March 2023). [CrossRef]
- Cardoso, C.E.D.; Almeida, J.C.; Lopes, C.B.; Trindade, T.; Vale, C.; Pereira, E. Recovery of Rare Earth Elements by Carbon-Based Nanomaterials—A Review. Nanomaterials 2019, 9, 814. [Google Scholar] [CrossRef] [PubMed]
- Stein, R.T.; Kasper, A.C.; Veit, H.M. Recovery of Rare Earth Elements Present in Mobile Phone Magnets with the Use of Organic Acids. Minerals 2022, 12, 668. [Google Scholar] [CrossRef]
- Yoldjian, G. The use of rare earths in ceramics. J. Less Common Met. 1985, 111, 17–22. [Google Scholar] [CrossRef]
- Haque, N.; Hughes, A.; Lim, S.; Vernon, C. Rare Earth Elements: Overview of Mining, Mineralogy, Uses, Sustainability and Environmental Impact. Resources 2014, 3, 614–635. [Google Scholar] [CrossRef]
- Castor, S.B.; Hedrick, J.B. Rare Earth Elements. Encycl. Inorg. Bioinorg. Chem. 2005, 769–792. [Google Scholar] [CrossRef]
- Suli, L.M.; Ibrahim, W.H.W.; Aziz, B.A.; Deraman, M.R.; Ismail, N.A. A Review of Rare Earth Mineral Processing Technology. Chem. Eng. Res. Bull. 2017, 19, 20. [Google Scholar] [CrossRef]
- Wills, B.A.; Finch, J.A. Wills’ Mineral Processing Technology: An Introduction to the Practical Aspects of Ore Treatment and Mineral Recovery; Butterworth-Heinemann: Oxford, UK, 2015. [Google Scholar]
- Schulz, B.; Merker, G.; Gutzmer, J. Automated SEM Mineral Liberation Analysis (MLA) with Generically Labelled EDX Spectra in the Mineral Processing of Rare Earth Element Ores. Minerals 2019, 9, 527. [Google Scholar] [CrossRef]
- Free, M.L. Hydrometallurgy; Springer International Publishing: Cham, Switzerland, 2022. [Google Scholar] [CrossRef]
- Hidayah, N.N.; Abidin, S.Z. The evolution of mineral processing in extraction of rare earth elements using solid-liquid extraction over liquid-liquid extraction: A review. Miner. Eng. 2017, 112, 103–113. [Google Scholar] [CrossRef]
- Yang, Y.; Walton, A.; Sheridan, R.; Güth, K.; Gauß, R.; Gutfleisch, O.; Buchert, M.; Steenari, B.-M.; Van Gerven, T.; Jones, P.T.; et al. REE Recovery from End-of-Life NdFeB Permanent Magnet Scrap: A Critical Review. J. Sustain. Met. 2017, 3, 122–149. [Google Scholar] [CrossRef]
- Traore, M.; Gong, A.; Wang, Y.; Qiu, L.; Bai, Y.; Zhao, W.; Liu, Y.; Chen, Y.; Liu, Y.; Wu, H.; et al. Research progress of rare earth separation methods and technologies. J. Rare Earths 2023, 41, 182–189. [Google Scholar] [CrossRef]
- Sarswat, P.K.; Free, M.L. Frequency and atomic mass based selective electrochemical recovery of rare earth metals and isotopes. Electrochimica Acta 2016, 219, 435–446. [Google Scholar] [CrossRef]
- Sarswat, P.K.; Podder, P.; Zhang, Z.; Free, M.L. Autogenous acid production using a regulated bio-oxidation method for economical recovery of REEs and critical metals from coal-based resources. Appl. Surf. Sci. Adv. 2022, 11, 100283. [Google Scholar] [CrossRef]
- Sarswat, P.K.; Zhang, Z.; Free, M.L. Rare Earth Elements Extraction from Coal Waste Using a Biooxidation Approach. In Rare Metal Technology; Springer International Publishing: Cham, Switzerland, 2021; pp. 211–216. [Google Scholar] [CrossRef]
- Free, M.L.; Ilunga, J.K.; Podder, P.; Sarswat, P.K. Assessing and Improving Biooxidation for Acid Generation and Rare Earth Element Extraction. Processes 2023, 11, 1005. [Google Scholar] [CrossRef]
- Hovey, J.L.; Dittrich, T.M.; Allen, M.J. Coordination chemistry of surface-associated ligands for solid–liquid adsorption of rare-earth elements. J. Rare Earths 2023, 41, 1–18. [Google Scholar] [CrossRef]
- El Ouardi, Y.; Virolainen, S.; Mouele, E.S.M.; Laatikainen, M.; Repo, E.; Laatikainen, K. The recent progress of ion exchange for the separation of rare earths from secondary resources—A review. Hydrometallurgy 2023, 218, 106047. [Google Scholar] [CrossRef]
- Pearson, R.G. Hard and soft acids and bases, HSAB, part I: Fundamental principles. J. Chem. Educ. 1968, 45, 581–587. [Google Scholar] [CrossRef]
- Marcus, Y. Thermodynamics of solvation of ions. Part 5.—Gibbs free energy of hydration at 298.15 K. J. Chem. Soc. Faraday Trans. 1991, 87, 2995–2999. [Google Scholar] [CrossRef]
- Moeller, T. The Chemistry of the Lanthanides; Elsevier: Amsterdam, The Netherlands, 1973. [Google Scholar] [CrossRef]
- Wall, F. Rare Earth Elements. In Encyclopedia of Geology; Elsevier: Amsterdam, The Netherlands, 2021; pp. 680–693. [Google Scholar] [CrossRef]
- James, C.; Grant, A.J. The separation of the rare earths giving the more soluble double sulfates from brazilian monazite sand. J. Am. Chem. Soc. 1916, 38, 41–47. [Google Scholar] [CrossRef]
- Ishino, T. Separation of Rare Earth Elements. J. Soc. Mater. Sci. Jpn. 1963, 12, 559–571. [Google Scholar] [CrossRef]
- Zhao, X.; Wong, M.; Mao, C.; Trieu, T.X.; Zhang, J.; Feng, P.; Bu, X. Size-Selective Crystallization of Homochiral Camphorate Metal–Organic Frameworks for Lanthanide Separation. J. Am. Chem. Soc. 2014, 136, 12572–12575. [Google Scholar] [CrossRef]
- Moeller, T.; Kremers, H.E. Observations on Rare Earths Extraction of Ytterbium from Rare Earth Mixtures with Sodium Amalgam. Ind. Eng. Chem. Anal. Ed. 1945, 17, 798–800. [Google Scholar] [CrossRef]
- Furukawa, H.; Cordova, K.E.; O’Keeffe, M.; Yaghi, O.M. The Chemistry and Applications of Metal-Organic Frameworks. Science 2013, 341, 1230444. [Google Scholar] [CrossRef]
- Yin, X.; Wang, Y.; Bai, X.; Wang, Y.; Chen, L.; Xiao, C.; Diwu, J.; Du, S.; Chai, Z.; Albrecht-Schmitt, T.E.; et al. Rare earth separations by selective borate crystallization. Nat. Commun. 2017, 8, 14438. [Google Scholar] [CrossRef]
- Willauer, A.R.; Palumbo, C.T.; Fadaei-Tirani, F.; Zivkovic, I.; Douair, I.; Maron, L.; Mazzanti, M. Accessing the +IV Oxidation State in Molecular Complexes of Praseodymium. J. Am. Chem. Soc. 2020, 142, 5538–5542. [Google Scholar] [CrossRef] [PubMed]
- Palumbo, C.T.; Zivkovic, I.; Scopelliti, R.; Mazzanti, M. Molecular Complex of Tb in the +4 Oxidation State. J. Am. Chem. Soc. 2019, 141, 9827–9831. [Google Scholar] [CrossRef]
- Jamrack, W. Rare Metal Extraction by Chemical Engineering Techniques; Elsevier: Amsterdam, The Netherlands, 1963. [Google Scholar] [CrossRef]
- Chen, B.; He, M.; Zhang, H.; Jiang, Z.; Hu, B. Chromatographic Techniques for Rare Earth Elements Analysis. Phys. Sci. Rev. 2019, 2, 1–35. [Google Scholar] [CrossRef]
- Kumar, S.; Jain, S. History, Introduction, and Kinetics of Ion Exchange Materials. J. Chem. 2013, 2013, 1–13. [Google Scholar] [CrossRef]
- Free, M.L. Hydrometallurgy, Fundemetals and Applications; Springer Nature: Singapore, 2022. [Google Scholar]
- Hérès, X.; Blet, V.; Di Natale, P.; Ouaattou, A.; Mazouz, H.; Dhiba, D.; Cuer, F. Selective Extraction of Rare Earth Elements from Phosphoric Acid by Ion Exchange Resins. Metals 2018, 8, 682. [Google Scholar] [CrossRef]
- Mayer, S.W.; Freiling, E.C. Ion Exchange as a Separation Method. VI. Column Studies of the Relative Efficiencies of Various Complexing Agents for the Separation of Lighter Rare Earths. J. Am. Chem. Soc. 1953, 75, 5647–5649. [Google Scholar] [CrossRef]
- Page, M.J.; Soldenhoff, K.; Ogden, M.D. Comparative study of the application of chelating resins for rare earth recovery. Hydrometallurgy 2017, 169, 275–281. [Google Scholar] [CrossRef]
- Xie, F.; Zhang, T.A.; Dreisinger, D.; Doyle, F. A critical review on solvent extraction of rare earths from aqueous solutions. Miner. Eng. 2014, 56, 10–28. [Google Scholar] [CrossRef]
- Binnemans, K.; Jones, P.T. Solvometallurgy: An Emerging Branch of Extractive Metallurgy. J. Sustain. Met. 2017, 3, 570–600. [Google Scholar] [CrossRef]
- Joshi, D.R.; Adhikari, N. An Overview on Common Organic Solvents and Their Toxicity. J. Pharm. Res. Int. 2019, 28, 1–18. [Google Scholar] [CrossRef]
- Sun, X.; Ji, Y.; Hu, F.; He, B.; Chen, J.; Li, D. The inner synergistic effect of bifunctional ionic liquid extractant for solvent extraction. Talanta 2010, 81, 1877–1883. [Google Scholar] [CrossRef]
- Abbott, A.P.; Frisch, G.; Gurman, S.J.; Hillman, A.R.; Hartley, J.; Holyoak, F.; Ryder, K.S. Ionometallurgy: Designer redox properties for metal processing. Chem. Commun. 2011, 47, 10031–10033. [Google Scholar] [CrossRef] [PubMed]
- Arrachart, G.; Couturier, J.; Dourdain, S.; Levard, C.; Pellet-Rostaing, S. Recovery of Rare Earth Elements (REEs) Using Ionic Solvents. Processes 2021, 9, 1202. [Google Scholar] [CrossRef]
- Prodius, D.; Mudring, A.-V. Rare earth metal-containing ionic liquids. Coord. Chem. Rev. 2018, 363, 1–16. [Google Scholar] [CrossRef]
- Trinh, H.B.; Lee, J.-C.; Lee, J. Task-Specific Ionic Liquids for the Separation and Recovery of Rare Earth Elements; INC: New York, NY, USA, 2022. [Google Scholar] [CrossRef]
- Wang, K.; Adidharma, H.; Radosz, M.; Wan, P.; Xu, X.; Russell, C.K.; Tian, H.; Fan, M.; Yu, J. Recovery of rare earth elements with ionic liquids. Green Chem. 2017, 19, 4469–4493. [Google Scholar] [CrossRef]
- Khodakarami, M.; Alagha, L. Separation and recovery of rare earth elements using novel ammonium-based task-specific ionic liquids with bidentate and tridentate O-donor functional groups. Sep. Purif. Technol. 2019, 232, 115952. [Google Scholar] [CrossRef]
- Lei, Z.; Chen, B.; Koo, Y.-M.; MacFarlane, D.R. Introduction: Ionic Liquids. Chem. Rev. 2017, 117, 6633–6635. [Google Scholar] [CrossRef]
- Kaur, G.; Kumar, H.; Singla, M. Diverse applications of ionic liquids: A comprehensive review. J. Mol. Liq. 2022, 351, 118556. [Google Scholar] [CrossRef]
- Micheau, C.; Arrachart, G.; Turgis, R.; Lejeune, M.; Draye, M.; Michel, S.; Legeai, S.; Pellet-Rostaing, S. Ionic Liquids as Extraction Media in a Two-Step Eco-Friendly Process for Selective Tantalum Recovery. ACS Sustain. Chem. Eng. 2020, 8, 1954–1963. [Google Scholar] [CrossRef]
- Seddon, K.R.; Stark, A.; Torres, M.-J. Influence of chloride, water, and organic solvents on the physical properties of ionic liquids. Pure Appl. Chem. 2000, 72, 2275–2287. [Google Scholar] [CrossRef]
- Widegren, J.A.; Laesecke, A.; Magee, J.W. The effect of dissolved water on the viscosities of hydrophobic room-temperature ionic liquids. Chem. Commun. 2005, 72, 1610–1612. [Google Scholar] [CrossRef]
- Huddleston, J.G.; Visser, A.E.; Reichert, W.M.; Willauer, H.D.; Broker, G.A.; Rogers, R.D. Characterization and comparison of hydrophilic and hydrophobic room temperature ionic liquids incorporating the imidazolium cation. Green Chem. 2001, 3, 156–164. [Google Scholar] [CrossRef]
- Toh, S.L.I.; McFarlane, J.; Tsouris, C.; DePaoli, D.W.; Luo, H.; Dai, S. Room-Temperature Ionic Liquids in Liquid–Liquid Extraction: Effects of Solubility in Aqueous Solutions on Surface Properties. Solvent Extr. Ion Exch. 2006, 24, 33–56. [Google Scholar] [CrossRef]
- Jacquemin, J.; Husson, P.; Padua, A.A.H.; Majer, V. Density and viscosity of several pure and water-saturated ionic liquids. Green Chem. 2006, 8, 172–180. [Google Scholar] [CrossRef]
- Zuo, Y.; Liu, Y.; Chen, J.; Li, D.Q. The Separation of Cerium(IV) from Nitric Acid Solutions Containing Thorium(IV) and Lanthanides(III) Using Pure [C8mim]PF6 as Extracting Phase. Ind. Eng. Chem. Res. 2008, 47, 2349–2355. [Google Scholar] [CrossRef]
- Hoogerstraete, T.V.; Onghena, B.; Binnemans, K. Homogeneous Liquid–Liquid Extraction of Rare Earths with the Betaine—Betainium Bis(trifluoromethylsulfonyl)imide Ionic Liquid System. Int. J. Mol. Sci. 2013, 14, 21353–21377. [Google Scholar] [CrossRef]
- Rout, A.; Kotlarska, J.; Dehaen, W.; Binnemans, K. Liquid–liquid extraction of neodymium(iii) by dialkylphosphate ionic liquids from acidic medium: The importance of the ionic liquid cation. Phys. Chem. Chem. Phys. 2013, 15, 16533–16541. [Google Scholar] [CrossRef] [PubMed]
- Dupont, D.; Raiguel, S.; Binnemans, K. Sulfonic acid functionalized ionic liquids for dissolution of metal oxides and solvent extraction of metal ions. Chem. Commun. 2015, 51, 9006–9009. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Guo, W.; Liu, D.; Xu, J. Rational design of novel carboxylic acid functionalized phosphonium based ionic liquids as high-performance extractants for rare earths. J. Rare Earths 2021, 39, 1435–1441. [Google Scholar] [CrossRef]
- Chen, L.; Wu, Y.; Dong, H.; Meng, M.; Li, C.; Yan, Y.; Chen, J. An overview on membrane strategies for rare earths extraction and separation. Sep. Purif. Technol. 2018, 197, 70–85. [Google Scholar] [CrossRef]
- Bartsch, R.A.; Way, J.D. Chemical Separations with Liquid Membranes: An Overview; American Chemical Society: Washington, DC, USA, 1996; Volume 642. [Google Scholar] [CrossRef]
- Kecili, R.; Hussain, C.M. Mechanism of Adsorption on Nanomaterials; Elsevier Inc.: Amsterdam, The Netherlands, 2018. [Google Scholar] [CrossRef]
- Talan, D.; Huang, Q. A review of environmental aspect of rare earth element extraction processes and solution purification techniques. Miner. Eng. 2022, 179, 107430. [Google Scholar] [CrossRef]
- Anastopoulos, I.; Bhatnagar, A.; Lima, E.C. Adsorption of rare earth metals: A review of recent literature. J. Mol. Liq. 2016, 221, 954–962. [Google Scholar] [CrossRef]
- Cheng, Y.-L. We are IntechOpen, the world’s leading publisher of Open Access books Built by scientists, for scientists TOP 1%. Intech 2016, 11, 13. [Google Scholar] [CrossRef]
- Brião, G.D.V.; da Silva, M.G.; Vieira, M.G.A. Expanded vermiculite as an alternative adsorbent for the dysprosium recovery. J. Taiwan Inst. Chem. Eng. 2021, 127, 228–235. [Google Scholar] [CrossRef]
- Zhou, J.; Liu, H.; Liu, D.; Yuan, P.; Bu, H.; Du, P.; Fan, W.; Li, M. Sorption/desorption of Eu(III) on halloysite and kaolinite. Appl. Clay Sci. 2022, 216, 106356. [Google Scholar] [CrossRef]
- Vasylechko, V.; Stechynska, E.; Stashkiv, O.; Gryshchouk, G.; Patsay, I. Sorption of Neodymium and Gadolinium on Transcarpathian Clinoptilolite. Acta Phys. Pol. A 2018, 133, 794–797. [Google Scholar] [CrossRef]
- Liu, E.; Xu, X.; Zheng, X.; Zhang, F.; Liu, E.; Li, C. An ion imprinted macroporous chitosan membrane for efficiently selective adsorption of dysprosium. Sep. Purif. Technol. 2017, 189, 288–295. [Google Scholar] [CrossRef]
- Zheng, X.; Liu, E.; Zhang, F.; Dai, J.; Yan, Y.; Li, C. Selective adsorption and separation of gadolinium with three-dimensionally interconnected macroporous imprinted chitosan films. Cellulose 2017, 24, 977–988. [Google Scholar] [CrossRef]
- Ramasamy, D.L.; Porada, S.; Sillanpää, M. Marine algae: A promising resource for the selective recovery of scandium and rare earth elements from aqueous systems. Chem. Eng. J. 2019, 371, 759–768. [Google Scholar] [CrossRef]
- Zhou, H.; Long, J.; Yaghi, O. Introduction to Metal—Organic Frameworks. Chem. Rev. 2012, 112, 673–674. [Google Scholar] [CrossRef]
- Lee, Y.-R.; Yu, K.; Ravi, S.; Ahn, W.-S. Selective Adsorption of Rare Earth Elements over Functionalized Cr-MIL-101. ACS Appl. Mater. Interfaces 2018, 10, 23918–23927. [Google Scholar] [CrossRef]
- Fiket, Ž.; Galović, A.; Medunić, G.; Turk, M.F.; Ivanić, M.; Dolenec, M.; Biljan, I.; Šoster, A.; Kniewald, G. Adsorption of Rare Earth Elements from Aqueous Solutions Using Geopolymers. IECG 2018 2018, 2, 567. [Google Scholar] [CrossRef]
- Ravi, S.; Lee, Y.-R.; Yu, K.; Ahn, J.-W.; Ahn, W.-S. Benzene triamido-tetraphosphonic acid immobilized on mesoporous silica for adsorption of Nd3+ ions in aqueous solution. Microporous Mesoporous Mater. 2018, 258, 62–71. [Google Scholar] [CrossRef]
- Liu, E.; Chen, L.; Dai, J.; Wang, Y.; Li, C.; Yan, Y. Fabrication of phosphate functionalized chiral nematic mesoporous silica films for the efficient and selective adsorption of lanthanum ions. J. Mol. Liq. 2019, 277, 786–793. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).