
Digital Twin Based Design and Experimental Validation of a Continuous Peptide Polishing Step


 , and
, and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Feed Mixtures, Buffers and Stationary Phases
2.2. Batch Chromatography
3. Chromatography Modeling
3.1. General Rate Model
3.1.1. Mass Balance of Mobile Phase
3.1.2. Mass Balance of Stationary Phase
3.1.3. Adsorption Equilibrium
3.2. Model Parameter Determination
3.2.1. Fluid Dynamics
3.2.2. Adsorption Equilibrium
3.2.3. Mass Transport
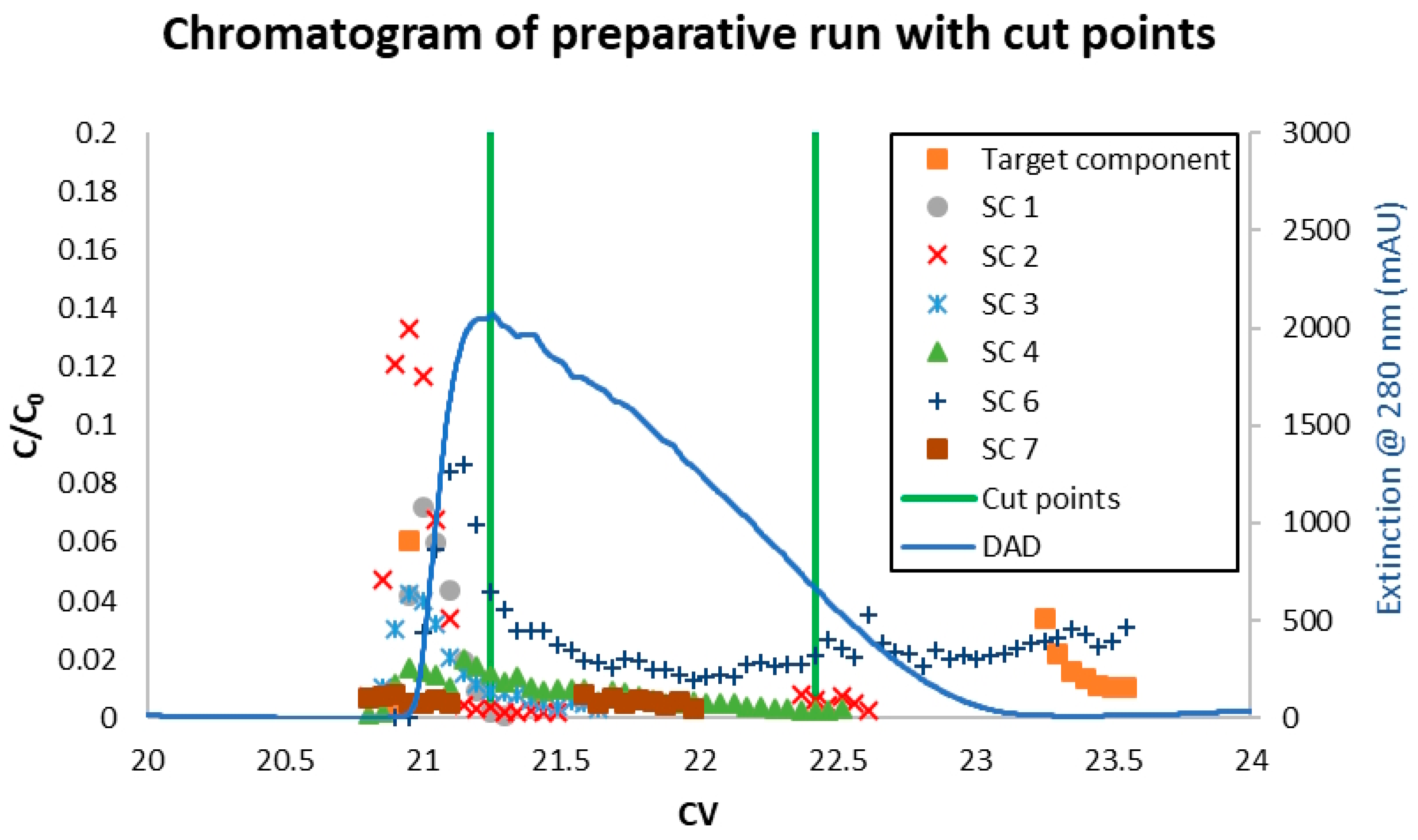
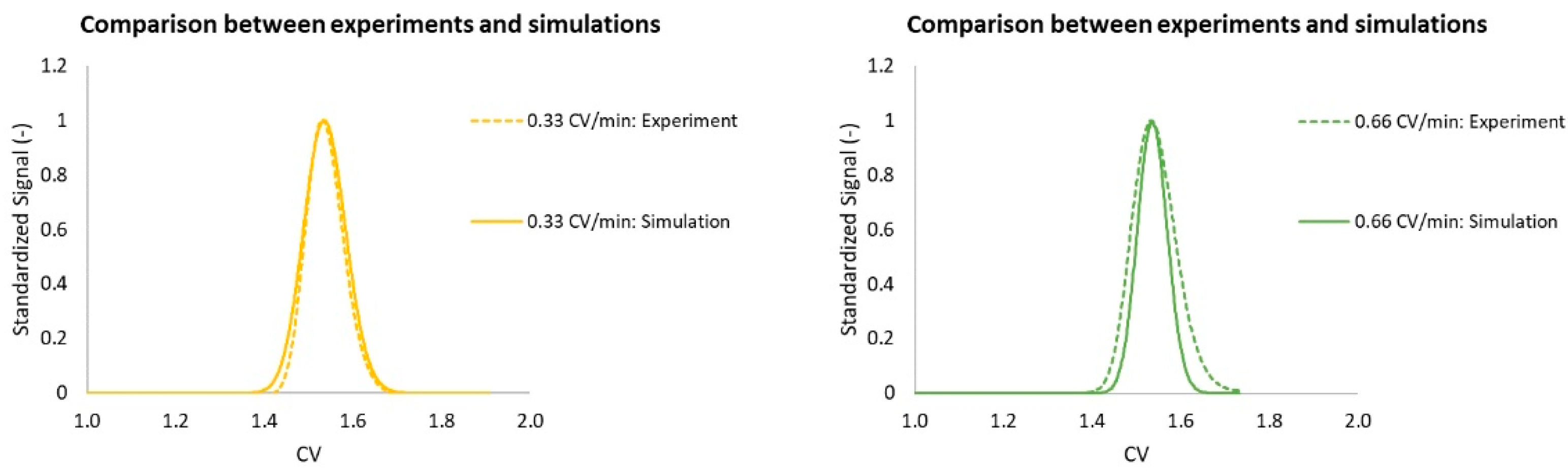
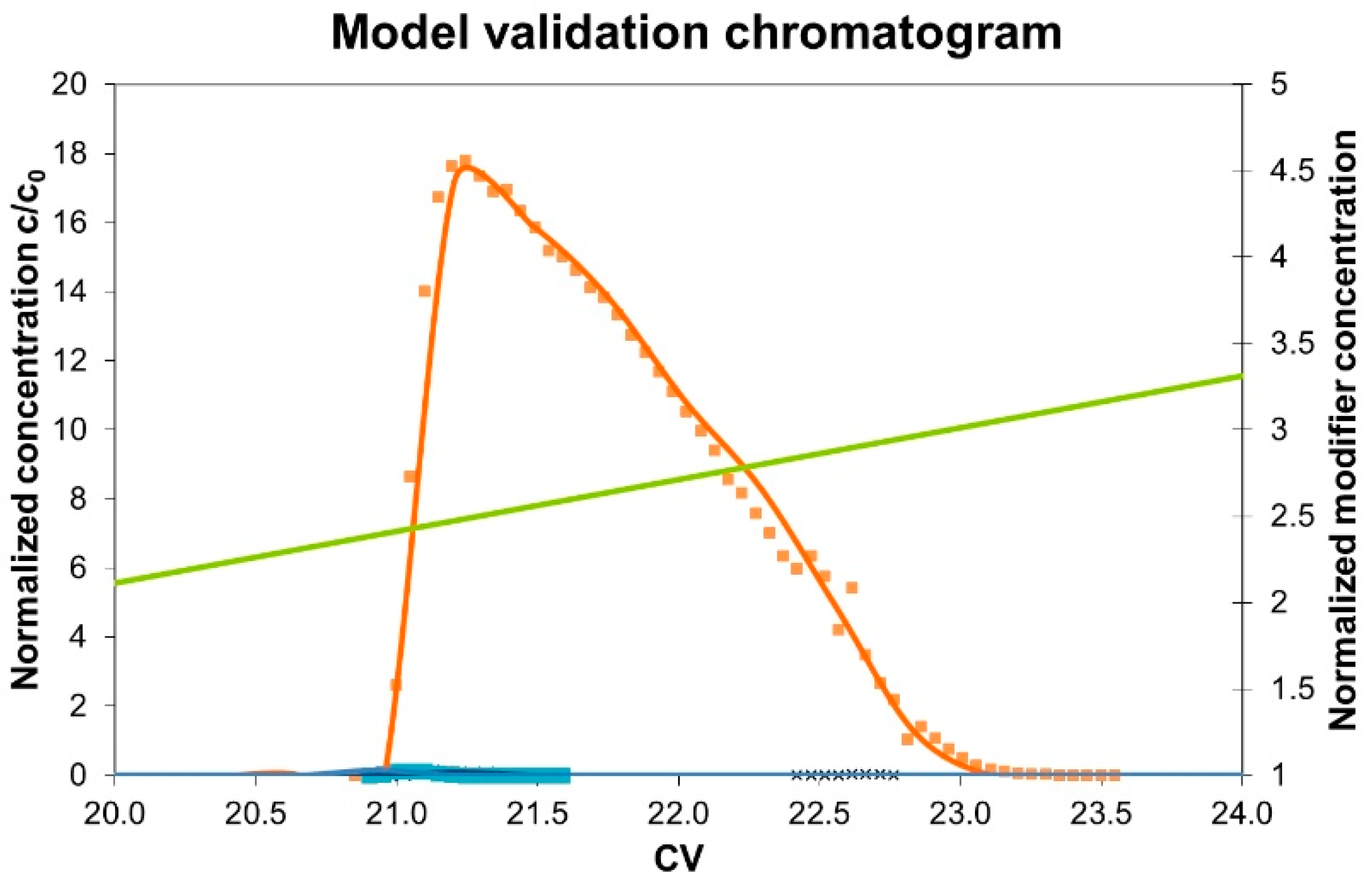
3.3. Model Validation
4. Results and Discussion
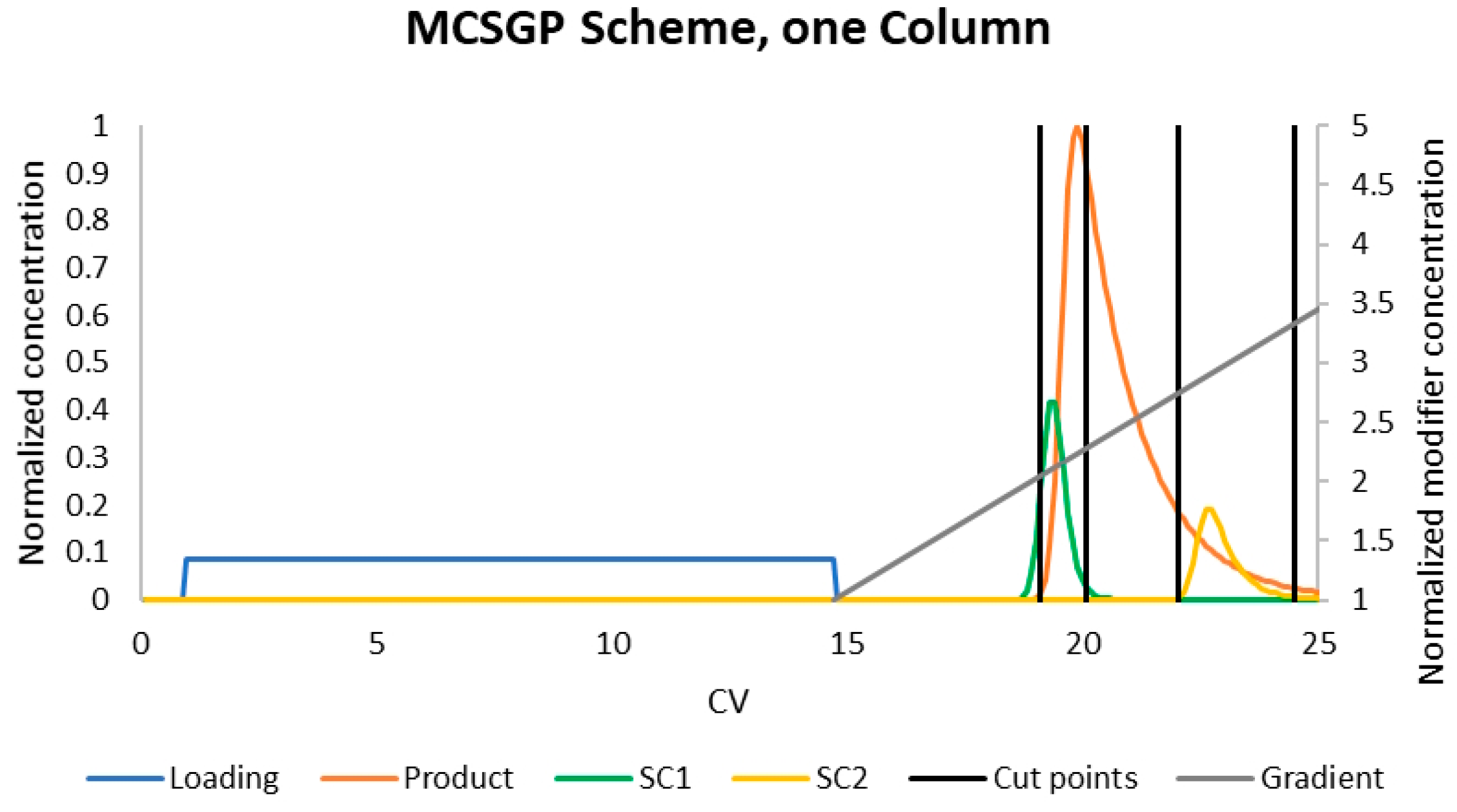
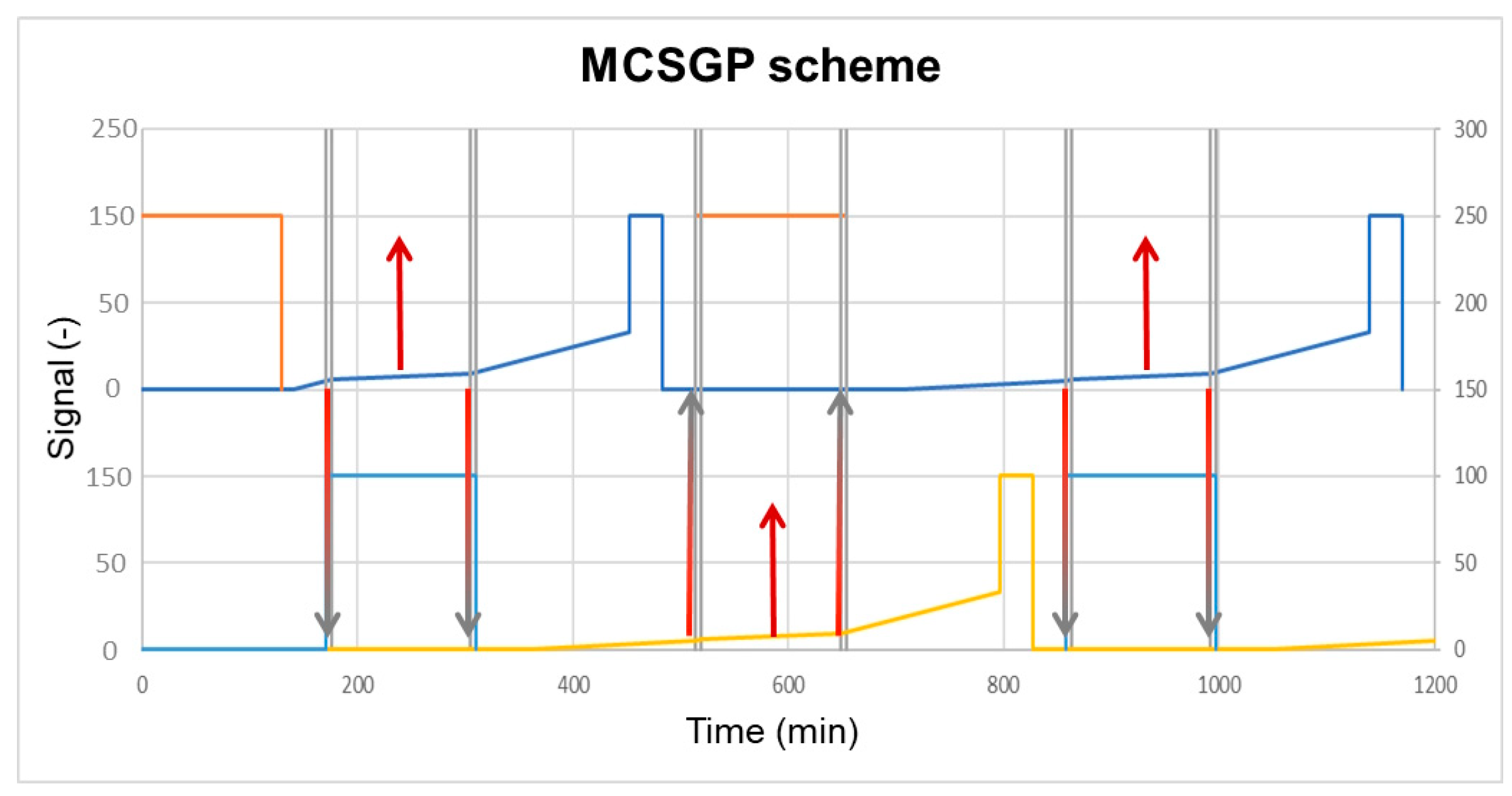
4.1. MCSGP
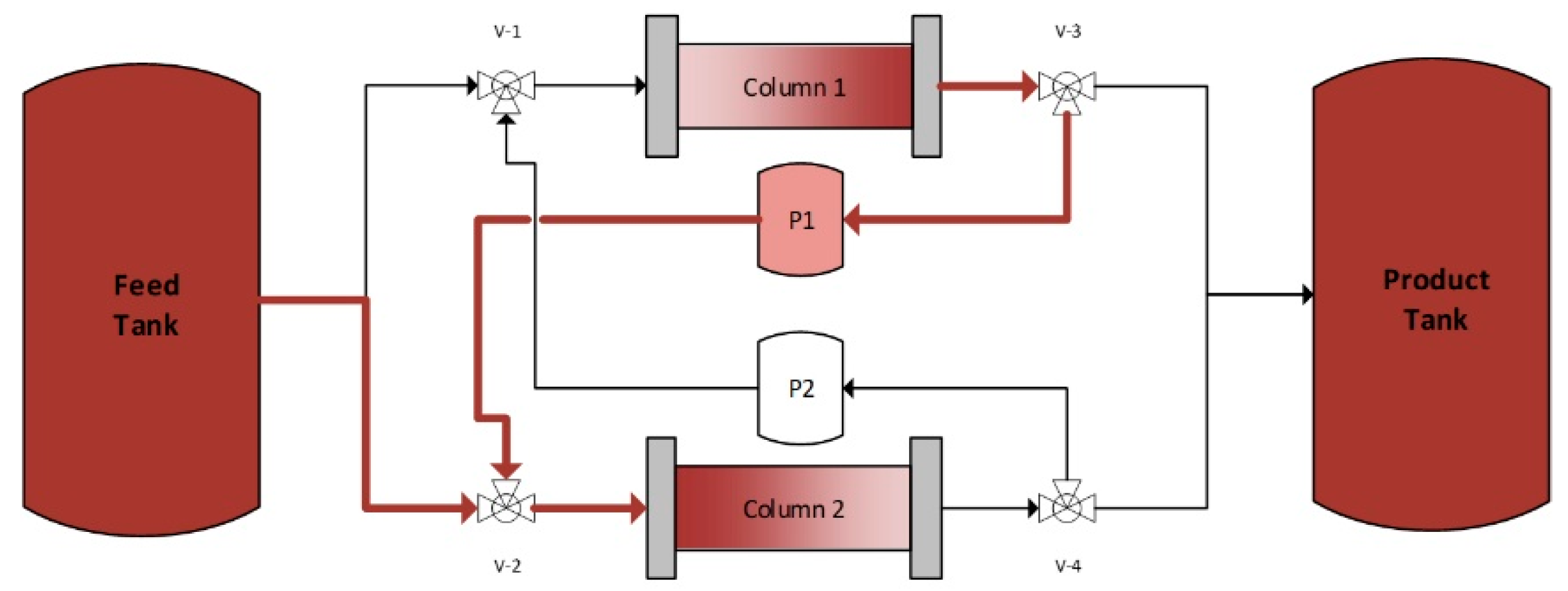
4.2. Continuous Twin Column Chromatography (CTCC)
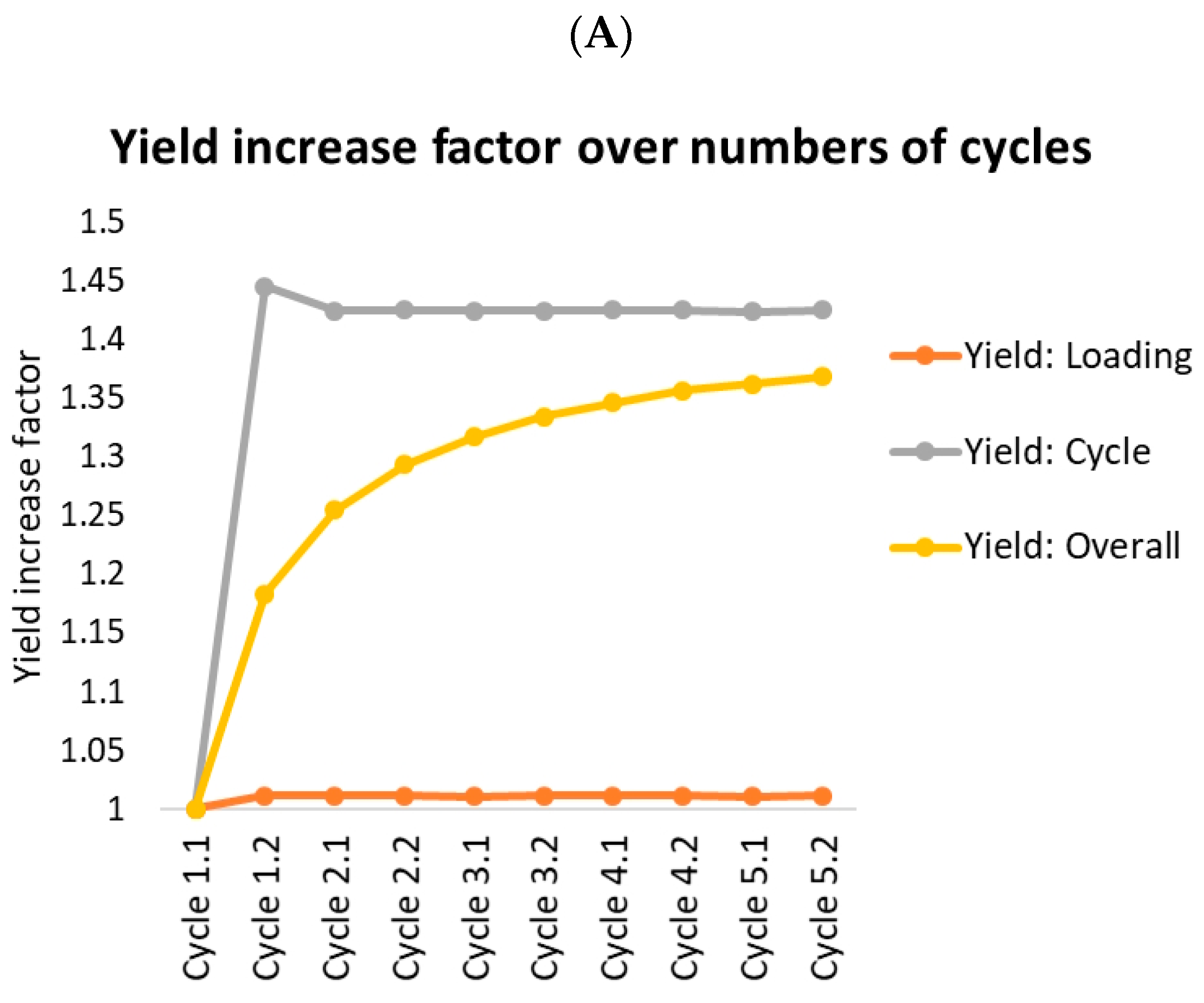
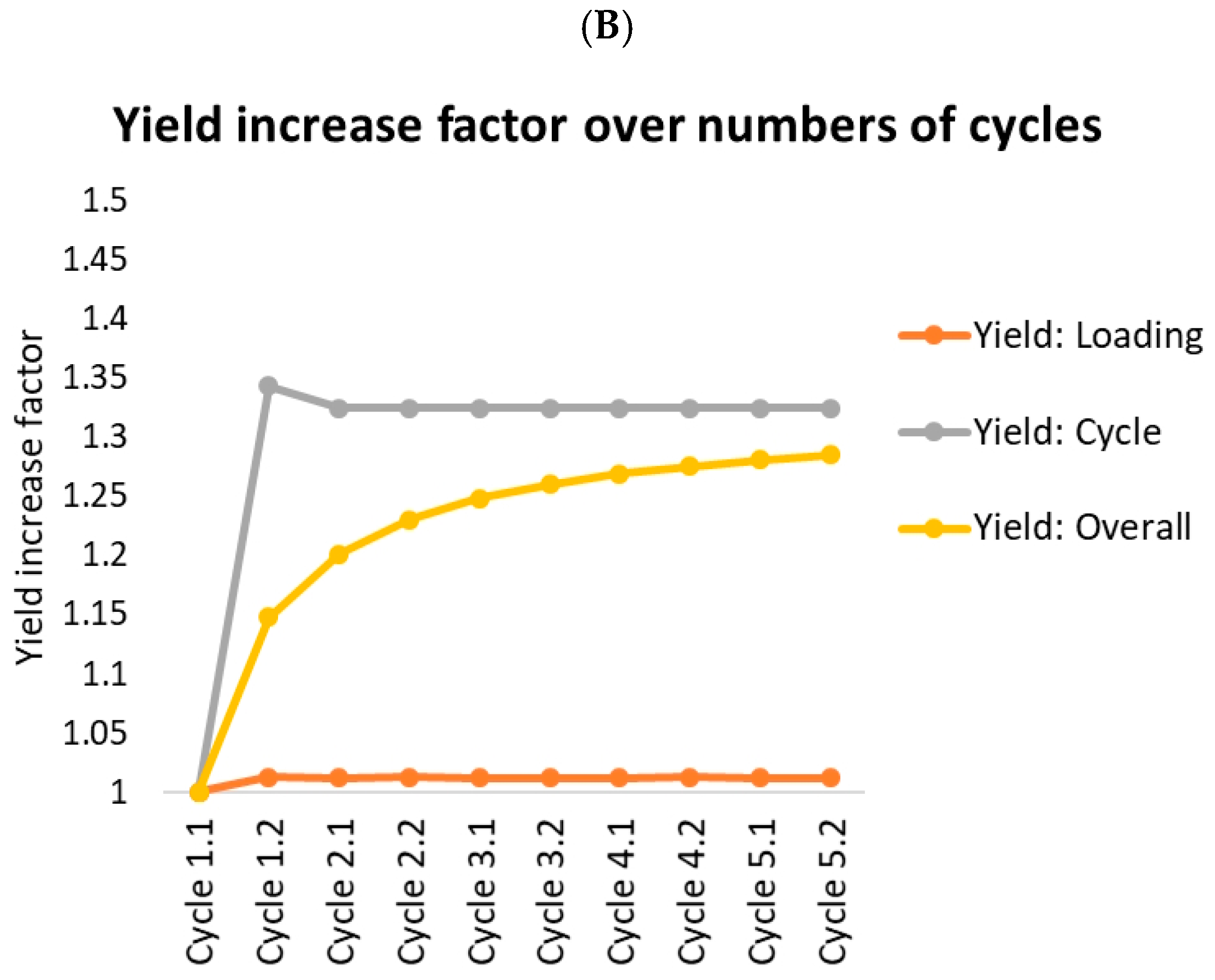
- Yield: Loading (orange lines) indicates the amount of product in product fraction compared to the overall amount loaded (feed plus reloading fractions) in this cycle. This should roughly resemble the batch yield.
- Yield: Cycle (gray) indicates the amount of product in product fraction compared to the amount of feed loaded in this cycle. Option A reaches 100% after the first cycle. There is no product lost at all. Option B loses 7% in fraction 3.
- Yield: Overall (yellow) indicates the overall amount of product gained compared to the overall amount of feed loaded. This starts at the batch yield and approaches the cycle yield, which it would reach after an infinite number of cycles. The gap between the cycle yield and the overall yield is caused by the amount of product stored in the fractions. Since this amount stays roughly the same, the yield loss caused by this becomes more and more unimportant compared to the overall amount of protein produced.
4.3. Experimental Validation
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Abbreviations
| (g/L) | Concentration of component i | |
| (g/L) | Concentration of component i inside the pores | |
| CTCC | Continuous Twin Column Chromatography | |
| CV | Column Volume | |
| (cm2/s) | Axial dispersion coefficient | |
| Deff | (cm2/s) | Effective diffusion coefficient |
| (cm2/s) | Molecular diffusion coefficient | |
| (cm) | Particle diameter | |
| Dp,i | (cm2/s) | Pore diffusion coefficient |
| DS,i | (cm2/s) | Surface diffusion coefficient |
| (-) | Porosity | |
| (-) | Voidage | |
| Hi | (-) | Henry coefficient of component i |
| Ki | (L/g) | Langmuir coefficient of component i |
| (cm/s) | Effective mass transport coefficient | |
| kf | (cm/s) | Mass transport coefficient |
| l | (cm) | Length |
| MCSGP | Multicolumn Countercurrent Solvent Gradient Purification | |
| PAT | Process Analytical Technology | |
| (-) | Peclet-Number | |
| qi | (g/L) | Loading of component i |
| qmax,i | (g/L) | Maximum loading capacity of component i |
| r | (cm) | Radius |
| Re | (-) | Reynolds-Number |
| (cm) | Particle Radius | |
| (-) | Sherwood-Number | |
| t | (s); (min) | Time |
| (s); (min) | Mean residence time | |
| (cm/s) | Interstitial velocity | |
| v | (cm/s) | Velocity |
| (mL/min) | Volumetric flow | |
| (mL) | Volume of column | |
| (mg/cm·s) | Dynamic viscosity | |
| (g/L) | Density | |
| (s2) | Variance |
References
- Carta, G.; Jungbauer, A. Protein Chromatography: Process Development and Scale-Up; Wiley-VCH: Weinheim, Germany, 2010; ISBN 978-3-527-31819-3. [Google Scholar]
- Scopes, R.K. Protein Purification: Principles and Practice, 3rd ed.; Springer: New York, NY, USA, 1994; ISBN 978-1-4757-2333-5. [Google Scholar]
- Subramanian, G. (Ed.) Biopharmaceutical Production Technology; Wiley-VCH: Weinheim, Germany, 2012; ISBN 9783527653096. [Google Scholar]
- Kornecki, M.; Mestmäcker, F.; Zobel-Roos, S.; Heikaus de Figueiredo, L.; Schlüter, H.; Strube, J. Host Cell Proteins in Biologics Manufacturing: The Good, the Bad, and the Ugly. Antibodies 2017, 6, 13. [Google Scholar] [CrossRef]
- Gronemeyer, P.; Strube, J. Purification of Antibodies and their Fragments by ATPE and Precipitation: One Step towards a Chromatography-Free Manufacturing Process. Chem. Ing. Tech. 2015, 87, 1057–1058. [Google Scholar] [CrossRef]
- Subramanian, G. (Ed.) Continuous Biomanufacturing: Innovative Technologies and Methods; Wiley-VCH: Weinheim, Germany, 2018; ISBN 9783527699902. [Google Scholar]
- Strube, J. Technische Chromatographie: Auslegung, Optimierung, Betrieb und Wirtschaftlichkeit; Als Ms. gedr; Univ., Habil.-Schr.: Dortmund, Germany; Shaker: Aachen, Germany, 2000; ISBN 3826568974. [Google Scholar]
- Rodrigues, A. Simulated Moving Bed Technology: Principles, Design and Process Applications; Elsevier Science: Burlington, ON, Canada, 2015; ISBN 9780128020241. [Google Scholar]
- Kaspereit, M.; Seidel-Morgenstern, A. Auslegung der Regenerationszonen des SMB-Verfahrens. Chem. Ing. Tech. 2002, 74, 591–592. [Google Scholar] [CrossRef]
- Mazzotti, M. Equilibrium theory based design of simulated moving bed processes for a generalized Langmuir isotherm. J. Chromatogr. A 2006, 1126, 311–322. [Google Scholar] [CrossRef]
- Wellhoefer, M.; Sprinzl, W.; Hahn, R.; Jungbauer, A. Continuous processing of recombinant proteins: Integration of refolding and purification using simulated moving bed size-exclusion chromatography with buffer recycling. J. Chromatogr. A 2014, 1337, 48–56. [Google Scholar] [CrossRef] [PubMed]
- Aumann, L.; Morbidelli, M. A continuous multicolumn countercurrent solvent gradient purification (MCSGP) process. Biotechnol. Bioeng. 2007, 98, 1043–1055. [Google Scholar] [CrossRef]
- de Luca, C.; Lievore, G.; Bozza, D.; Buratti, A.; Cavazzini, A.; Ricci, A.; Macis, M.; Cabri, W.; Felletti, S.; Catani, M. Downstream Processing of Therapeutic Peptides by Means of Preparative Liquid Chromatography. Molecules 2021, 26, 4688. [Google Scholar] [CrossRef]
- de Luca, C.; Felletti, S.; Lievore, G.; Buratti, A.; Vogg, S.; Morbidelli, M.; Cavazzini, A.; Catani, M.; Macis, M.; Ricci, A.; et al. From batch to continuous chromatographic purification of a therapeutic peptide through multicolumn countercurrent solvent gradient purification. J. Chromatogr. A 2020, 1625, 461304. [Google Scholar] [CrossRef]
- Aumann, L.; Stroehlein, G.; Muller-Spath, T.; Schenkel, B.; Morbidelli, M. Protein Peptide Purification using the Multicolumn Countercurrent Solvent Gradient Purification (MCSGP) Process. BioPharm Int. 2009, 22. [Google Scholar]
- Zobel-Roos, S.; Mouellef, M.; Ditz, R.; Strube, J. Distinct and Quantitative Validation Method for Predictive Process Modelling in Preparative Chromatography of Synthetic and Bio-Based Feed Mixtures Following a Quality-by-Design (QbD) Approach. Processes 2019, 7, 580. [Google Scholar] [CrossRef]
- Kaczmarski, K.; Cavazzini, A.; Szabelski, P.; Zhou, D.; Liu, X.; Guiochon, G. Application of the general rate model and the generalized Maxwell–Stefan equation to the study of the mass transfer kinetics of a pair of enantiomers. J. Chromatogr. A 2002, 962, 57–67. [Google Scholar] [CrossRef] [PubMed]
- Kaczmarski, K.; Gubernak, M.; Zhou, D.; Guiochon, G. Application of the general rate model with the Maxwell–Stefan equations for the prediction of the band profiles of the 1-indanol enantiomers. Chem. Eng. Sci. 2003, 58, 2325–2338. [Google Scholar] [CrossRef]
- Guiochon, G.; Felinger, A.; Shirazi, D.G.; Katti, A.M. Fundamentals of Preparative and Nonlinear Chromatography, 2nd ed.; Elsevier Academic Press: Amsterdam, The Netherlands, 2006. [Google Scholar]
- Zobel-Roos, S. Entwicklung, Modellierung und Validierung von Integrierten Kontinuierlichen Gegenstrom-Chromatographie-Prozessen; 1. Auflage; Shaker: Herzogenrath, Germany, 2018; ISBN 3844061878. [Google Scholar]
- Felinger, A.; Guiochon, G. Comparison of the Kinetic Models of Linear Chromatography. Chromatographia 2004, 60, S175–S180. [Google Scholar] [CrossRef]
- Piątkowski, W.; Antos, D.; Kaczmarski, K. Modeling of preparative chromatography processes with slow intraparticle mass transport kinetics. J. Chromatogr. A 2003, 988, 219–231. [Google Scholar] [CrossRef]
- Seidel-Morgenstern, A. Experimental determination of single solute and competitive adsorption isotherms. J. Chromatogr. A 2004, 1037, 255–272. [Google Scholar] [CrossRef]
- Asnin, L. Adsorption models in chiral chromatography. J. Chromatogr. A 2012, 1269, 3–25. [Google Scholar] [CrossRef] [PubMed]
- Blümel, C.; Kniep, H.; Seidel-Morgenstern, A. Measuring adsorption isotherms using a closed-loop perturbation method to minimize sample consumption. In Proceedings of the 6th International Conference of Fundamentals of Adsorption—FOA 6, Presqu’ile de Giens, France, 23–27 May 1998; Elsevier: Amsterdam, The Netherlands, 1998; pp. 449–454. [Google Scholar]
- Cavazzini, A.; Felinger, A.; Guiochon, G. Comparison between adsorption isotherm determination techniques and overloaded band profiles on four batches of monolithic columns. J. Chromatogr. A 2003, 1012, 139–149. [Google Scholar] [CrossRef] [PubMed]
- Ching, C.B.; Chu, K.H.; Ruthven, D.M. A study of multicomponent adsorption equilibria by liquid chromatography. AIChE J. 1990, 36, 275–281. [Google Scholar] [CrossRef]
- Gamba, G.; Rota, R.; Storti, G.; Carra, S.; Morbidelli, M. Absorbed solution theory models for multicomponent adsorption equilibria. AIChE J. 1989, 35, 959–966. [Google Scholar] [CrossRef]
- Hu, X.; Do, D.D. Comparing various multicomponent adsorption equilibrium models. AIChE J. 1995, 41, 1585–1592. [Google Scholar] [CrossRef]
- Heinonen, J.; Rubiera Landa, H.O.; Sainio, T.; Seidel-Morgenstern, A. Use of Adsorbed Solution theory to model competitive and co-operative sorption on elastic ion exchange resins. Sep. Purif. Technol. 2012, 95, 235–247. [Google Scholar] [CrossRef]
- Emerton, D.A. Profitability in the Biosimilars Market: Can You Translate Scientific Excellence into a Healthy Commercial Return? BioProcess Int. 2013, 11, 6–23. [Google Scholar]
- Erto, A.; Lancia, A.; Musmarra, D. A modelling analysis of PCE/TCE mixture adsorption based on Ideal Adsorbed Solution Theory. Sep. Purif. Technol. 2011, 80, 140–147. [Google Scholar] [CrossRef]
- Myers, A.L.; Prausnitz, J.M. Thermodynamics of mixed-gas adsorption. AIChE J. 1965, 11, 121–127. [Google Scholar] [CrossRef]
- Costa, E.; Calleja, G.; Marron, C.; Jimenez, A.; Pau, J. Equilibrium adsorption of methane, ethane, ethylene, and propylene and their mixtures on activated carbon. J. Chem. Eng. Data 1989, 34, 156–160. [Google Scholar] [CrossRef]
- Brooks, C.A.; Cramer, S.M. Steric mass-action ion exchange: Displacement profiles and induced salt gradients. AIChE J. 1992, 38, 1969–1978. [Google Scholar] [CrossRef]
- Langmuir, I. The adsorption of gases on plane surfaces of glass, mica and platinum. J. Am. Chem. Soc. 1918, 40, 1361–1403. [Google Scholar] [CrossRef]
- DePhillips, P.; Lenhoff, A.M. Pore size distributions of cation-exchange adsorbents determined by inverse size-exclusion chromatography. J. Chromatogr. A 2000, 883, 39–54. [Google Scholar] [CrossRef]
- Goto, M.; McCoy, B.J. Inverse size-exclusion chromatography for distributed pore and solute sizes. Chem. Eng. Sci. 2000, 55, 723–732. [Google Scholar] [CrossRef]
- Levenspiel, O. Chemical Reaction Engineering, 3rd ed.; Wiley: New York, NY, USA, 1999; ISBN 9780471254249. [Google Scholar]
- Jacobson, J.M.; Frenz, J.H.; Horvath, C.G. Measurement of competitive adsorption isotherms by frontal chromatography. Ind. Eng. Chem. Res. 1987, 26, 43–50. [Google Scholar] [CrossRef]
- Strube, J.; Zobel-Roos, S. Preparative Chromatography for (Bio)Pharmaceuticals: Digital Multi-Purpose Plants for Fast and Flexible Process Solutions. Available online: https://analyticalscience.wiley.com/do/10.1002/was.00080322 (accessed on 15 March 2022).









Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zobel-Roos, S.; Vetter, F.; Scheps, D.; Pfeiffer, M.; Gunne, M.; Boscheinen, O.; Strube, J. Digital Twin Based Design and Experimental Validation of a Continuous Peptide Polishing Step. Processes 2023, 11, 1401. https://doi.org/10.3390/pr11051401
Zobel-Roos S, Vetter F, Scheps D, Pfeiffer M, Gunne M, Boscheinen O, Strube J. Digital Twin Based Design and Experimental Validation of a Continuous Peptide Polishing Step. Processes. 2023; 11(5):1401. https://doi.org/10.3390/pr11051401
Chicago/Turabian StyleZobel-Roos, Steffen, Florian Vetter, Daniel Scheps, Marcus Pfeiffer, Matthias Gunne, Oliver Boscheinen, and Jochen Strube. 2023. "Digital Twin Based Design and Experimental Validation of a Continuous Peptide Polishing Step" Processes 11, no. 5: 1401. https://doi.org/10.3390/pr11051401
APA StyleZobel-Roos, S., Vetter, F., Scheps, D., Pfeiffer, M., Gunne, M., Boscheinen, O., & Strube, J. (2023). Digital Twin Based Design and Experimental Validation of a Continuous Peptide Polishing Step. Processes, 11(5), 1401. https://doi.org/10.3390/pr11051401