Special Issue of “Synthesis, Biological Evaluation and Molecular Modeling of Enzyme Inhibitors”




{kind=link}
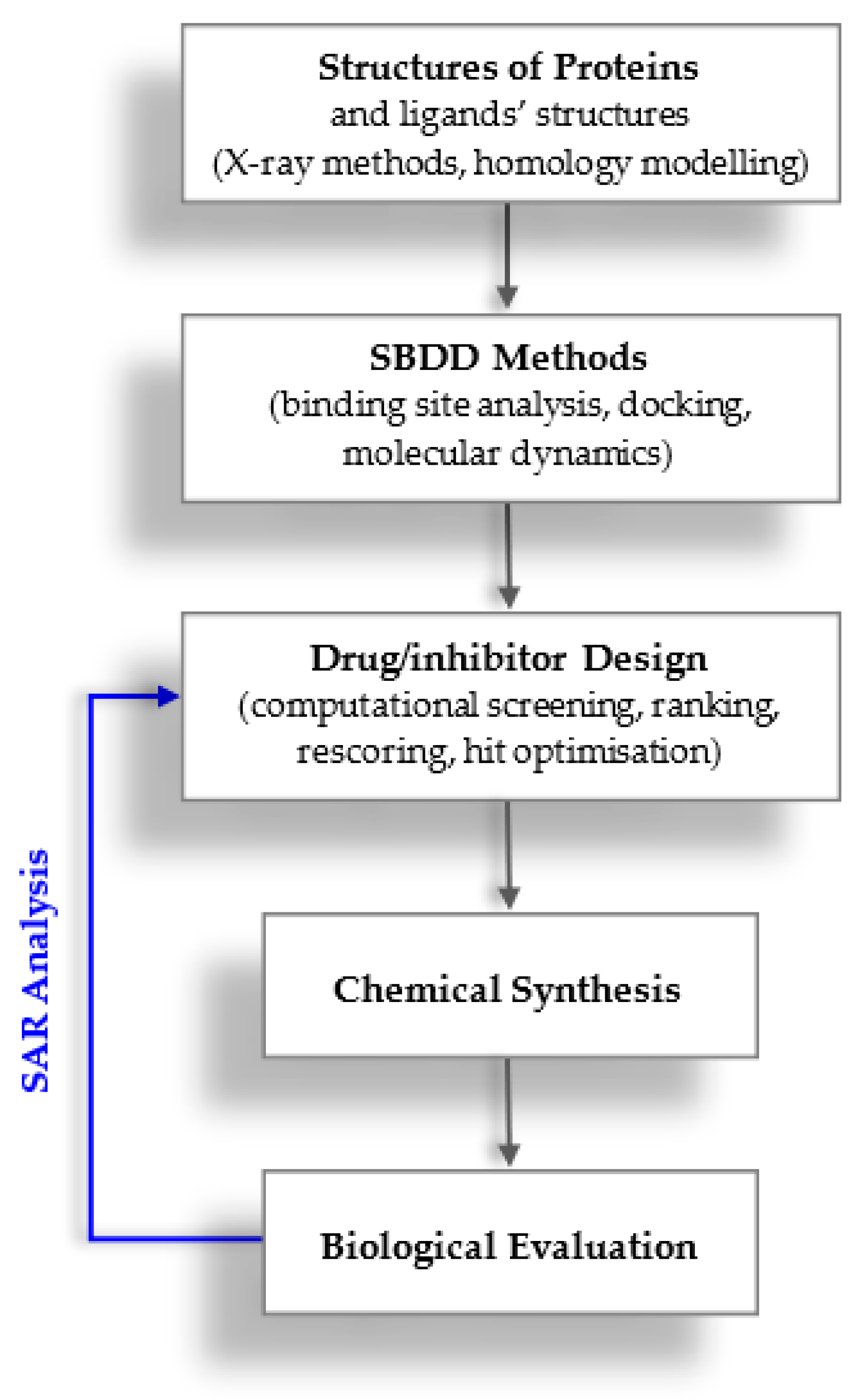
1. Introduction
2. An Overview of Published Articles
3. Conclusions
Author Contributions
Conflicts of Interest
List of Contributions
- Mikstacka, R.; Dutkiewicz, Z. New Perspectives of CYP1B1 Inhibition in the Light of Molecular Studies. Processes 2021, 9, 817.
- Richert, M.; Mikstacka, R.; Walczyk, M.; Cieślak, M.J.; Kaźmierczak-Barańska, J.; Królewska-Golińska, K.; Muzioł, T.M.; Biniak, S. Gold(I) Complexes with P-Donor Ligands and Their Biological Evaluation. Processes 2021, 9, 2100.
- Xu, M.; Zhang, L.; Zhao, F.; Wang, J.; Zhao, B.; Zhou, Z.; Han, Y. Cloning and Expression of Levansucrase Gene of Bacillus velezensis BM-2 and Enzymatic Synthesis of Levan. Processes 2021, 9, 317.
- Li, W.-T.; Chuang, Y.-H.; Liao, J.-H.; Hsieh, J.-F. Synthesis of 2-(4-hydroxyphenyl)ethyl 3,4,5-Trihydroxybenzoate and Its Inhibitory Effect on Sucrase and Maltase. Processes 2020, 8, 1603.
- Shokeer, A.; Ismail, A.; Hegazy, U.M.; Kolm, R.H.; Mannervik, B. Mutational Analysis of the Binding of Alternative Substrates and Inhibitors to the Active Site of Human Glutathione Transferase P1-1. Processes 2020, 8, 1232.
References
- Geronikaki, A. Recent Trends in Enzyme Inhibition and Activation in Drug Design. Molecules 2020, 26, 17. [Google Scholar] [CrossRef]
- Rufer, A.C. Drug discovery for enzymes. Drug Discov. Today 2021, 26, 875–886. [Google Scholar] [CrossRef]
- Talevi, A. Computer-Aided Drug Discovery and Design: Recent Advances and Future Prospects. Methods Mol. Biol. 2023, 2714, 1–20. [Google Scholar] [CrossRef]
- Zhang, Y.; Luo, M.; Wu, P.; Wu, S.; Lee, T.Y.; Bai, C. Application of Computational Biology and Artificial Intelligence in Drug Design. Int. J. Mol. Sci. 2022, 23, 13568. [Google Scholar] [CrossRef]
- Prieto-Martínez, F.D.; Arciniega, M.; Medina-Franco, J.L. Molecular docking: Current advances and challenges. Tip Rev. Espec. Cienc. Quím. Biol. 2018, 21, 65–87. [Google Scholar] [CrossRef]
- Śledź, P.; Caflisch, A. Protein structure-based drug design: From docking to molecular dynamics. Curr. Opin. Struct. Biol. 2018, 48, 93–102. [Google Scholar] [CrossRef] [PubMed]
- Salmaso, V.; Moro, S. Bridging Molecular Docking to molecular dynamics in exploring ligand-protein recognition process: An overview. Front. Pharmacol. 2018, 9, 923. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Ma, J.; Li, M.; Zhang, Y.; Jiang, B.; Zhao, X.; Huai, C.; Shen, L.; Zhang, N.; He, L.; et al. Cytochrome P450 Enzymes and Drug Metabolism in Humans. Int. J. Mol. Sci. 2021, 22, 12808. [Google Scholar] [CrossRef]
- Manikandan, P.; Nagini, S. Cytochrome P450 Structure, Function and Clinical Significance: A Review. Curr. Drug Targets 2018, 19, 38–54. [Google Scholar] [CrossRef] [PubMed]
- Zou, T.; Lum, C.T.; Lok, C.-N.; Zhang, J.-J.; Che, C.-M. Chemical biology of anticancer gold(III) and gold(I) complexes. Chem. Soc. Rev. 2015, 44, 8786–8801. [Google Scholar] [CrossRef]
- Mohammadi, F.; Soltani, A.; Ghahremanloo, A.; Javid, H.; Hashemy, S.I. The thioredoxin system and cancer therapy: A review. Cancer Chemother. Pharmacol. 2019, 84, 925–935. [Google Scholar] [CrossRef] [PubMed]
- Netrusov, A.I.; Liyaskina, E.V.; Kurgaeva, I.V.; Liyaskina, A.U.; Yang, G.; Revin, V.V. Exopolysaccharides Producing Bacteria: A Review. Microorganisms 2023, 11, 1541. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Zhang, L.; Zhao, F.; Wang, J.; Zhao, B.; Zhou, Z.; Han, Y. Cloning and Expression of Levansucrase Gene of Bacillus velezensis BM-2 and Enzymatic Synthesis of Levan. Processes 2021, 9, 317. [Google Scholar] [CrossRef]
- Mikstacka, R.; Dutkiewicz, Z. New Perspectives of CYP1B1 Inhibition in the Light of Molecular Studies. Processes 2021, 9, 817. [Google Scholar] [CrossRef]
- Li, W.-T.; Chuang, Y.-H.; Liao, J.-H.; Hsieh, J.-F. Synthesis of 2-(4-hydroxyphenyl)ethyl 3,4,5-Trihydroxybenzoate and Its Inhibitory Effect on Sucrase and Maltase. Processes 2020, 8, 1603. [Google Scholar] [CrossRef]
- Shokeer, A.; Ismail, A.; Hegazy, U.M.; Kolm, R.H.; Mannervik, B. Mutational Analysis of the Binding of Alternative Substrates and Inhibitors to the Active Site of Human Glutathione Transferase P1-1. Processes 2020, 8, 1232. [Google Scholar] [CrossRef]
- Li, J.; Fu, A.; Zhang, L. An overview of scoring functions used for protein–ligand interactions in molecular docking. Interdiscip. Sci. Comput. Sci. 2019, 11, 320–328. [Google Scholar] [CrossRef]
- Fan, M.; Wang, J.; Jiang, H.; Feng, Y.; Mahdavi, M.; Madduri, K.; Kandemir, M.T.; Dokholyan, N.V. GPU-accelerated flexible molecular docking. J. Phys. Chem. B 2021, 125, 1049–1060. [Google Scholar] [CrossRef]
- Solis-Vasquez, L.; Tillack, A.F.; Santos-Martins, D.; Koch, A.; LeGrand, S.; Forli, S. Benchmarking the performance of irregular computations in AutoDock-GPU molecular docking. Parallel Comput. 2022, 109, 102861. [Google Scholar] [CrossRef]
- Yu, Y.; Cai, C.; Wang, J.; Bo, Z.; Zhu, Z.; Zheng, H. Uni-Dock: GPU-accelerated docking enables ultralarge virtual screening. J. Chem. Theory Comput. 2023, 19, 3336–3345. [Google Scholar] [CrossRef]
- Wang, J.; Arantes, P.R.; Bhattarai, A.; Hsu, R.V.; Pawnikar, S.; Huang, Y.M.; Palermo, G.; Miao, Y. Gaussian accelerated molecular dynamics: Principles and applications. WIREs Comput. Mol. Sci. 2021, 11, e1521. [Google Scholar] [CrossRef]
- Bernardi, R.C.; Melo, M.C.R.; Schulten, K. Enhanced sampling techniques in molecular dynamics simulations of Biological Systems. Biochim. Biophys. Acta-Gen. Subj. 2015, 1850, 872–877. [Google Scholar] [CrossRef]
- Rydzewski, J.; Nowak, W. Ligand diffusion in proteins via enhanced sampling in molecular dynamics. Phys. Life Rev. 2017, 22–23, 58–74. [Google Scholar] [CrossRef] [PubMed]
- Kuzmanic, A.; Bowman, G.R.; Juarez-Jimenez, J.; Michel, J.; Gervasio, F.L. Investigating cryptic binding sites by molecular dynamics simulations. Acc. Chem. Res. 2020, 53, 654–661. [Google Scholar] [CrossRef] [PubMed]
- Nerenberg, P.S.; Head-Gordon, T. New developments in force fields for biomolecular simulations. Curr. Opin. Struct. Biol. 2018, 49, 129–138. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Shi, D.; Zhou, S.; Liu, H.; Liu, H.; Yao, X. Molecular dynamics simulations and novel drug discovery. Expert Opin. Drug Discov. 2017, 13, 23–37. [Google Scholar] [CrossRef] [PubMed]
- Pinzi, L.; Rastelli, G. Molecular docking: Shifting paradigms in drug discovery. Int. J. Mol. Sci. 2019, 20, 4331. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mikstacka, R.; Dutkiewicz, Z.; Wierzchowski, M. Special Issue of “Synthesis, Biological Evaluation and Molecular Modeling of Enzyme Inhibitors”. Processes 2023, 11, 3128. https://doi.org/10.3390/pr11113128
Mikstacka R, Dutkiewicz Z, Wierzchowski M. Special Issue of “Synthesis, Biological Evaluation and Molecular Modeling of Enzyme Inhibitors”. Processes. 2023; 11(11):3128. https://doi.org/10.3390/pr11113128
Chicago/Turabian StyleMikstacka, Renata, Zbigniew Dutkiewicz, and Marcin Wierzchowski. 2023. "Special Issue of “Synthesis, Biological Evaluation and Molecular Modeling of Enzyme Inhibitors”" Processes 11, no. 11: 3128. https://doi.org/10.3390/pr11113128
APA StyleMikstacka, R., Dutkiewicz, Z., & Wierzchowski, M. (2023). Special Issue of “Synthesis, Biological Evaluation and Molecular Modeling of Enzyme Inhibitors”. Processes, 11(11), 3128. https://doi.org/10.3390/pr11113128