
Experimental Determination on the Deactivation Kinetics of Residue Hydroprocessing in a Two-Stage Fixed Bed Loaded with HDM and HDS Catalysts
 ,
, 
Abstract
:
1. Introduction
2. Experimental
2.1. Properties of Catalyst and Residue
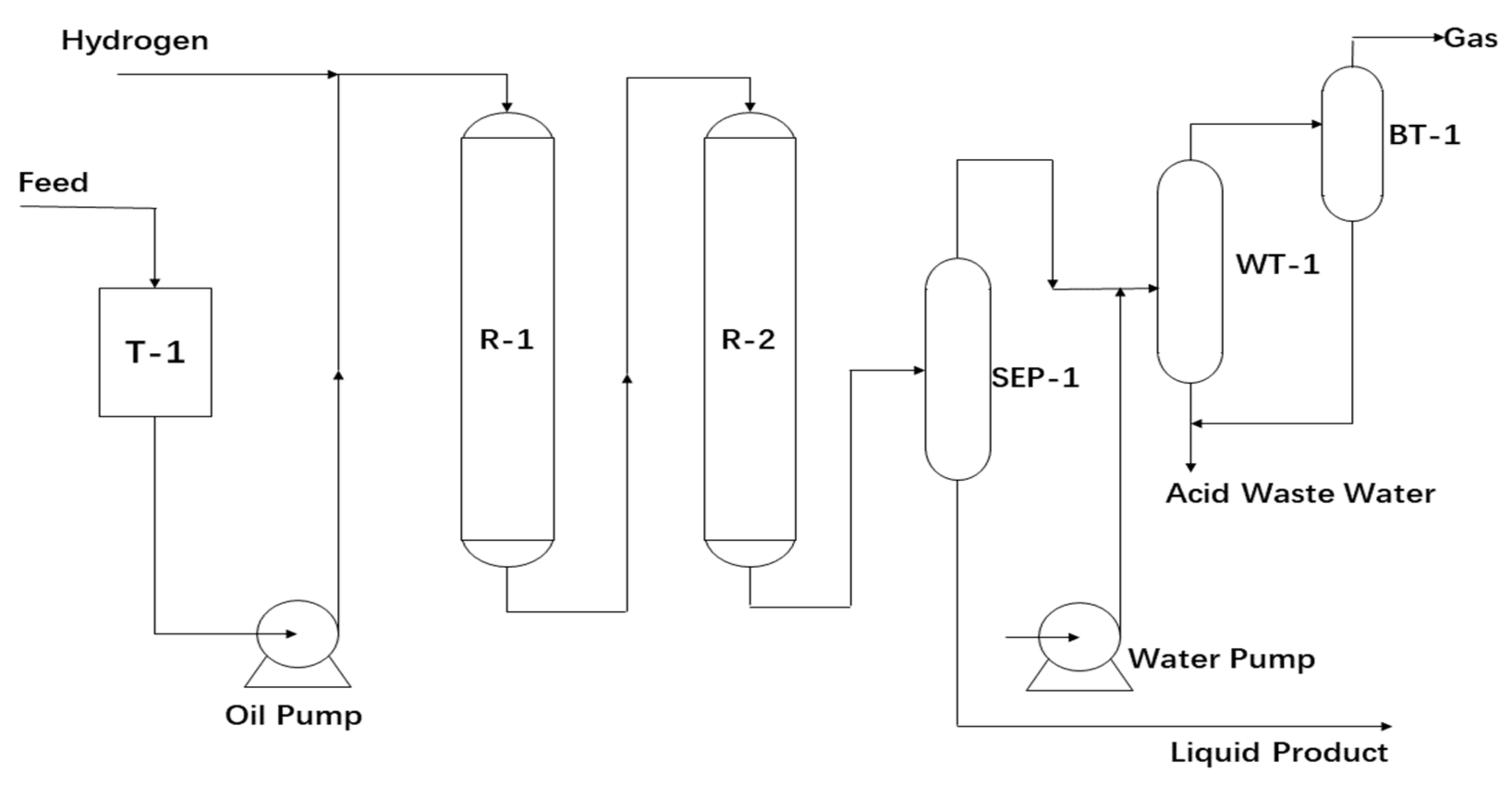
2.2. Apparatus
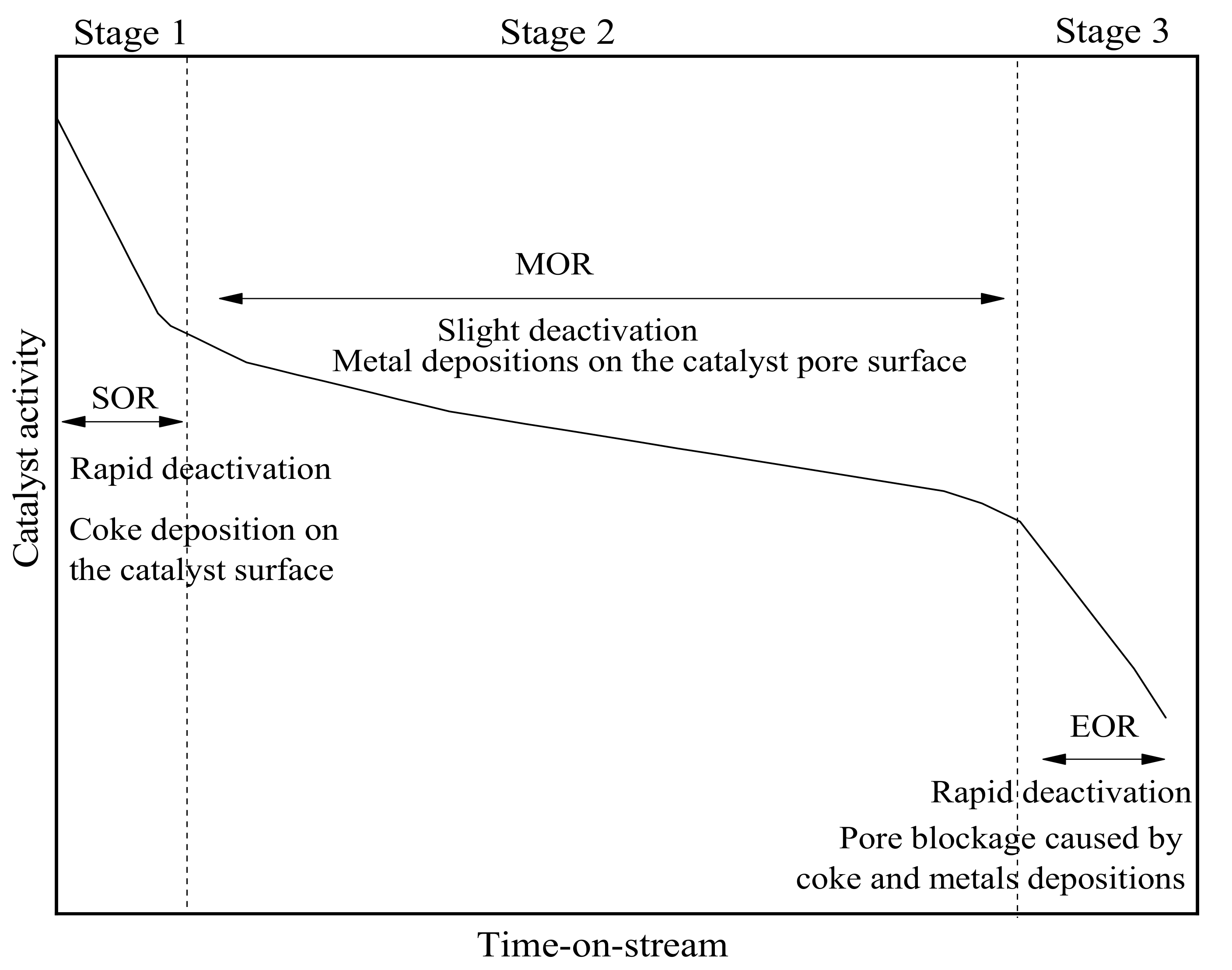
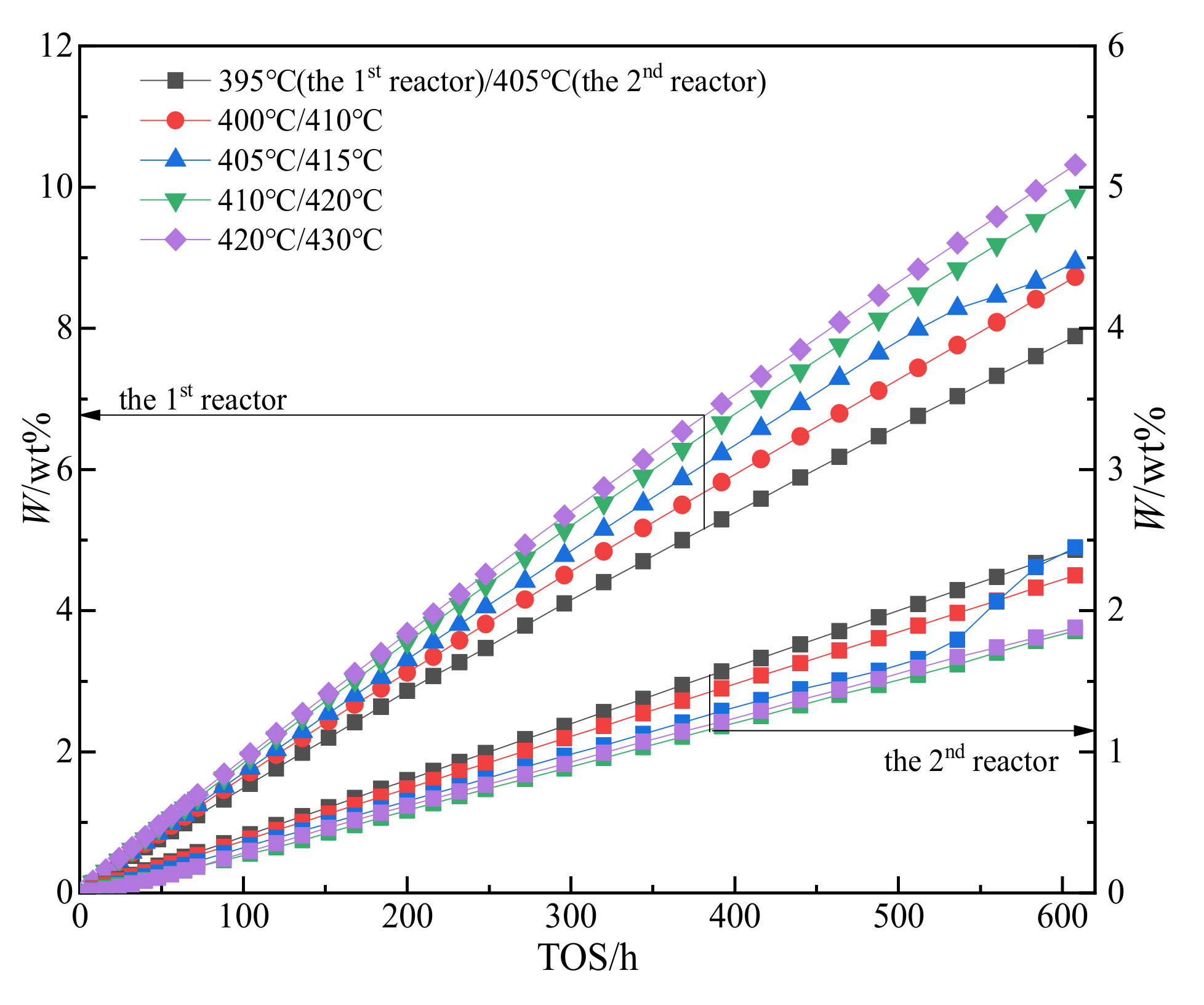
2.3. Experimental Study on Catalyst Deactivation
2.4. Experiments on Hydrogenation Kinetics
3. Results and Discussion
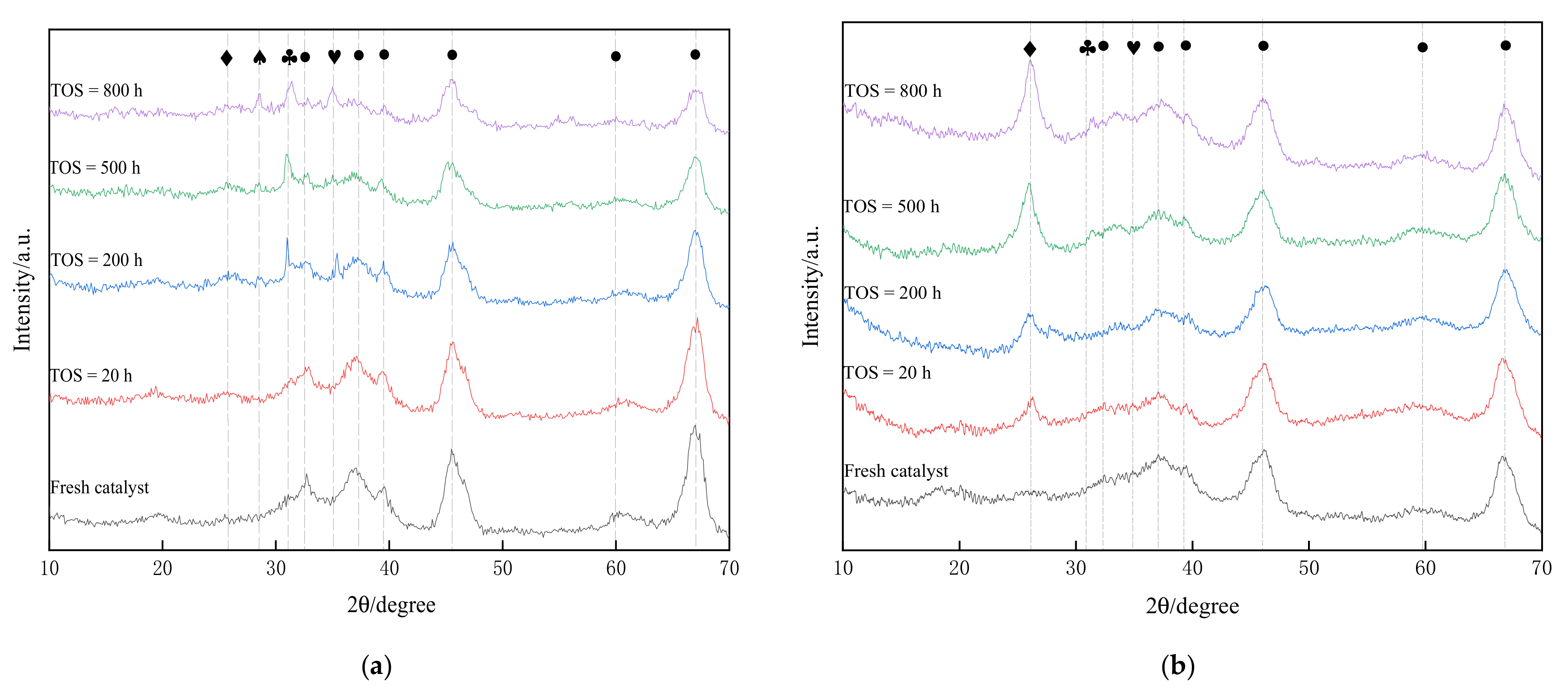
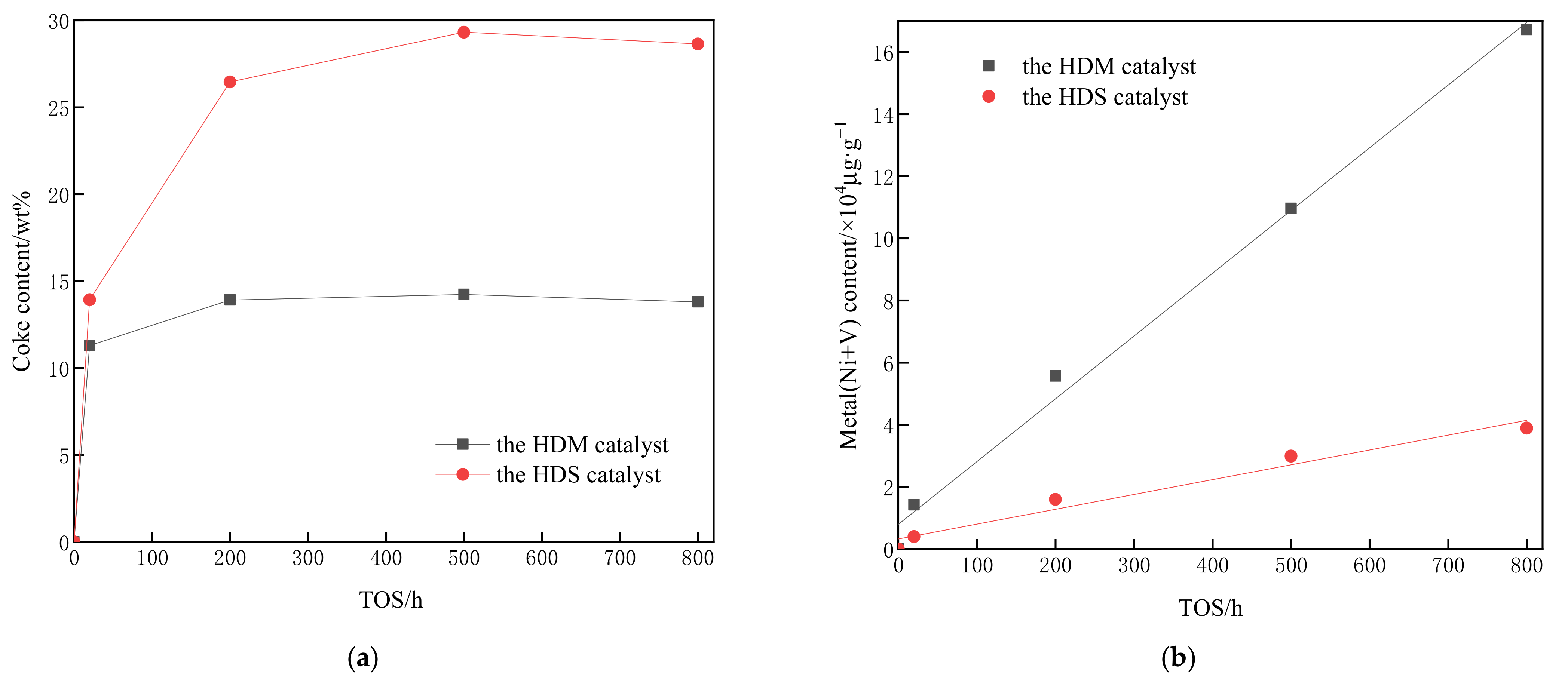
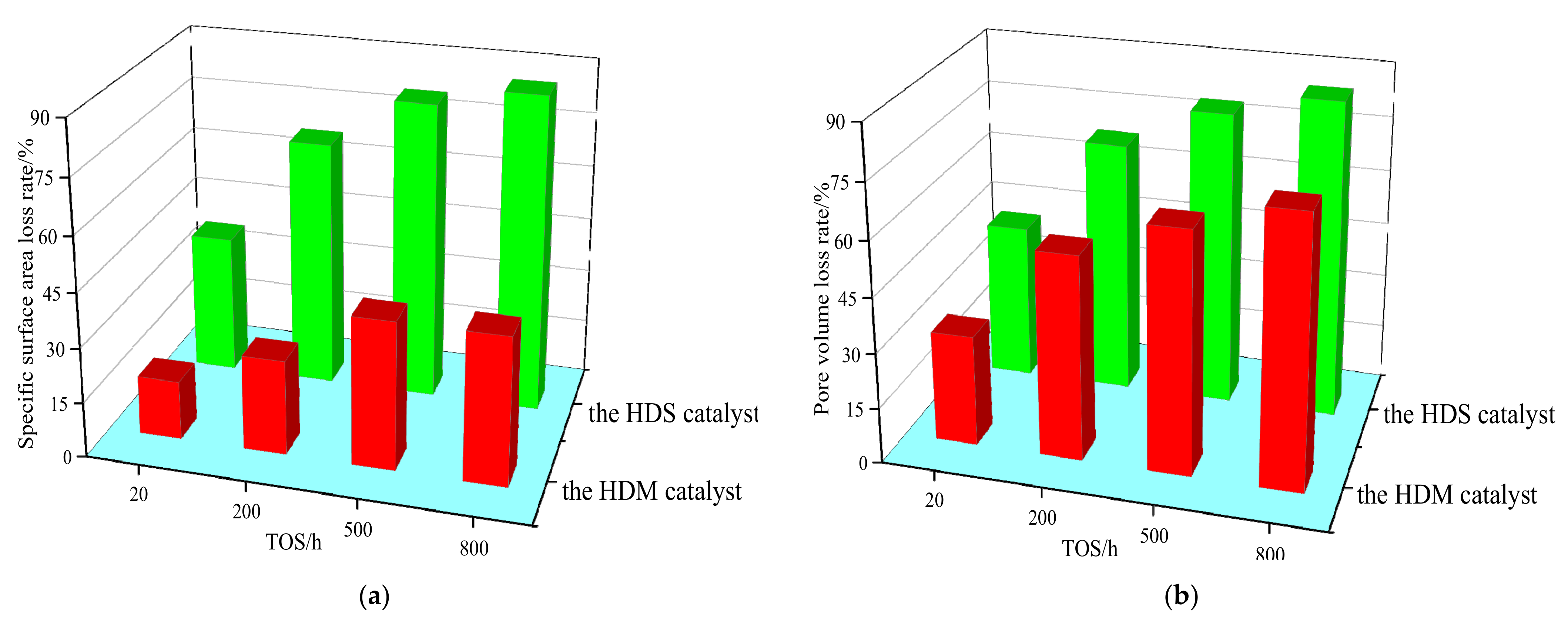
3.1. Characterization of HDM and HDS Catalysts
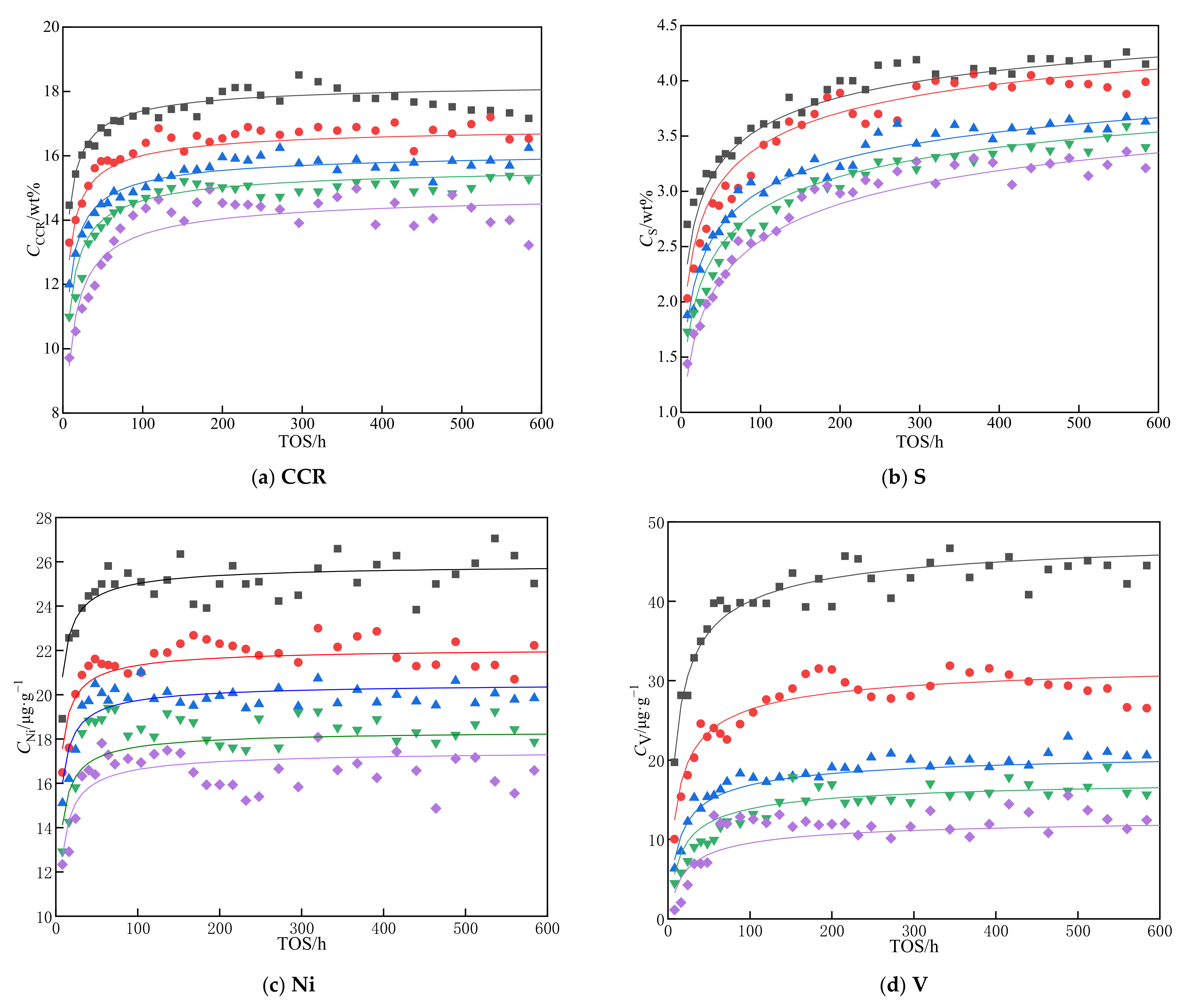
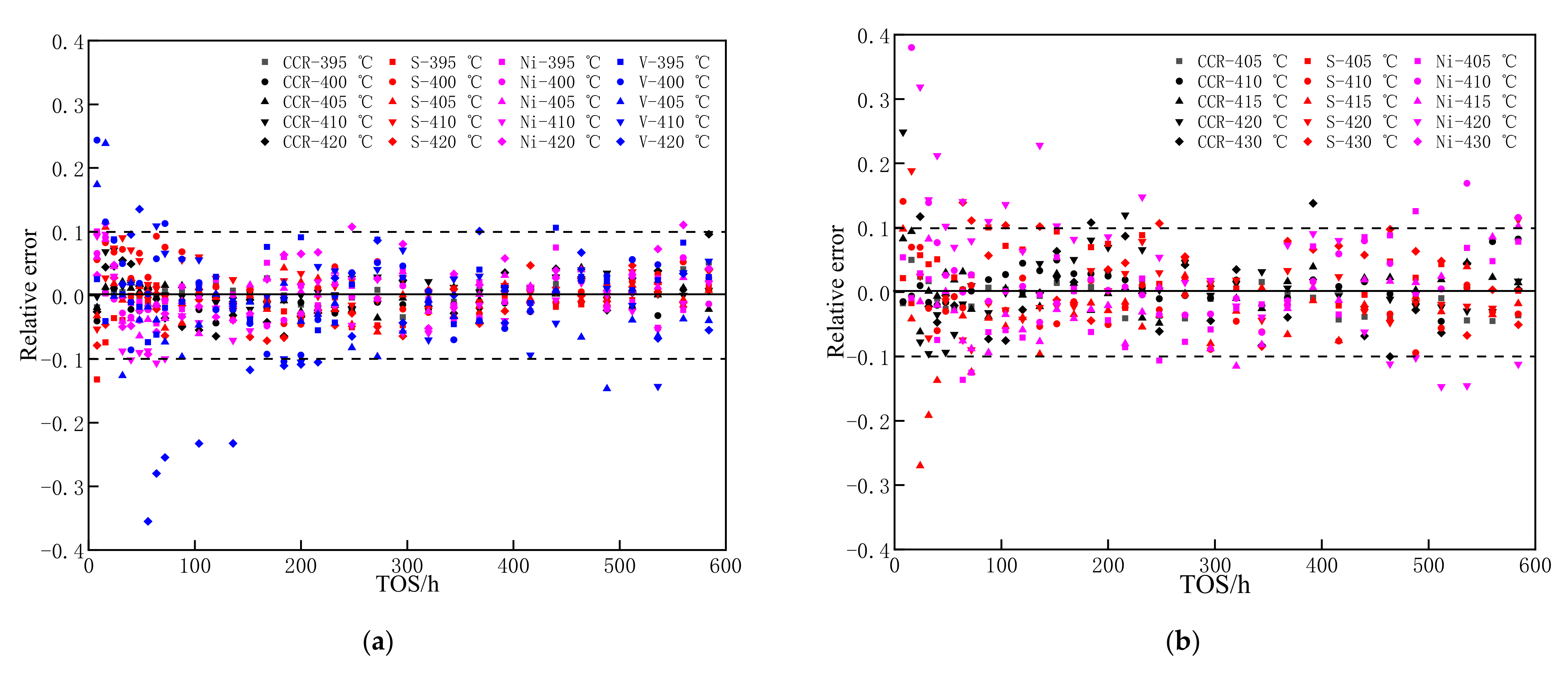
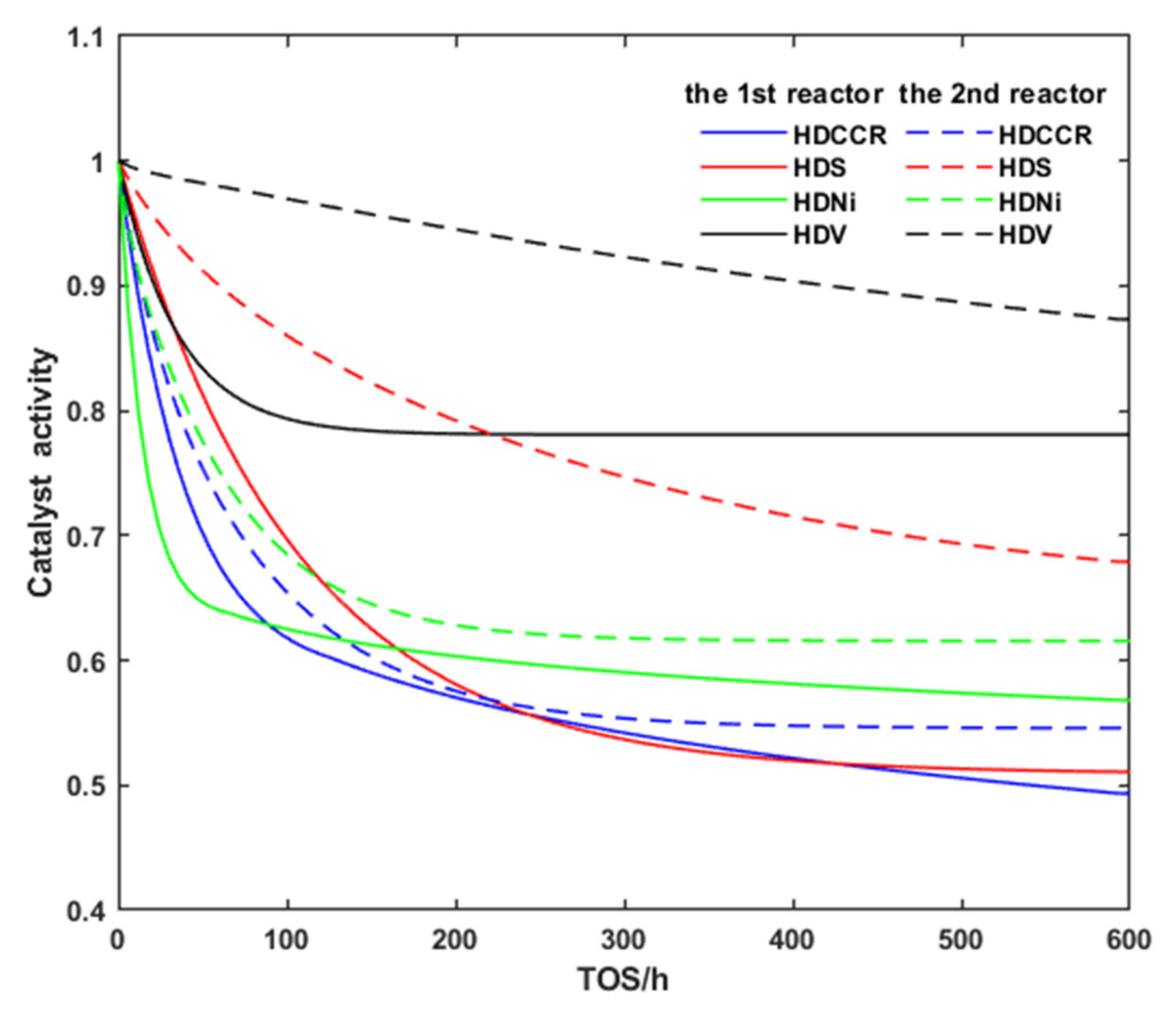
3.2. Deactivation Model of Residue Hydrotreating Catalysts
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sahu, R.; Song, B.J.; Im, J.S.; Jeon, Y.P.; Lee, C.W. A review of recent advances in catalytic hydrocracking of heavy residues. J. Ind. Eng. Chem. 2015, 27, 12–24. [Google Scholar] [CrossRef]
- Ali, M.F.; Abbas, S. A review of methods for the demetallization of residual fuel oils. Fuel Process. Technol. 2006, 87, 573–584. [Google Scholar] [CrossRef]
- Lee, D.K.; Lee, I.C.; Woo, S.I. Effects of transition metal addition to CoMo/γ-Al2O3 catalyst on the hydrotreating reactions of atmospheric residual oil. Appl. Catal. A Gen. 1994, 109, 195–210. [Google Scholar] [CrossRef]
- Novaes, L.D.; Pacheco, M.E.; Salim, V.M.M.; de Resende, N.S. Accelerated deactivation studies of hydrotreating catalysts in pilot unit. Appl. Catal. A-Gen. 2017, 548, 114–121. [Google Scholar] [CrossRef]
- Bartholomew, C.H. Mechanisms of catalyst deactivation. Appl. Catal. A-Gen. 2001, 212, 17–60. [Google Scholar] [CrossRef]
- Moulijn, J.A.; Diepen, A.E.V.; Kapteijn, F. Catalyst deactivation: Is it predictable? What to do? Appl. Catal. A-Gen. 2001, 212, 3–16. [Google Scholar] [CrossRef]
- Prajapati, R.; Kohli, K.; Maity, S.K.; Garg, M.O. Coking propensity during hydroprocessing of vacuum residues, deasphalted oils, and asphaltenes. Fuel 2017, 203, 514–521. [Google Scholar] [CrossRef]
- Rodriguez, E.; Felix, G.; Ancheyta, J.; Trejo, F. Modeling of hydrotreating catalyst deactivation for heavy oil hydrocarbons. Fuel 2018, 225, 118–133. [Google Scholar] [CrossRef]
- Maity, S.K.; Blanco, E.; Ancheyta, J.; Alonso, F.; Fukuyama, H. Early stage deactivation of heavy crude oil hydroprocessing catalysts. Fuel 2012, 100, 17–23. [Google Scholar] [CrossRef]
- Centeno, G.; Ancheyta, J.; Alvarez, A.; Marroquin, G.; Alonso, F.; Castillo, A. Effect of different heavy feedstocks on the deactivation of a commercial hydrotreating catalyst. Fuel 2012, 100, 73–79. [Google Scholar] [CrossRef]
- Elizalde, I.; Ancheyta, J. Application of a three-stage approach for modeling the complete period of catalyst deactivation during hydrotreating of heavy oil. Fuel 2014, 138, 45–51. [Google Scholar] [CrossRef]
- Torres, M.P.; Ancheytaa, J.; Martínez, J. Deactivation of a hydrotreating catalyst in a bench-scale continuous stirred tank reactor at different operating conditions. Fuel 2018, 234, 326–334. [Google Scholar] [CrossRef]
- Elizalde, I.; Ancheyta, J. Modeling the deactivation by metal deposition of heavy oil hydrotreating catalyst. Catal. Today 2014, 220–222, 221–227. [Google Scholar] [CrossRef]
- Marafi, A.; Stanislaus, A.; Furimsky, E. Kinetics and Modeling of Petroleum Residues Hydroprocessing. Catal. Rev.-Sci. Eng. 2010, 52, 204–324. [Google Scholar] [CrossRef]
- Ancheyta, J.; Betancourt, G.; Centeno, G.; Marroquin, G. Catalyst deactivation during hydroprocessing of Maya heavy crude oil. (II) Effect of temperature during time-on-stream. Energy Fuels 2003, 17, 462–467. [Google Scholar] [CrossRef]
- Elizalde, I.; Ancheyta, J. Modeling catalyst deactivation during hydrocracking of atmospheric residue by using the continuous kinetic lumping model. Fuel Process. Technol. 2014, 123, 114–121. [Google Scholar] [CrossRef]
- Calderon, C.J.; Ancheyta, J. Modeling of CSTR and SPR small-scale isothermal reactors for heavy oil hydrocracking and hydrotreating. Fuel 2018, 216, 852–860. [Google Scholar] [CrossRef]
- Rana, M.S.; Samano, V.; Ancheyta, J.; Diaz, J.A.I. A review of recent advances on process technologies for upgrading of heavy oils and residua. Fuel 2007, 86, 1216–1231. [Google Scholar] [CrossRef]
- Corella, J.; Monzon, A. Modeling of the deactivation kinetics of solid catalysts by two or more simultaneous and different causes. Ind. Eng. Chem. Res. 1988, 27, 415–421. [Google Scholar] [CrossRef]
- Cui, Q.Y.; Ma, X.L.; Nakano, K.; Nakabayashi, K.; Miyawaki, J.; Al-Mutairi, A.; Marafi, A.M.J.; Park, J.I.; Yoon, S.H.; Mochida, I. Hydrotreating Reactivities of Atmospheric Residues and Correlation with Their Composition and Properties. Energy Fuels 2018, 32, 6726–6736. [Google Scholar] [CrossRef]
- Callejas, M.A.; Martinez, M.T.; Blasco, T.; Sastre, E. Coke characterisation in aged residue hydrotreating catalysts by solid-state C-13-NMR spectroscopy and temperature-programmed oxidation. Appl. Catal. A-Gen. 2001, 218, 181–188. [Google Scholar] [CrossRef]
- Han, K.; Dai, L.; Hong, N. Research progress on deactivation of fixed bed residue hydrogenating catalysts at the initial stage of operation. Chem. Ind. Eng. Prog. 2017, 36, 211–220. [Google Scholar]
- Al-Dalama, K.; Stanislaus, A. Comparison between deactivation pattern of catalysts in fixed-bed and ebullating-bed residue hydroprocessing units. Chem. Eng. J. 2006, 120, 33–42. [Google Scholar] [CrossRef]
- Alvarez, A.; Ancheyta, J.; Centeno, G.; Marroquin, G. A modeling study of the effect of reactor configuration on the cycle length of heavy oil fixed-bed hydroprocessing. Fuel 2011, 90, 3551–3560. [Google Scholar] [CrossRef]
- Ramirez, S.; Leyva, C.; Ancheyta, J.; Centeno, G. On the importance of calculating fresh-basis catalyst composition from spent catalyst analysis. Fuel 2009, 88, 2311–2314. [Google Scholar] [CrossRef]
- Torres-Mancera, P.; Rayo, P.; Ancheyta, J.; Marroquin, G.; Centeno, G.; Alonso, F. Catalyst deactivation pattern along a residue hydrotreating bench-scale reactor. Catal. Today 2014, 220–222, 153–158. [Google Scholar] [CrossRef]
- Ancheyta, J.; López Isunza, F. Analysis of Deactivation Models Based on Time-on-Stream (TOS) Theory for Fluid Catalytic Cracking Process. J. Mex. Chem. Soc. 2000, 44, 183–187. [Google Scholar]
- Khang, S.J.; Mosby, J.F. Catalyst deactivation due to deposition of reaction products in macropores during hydroprocessing of petroleum residuals. Ind. Eng. Chem. Process Des. Dev. 1986, 25, 437–442. [Google Scholar] [CrossRef]
- Sámano, V.; Rana, S.M.; Ancheyta, J. An easy approach based on textural properties to evaluate catalyst deactivation during heavy oil hydrotreating. Catal. Commun. 2020, 133, 105823. [Google Scholar] [CrossRef]
- Sámano, V.; Tirado, A.; Félix, G.; Ancheyta, J. Revisiting the importance of appropriate parameter estimation based on sensitivity analysis for developing kinetic models. Fuel 2020, 267, 117113. [Google Scholar] [CrossRef]
- Maritnez, J.; Ancheyta, J. Modeling the kinetics of parallel thermal and catalytic hydrotreating of heavy oil. Fuel 2014, 138, 27–36. [Google Scholar] [CrossRef]
- Kohli, K.; Prajapati, R.; Maity, S.K.; Sau, M.; Sharma, B.K. Deactivation of a hydrotreating catalyst during hydroprocessing of synthetic crude by metal bearing compounds. Fuel 2019, 243, 579–589. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalysts | FEM-10 | FES-30 |
|---|---|---|
| Type | Demetallization | Desulfurization |
| Shape | Spherical | Spherical |
| Diameter of particles (mm) | 0.4–0.5 | 0.4–0.5 |
| Average pore size (nm) | 22 | 12 |
| VPore (mL/g) | 0.7 | 0.613 |
| SBET (m2/g) | 120 | 182 |
| Mo (wt.%) Ni (wt.%) | 6.54 13.46 | 1.97 2.81 |
| API gravity | 9.56 |
| Total sulfur (wt.%) | 5.72 |
| CCR (wt.%) | 22.85 |
| Ni (μg/g) | 43.35 |
| V (μg/g) | 141.5 |
| Distillation (°C) | |
| IBP/10 w% | 349.6/527.0 |
| 30/50 w% | 588.6/641.2 |
| 70/80.8 w% | 706.2/749.8 |
| Item | Temperature (°C) | Pressure (MPa) | LHSV (h−1) | H2/Oil Ratio (std m3/m3) | |
|---|---|---|---|---|---|
| 1st Reactor | 2nd Reactor | ||||
| 1 | 395 | 405 | 15.0 | 0.8 | 500 |
| 2 | 400 | 410 | 15.0 | 0.8 | 500 |
| 3 | 405 | 415 | 15.0 | 0.8 | 500 |
| 4 | 410 | 420 | 15.0 | 0.8 | 500 |
| 5 | 420 | 430 | 15.0 | 0.8 | 500 |
| Parameters | HDCCR | HDS | HDNi | HDV |
|---|---|---|---|---|
| 9.4324 × 106 | 2.0883 × 105 | 3.1355 × 105 | 3.2462 × 105 | |
| Ea,i/KJ·mol−1 | 112.83 | 89.01 | 84.05 | 68.76 |
| 1.199 | 0.83655 | 1.139463 | 1.0058 | |
| 0.082969 | 1.5838 | 6.6116 | 0.93829 | |
| 5.27834 | 2.3581 | 2.7420 | 1.6161 | |
| 4.3251 | 0.76 | 1.69649 | 0.015411 |
| Parameters | HDCCR | HDS | HDNi | HDV |
|---|---|---|---|---|
| 4.63227 × 105 | 3.16620 × 105 | 1.1392 × 105 | 6.0785 × 104 | |
| Ea,i/KJ·mol−1 | 145.25 | 129.19 | 128.83 | 114.40 |
| 0.98219 | 0.80902 | 1.2696 | 0.794727 | |
| 0.053560 | 0.088090 | 0.114588 | 0.050319 | |
| 1.8923 | 1.2493 | 1.854820 | 0.60018 | |
| 0.41896 | 0.054360 | 0.70240 | 19.986 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, K.; Cao, Y.; Zhu, H.; Ge, H.; Yang, T.; Zhou, Z.; Cheng, Z. Experimental Determination on the Deactivation Kinetics of Residue Hydroprocessing in a Two-Stage Fixed Bed Loaded with HDM and HDS Catalysts. Processes 2022, 10, 1822. https://doi.org/10.3390/pr10091822
Chen K, Cao Y, Zhu H, Ge H, Yang T, Zhou Z, Cheng Z. Experimental Determination on the Deactivation Kinetics of Residue Hydroprocessing in a Two-Stage Fixed Bed Loaded with HDM and HDS Catalysts. Processes. 2022; 10(9):1822. https://doi.org/10.3390/pr10091822
Chicago/Turabian StyleChen, Kaixuan, Yankai Cao, Huihong Zhu, Hailong Ge, Tao Yang, Zhiming Zhou, and Zhenmin Cheng. 2022. "Experimental Determination on the Deactivation Kinetics of Residue Hydroprocessing in a Two-Stage Fixed Bed Loaded with HDM and HDS Catalysts" Processes 10, no. 9: 1822. https://doi.org/10.3390/pr10091822
APA StyleChen, K., Cao, Y., Zhu, H., Ge, H., Yang, T., Zhou, Z., & Cheng, Z. (2022). Experimental Determination on the Deactivation Kinetics of Residue Hydroprocessing in a Two-Stage Fixed Bed Loaded with HDM and HDS Catalysts. Processes, 10(9), 1822. https://doi.org/10.3390/pr10091822