Hinokitiol Dysregulates Metabolism of Carcinoma Cell Lines and Induces Downregulation of HPV16E6 and E7 Oncogenes and p21 Upregulation in HPV Positive Cell Lines


Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Lines and Reagents
2.2. MTT-Assay for Cell Metabolic Activity and Cell Viability
2.3. Thymidine Incorporation Assay for Cell Proliferation
2.4. Cell Cycle Assay
2.5. RNA Extraction and qPCR
2.6. Statistical Analyses
3. Results
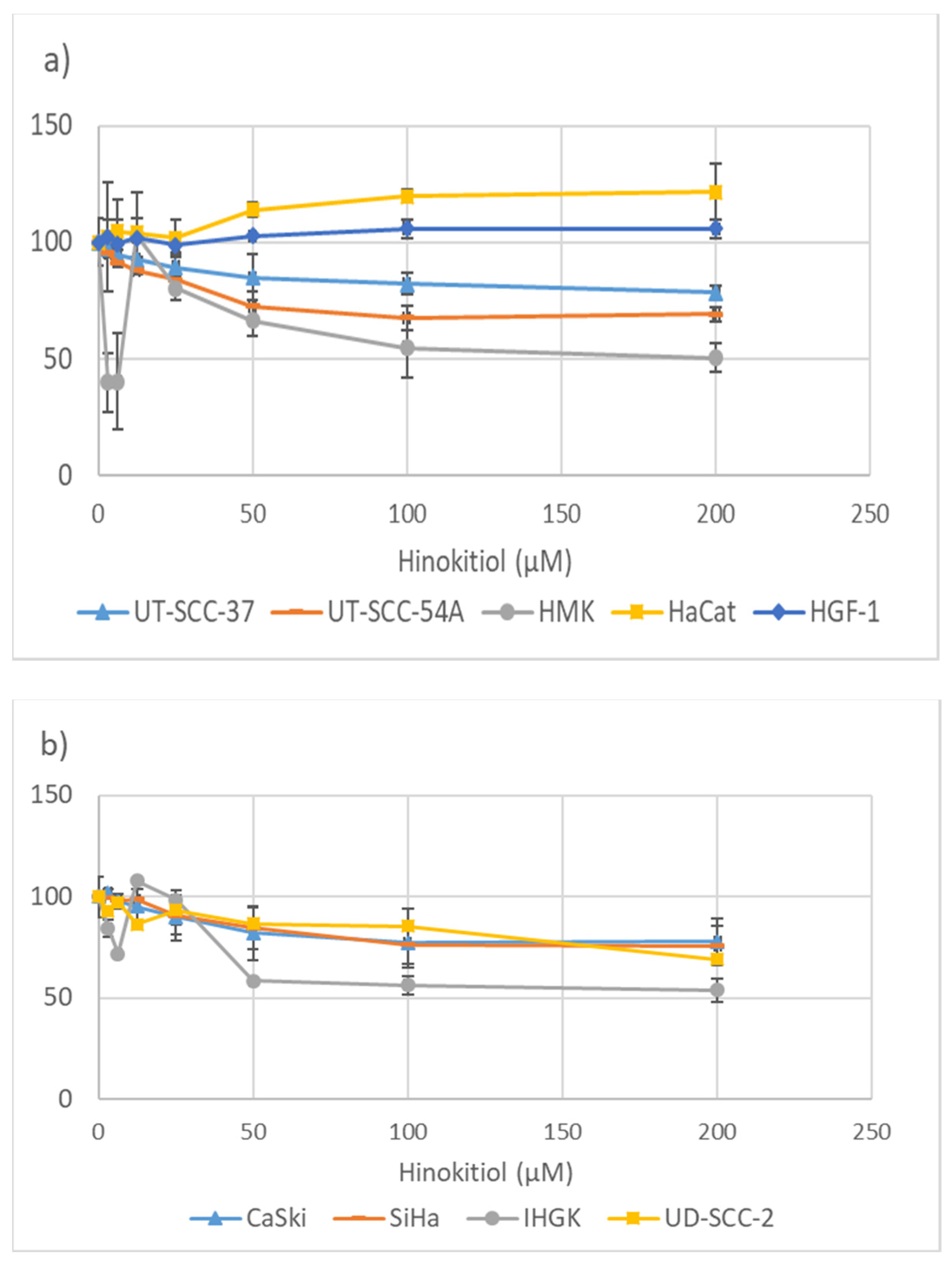
3.1. Hinokitiol and the Metabolic Activity of the Cells
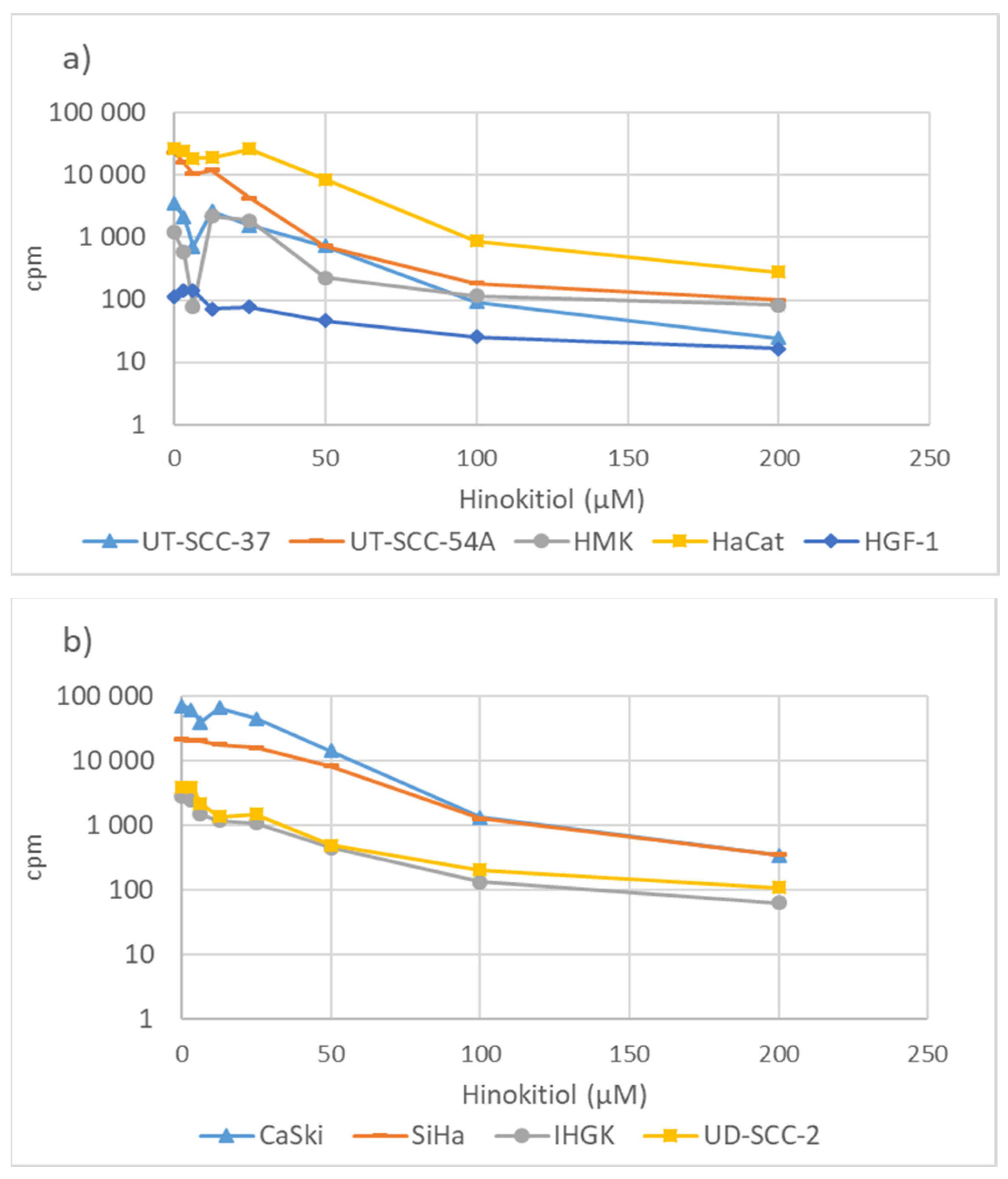
3.2. Hinokitiol and DNA Synthesis
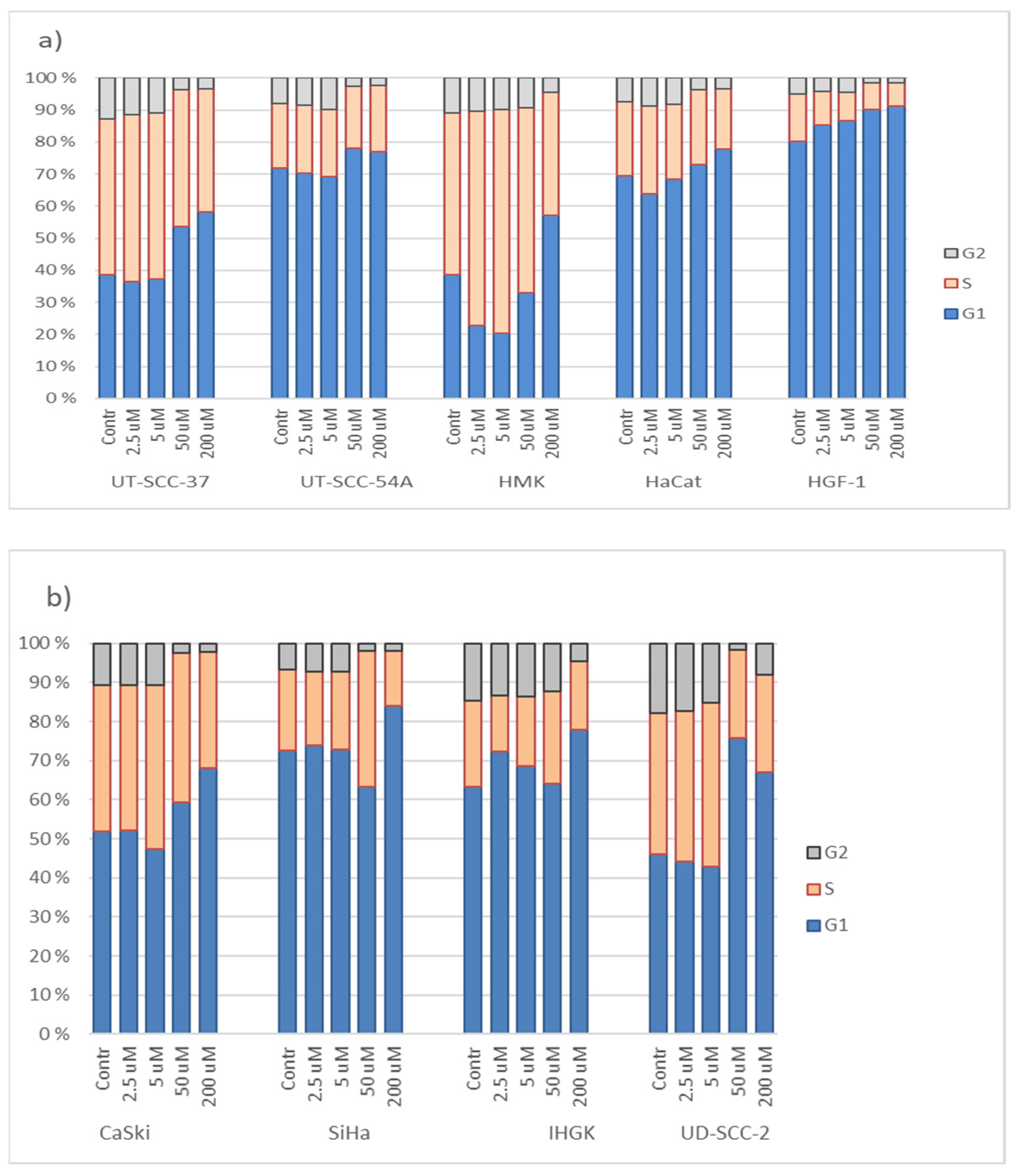
3.3. Hinokitiol and Cell Cycling
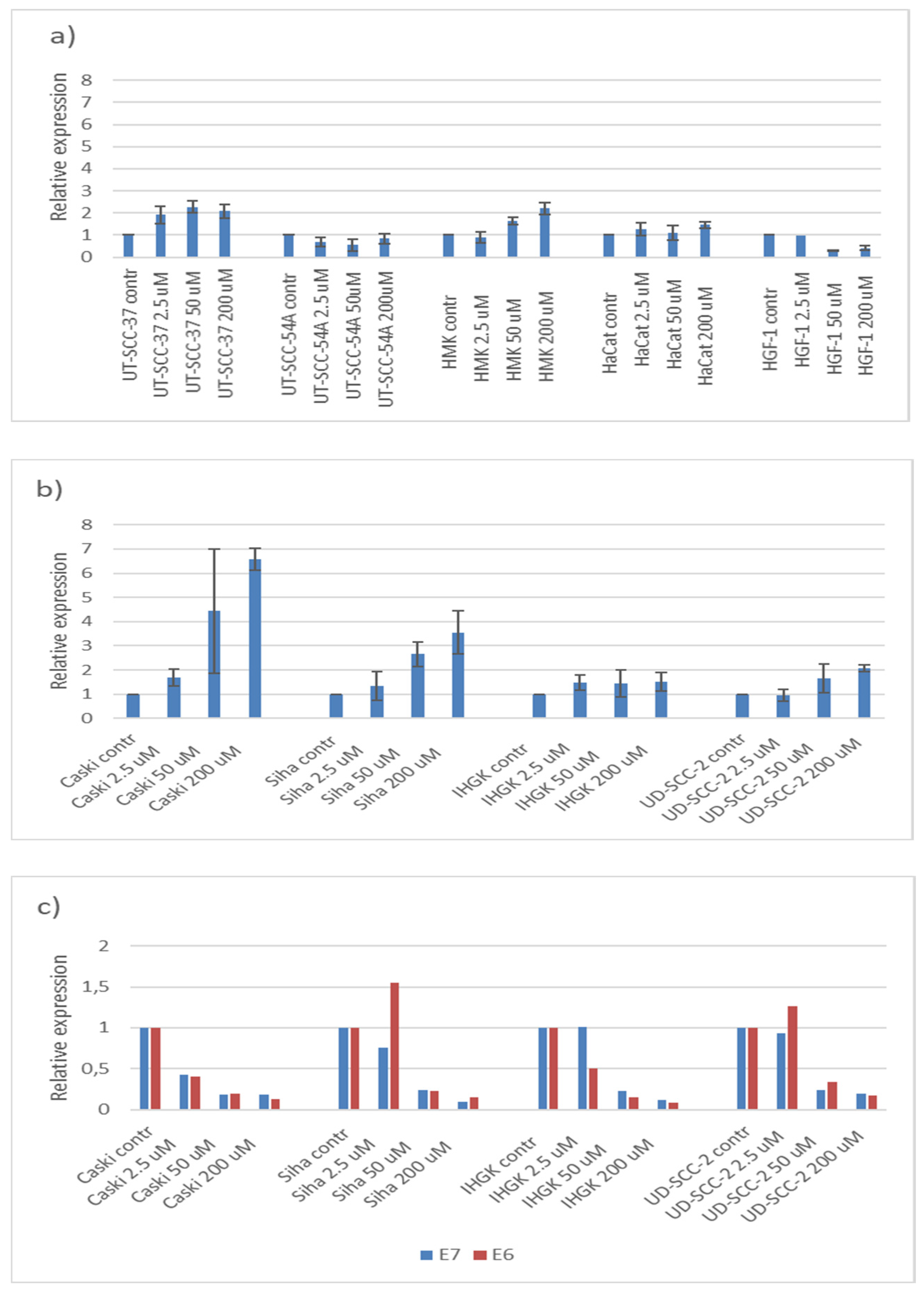
3.4. Hinokitiol, p21 and HPV16E6 and E7
4. Discussion
5. Conclusions
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Statement on Ethics
References
- Komatsu, K. Professor Tetsuo Nozoe and my tropylium ion chemistry. Chem. Rec. 2015, 15, 160–174. [Google Scholar] [CrossRef] [PubMed]
- Komaki, N.; Watanabe, T.; Ogasawara, A.; Sato, N.; Mikami, T.; Matsumoto, T. Antifungal mechanism of hinokitiol against Candida albicans. Biol. Pharm. Bull. 2008, 31, 735–737. [Google Scholar] [CrossRef] [PubMed]
- National Library of Medicine. National Center for Biotechnology Information. PubChem. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/Hinokitiol (accessed on 15 February 2022).
- Domon, H.; Hiyoshi, T.; Maekawa, T.; Yonezawa, D.; Tamura, H.; Kawabata, S.; Yanagihara, K.; Kimura, O.; Kunitomo, E.; Terao, Y. Antibacterial activity of hinokitiol against both antibiotic-resistant and -susceptible pathogenic bacteria that predominate in the oral cavity and upper airways. Microbiol. Immunol. 2019, 63, 213–222. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.J.; Lee, M.W.; Choi, J.S.; Lee, S.G.; Park, J.Y.; Kim, S.W. Inhibitory activity of hinokitiol against biofilm formation in fluconazole-resistant Candida species. PLoS ONE 2017, 12, e0171244. [Google Scholar] [CrossRef]
- Krenn, B.M.; Gaudernak, E.; Holzer, B.; Lanke, K.; Kuppeveld, F.J.M.V.; Seipelt, J. Antiviral activity of the zinc ionophores pyrithione and hinokitiol against picornavirus infections. J. Virol. 2009, 83, 58–64. [Google Scholar] [CrossRef]
- Shih, Y.H.; Chang, K.W.; Hsia, S.M.; Yu, C.C.; Fuh, L.J.; Chi, T.Y.; Shieh, T.M. In vitro antimicrobial and anticancer potential of hinokitiol against oral pathogens and oral cancer cell lines. Microbial. Res. 2013, 168, 254–262. [Google Scholar] [CrossRef]
- Ye, J.; Xu, Y.F.; Lou, L.X.; Jin, K.; Miao, Q.; Ye, X. Anti-inflammatory effects of hinokitiol on human corneal epithelial cells: An in vitro study. Eye 2015, 29, 964–971. [Google Scholar] [CrossRef]
- Budihas, S.R.; Gorshkova, I.; Gaidamakov, S.; Wamiru, A.; Bona, M.K.; Parniak, M.A.; Crouch, R.J.; McMahon, J.B.; Beutler, J.A.; Grice, S.F.J.L. Selective inhibition of HIV-1 reverse transcriptase-associated ribonuclease H activity by hydroxylated tropolones. Nucleic Acids Res. 2005, 33, 1249–1256. [Google Scholar] [CrossRef]
- Hu, Y.; Cheng, X.; Cao, F.; Huang, A.; Tavis, J.E. β-Thujaplicinol inhibits hepatitis B virus replication by blocking the viral ribonuclease H activity. Antiviral Res. 1998, 99, 221–229. [Google Scholar] [CrossRef]
- Miyamoto, D.; Kusagaya, Y.; Endo, N.; Sometani, A.; Takeo, S.; Suzuki, T.; Arima, Y.; Nakajima, K.; Suzuki, Y. β-Thujaplicin-copper chelates inhibit replication of human influenza viruses. Antiviral Res. 1998, 39, 89–100. [Google Scholar] [CrossRef]
- Hachlafi, N.E.; Lakdar, F.; Khouchlaa, A.; Bakrim, S.; Omari, N.E.; Balahbib, A.; Shariati, M.A.; Zengin, G.; Fikti-Benbrahim, K.; Orlando, G.; et al. Health Benefits and Pharmacological Properties of Hinokitiol. Processes 2021, 9, 1680. [Google Scholar] [CrossRef]
- Ko, J.; Bao, C.; Park, H.; Kim, M.; Choi, H.Y.; Kim, Y. β-Thujaplicin modulates estrogen receptor signaling and inhibits proliferation of human breast cancer cells. Biosci. Biotechnol. Biochem. 2015, 79, 1011–1017. [Google Scholar] [CrossRef][Green Version]
- Liu, S.; Yamauchi, H. Hinokitiol, a metal chelator derived from natural plants, suppresses cell growth and disrupts androgen receptor signaling in prostate carcinoma cell lines. Biochem. Biophys. Res. Commun. 2006, 351, 26–32. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, W.C.; Liao, Y.W.; Chen, P.N.; Lu, K.H.; Yu, C.C.; Hsieh, P.L. Hinokitiol suppresses cancer stemness and oncogenicity in glioma stem cells by Nrf2 regulation. Cancer Chemother. Pharmacol. 2017, 80, 411–419. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.C.; Chen, B.K.; Chen, P.H.; Chen, L.C. Hinokitiol induces cell death and inhibits epidermal growth factor-induced cell migration and signaling pathways in human cervical adenocarcinoma. Taiwan. J. Obstet. Gynecol. 2020, 59, 698–705. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; He, J.; Ye, X.; Zhu, J.; Hu, X.; Shen, M. β-Thujaplicin induces autophagic cell death, apoptosis, and cell cycle arrest through ROS-mediated Akt and p38/ERK MAPK signaling in human hepatocellular carcinoma. Cell Death Dis. 2019, 10, 255. [Google Scholar] [CrossRef]
- Doorbar, J. Molecular biology of human papillomavirus infection and cervical cancer. Clin. Sci. 2006, 110, 525–541. [Google Scholar] [CrossRef]
- Syrjänen, S.; Syrjänen, K. HPV in Head and Neck Carcinomas: Different HPV Profiles in Oropharyngeal Carcinomas—Why? Acta Cytol. 2019, 63, 124–142. [Google Scholar] [CrossRef]
- Fotopoulou, T.; Ćirić, A.; Kritsi, E.; Calhelha, R.C.; Ferreira, I.C.F.R.; Soković, M.; Zoumpoulakis, P.; Koufaki, M. Antimicrobial/Antibiofilm Activity and Cytotoxic Studies of β-Thujaplicin Derivatives. Arch. Pharm. 2016, 349, 698–709. [Google Scholar] [CrossRef]
- Balló, H.; Koldovsky, P.; Hoffmann, T.; Balz, V.; Hildebrandt, B.; Gerharz, C.D.; Bier, H. Establishment and characterization of four cell lines derived from human head and neck squamous cell carcinomas for an autologous tumor-fibroblast in vitro model. Anticancer Res. 1999, 19, 3827–3836. [Google Scholar]
- Ruutu, M.; Johansson, B.; Grenman, R.; Syrjänen, S. Two different global gene expression profiles in cancer cell lines established from etiologically different oral carcinomas. Oncol. Rep. 2005, 14, 1511–1517. [Google Scholar] [CrossRef] [PubMed]
- Oda, D.; Bigler, L.; Lee, P.; Blanton, R. HPV immortalization of human oral epithelial cells: A model for carcinogenesis. Exp. Cell Res. 1996, 226, 164–169. [Google Scholar] [CrossRef] [PubMed]
- Mäkelä, M.; Larjava, H.; Pirilä, E.; Maisi, P.; Salo, T.; Sorsa, T.; Uitto, V.J. Matrix metalloproteinase 2 (gelatinase A) is related to migration of keratinocytes. Exp. Cell Res. 1999, 251, 67–78. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Yamauchi, H. p27-Associated G1 arrest induced by hinokitiol in human malignant melanoma cells is mediated via down-regulation of pRb, Skp2 ubiquitin ligase, and impairment of Cdk2 function. Cancer Lett. 2009, 286, 240–249. [Google Scholar] [CrossRef] [PubMed]
- Turunen, A.; Syrjänen, S. Extracellular calcium regulates keratinocyte proliferation and HPV 16 E6 RNA expression in vitro. Apmis 2014, 122, 781–789. [Google Scholar] [CrossRef][Green Version]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCt method. Methods 2016, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Viale, A.; Franco, F.D.; Orleth, A.; Cambiaghi, V.; Giuliani, V.; Bossi, D.; Ronchini, C.; Ronzoni, S.; Muradore, I.; Monestiroli, S.; et al. Cell-cycle restriction limits DNA damage and maintains self-renewal of leukaemia stem cells. Nature 2009, 457, 51–56. [Google Scholar] [CrossRef]
- Basukala, O.; Banks, L. The Not-So-Good, the Bad and the Ugly: HPV E5, E6 and E7 Oncoproteins in the Orchestration of Carcinogenesis. Viruses 2021, 13, 1892. [Google Scholar] [CrossRef]
- Vats, A.; Trejo-Cerro, O.; Thomas, M.; Banks, L. Human papillomavirus E6 and E7: What remains? Tumour Virus Res. 2021, 11, 200213. [Google Scholar] [CrossRef]
- Tu, D.G.; Yu, Y.; Lee, C.H.; Kuo, Y.L.; Lu, Y.C.; Tu, C.W.; Chang, W.W. Hinokitiol inhibits vasculogenic mimicry activity of breast cancer stem/progenitor cells through proteasome-mediated degradation of epidermal growth factor receptor. Oncol. Lett. 2016, 11, 2934–2940. [Google Scholar] [CrossRef]
- Li, J.Y.; Liu, C.P.; Shiao, W.C.; Jayakumar, T.; Li, Y.S.; Chang, N.C.; Huang, S.Y.; Hsieh, C.-Y. Inhibitory effect of PDGF-BB and serum-stimulated responses in vascular smooth muscle cell proliferation by hinokitiol via up-regulation of p21 and p53. Arch. Med. Sci. 2018, 14, 579–587. [Google Scholar] [CrossRef] [PubMed]
- Li, L.H.; Wu, P.; Lee, J.Y.; Li, P.R.; Hsieh, W.Y.; Ho, C.C.; Ho, C.L.; Chen, W.J.; Wang, C.C.; Yen, M.-Y.; et al. Hinokitiol induces DNA damage and autophagy followed by cell cycle arrest and senescence in gefitinib-resistant lung adenocarcinoma cells. PLoS ONE 2019, 9, e104203. [Google Scholar] [CrossRef] [PubMed]
- Doorbar, J.; Egawa, N.; Griffin, H.; Kranjec, C.; Murakami, I. Human papillomavirus molecular biology and disease association. Rev. Med. Virol. 2015, 25, 2–23. [Google Scholar] [CrossRef]
- Wang-Johanning, F.; Lu, D.W.; Wang, Y.; Johnson, M.R.; Johanning, G.L. Quantitation of human papillomavirus 16 E6 and E7 DNA and RNA in residual material from ThinPrep Papanicolaou tests using real-time polymerase chain reaction analysis. Cancer 2002, 94, 2199–2210. [Google Scholar] [CrossRef]
- Shamloo, B.; Usluer, S. p21 in cancer research. Cancers 2019, 11, 1178. [Google Scholar] [CrossRef]
- Marhenke, S.; Buitrago-Molina, L.B.; Endig, J.; Orlik, J.; Schweitzer, N.; Klett, S.; Longerich, T.; Geffers, R.; Muñoz, A.S.S.; Dorrell, C.; et al. p21 promotes sustained liver regeneration and hepatocarcinogenesis in chronic cholestatic liver injury. Gut 2014, 63, 1501–1512. [Google Scholar] [CrossRef] [PubMed]
- Wei, K.C.; Chen, R.F.; Chen, Y.F.; Lin, C.H. Hinokitiol suppresses growth of B16 melanoma by activating ERK/MKP3/proteosome pathway to downregulate survivin expression. Toxicol. Appl. Pharmacol. 2019, 366, 35–45. [Google Scholar] [CrossRef] [PubMed]
- Ruutu, M.; Peitsaro, P.; Johansson, B.; Syrjänen, S. Transcriptional profiling of a human papillomavirus 33-positive squamous epithelial cell line which acquired a selective growth advantage after viral integration. Int. J. Cancer 2002, 100, 318–326. [Google Scholar] [CrossRef] [PubMed]
- Iha, K.; Suzuki, N.; Yoneda, M.; Takeshita, T.; Hirofuji, T. Effect of mouth cleaning with hinokitiol-containing gel on oral malodor: A randomized, open-label pilot study. Oral Surg. Oral Med. Oral Pathol. Oral Radiol. 2013, 116, 433–439. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Line | Origin | Organ | HPV Status HPV Copies/Cell | Acquired from or Reference |
|---|---|---|---|---|
| CaSki | Cervical carcinoma | uteri | HPV16 600 copies | ATCC (CRL-1550) |
| SiHa | Squamous cell carcinoma | cervix uteri | HPV16 2 copies | ATCC (HTB-35) |
| UD-SCC-2 | Squamous cell carcinoma | hypopharynx | HPV16 600 copies | Ballo et al., 1999 [21] |
| UT-SCC-37 | Squamous cell carcinoma | gingiva | HPV negative | Gift from Prof. R. Grenman. Turku University Hospital, Turku, Finland Ruutu et al., 2005 [22] |
| UT-SCC-54A | Squamous cell carcinoma | buccal mucosa | HPV negative | Gift from Prof. R. Grenman. Turku University Hospital, Finland Ruutu et al., 2005 [22] |
| IHGK | Keratinocyte | gingiva | transformed with HPV16 E6/E7 | Oda 1996 [23] |
| HMK | Spontaneously immortalized keratinocyte | gingiva | HPV negative | Mäkelä 1999 [24] |
| HaCat | Spontaneously immortalized keratinocyte | skin | HPV negative | ATCC |
| HGF-1 | Fibroblast | gingiva | HPV negative | Institute of Dentistry, University of Turku, Finland |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sääskilahti, E.; Syrjänen, S.; Loimaranta, V.; Louvanto, K. Hinokitiol Dysregulates Metabolism of Carcinoma Cell Lines and Induces Downregulation of HPV16E6 and E7 Oncogenes and p21 Upregulation in HPV Positive Cell Lines. Processes 2022, 10, 736. https://doi.org/10.3390/pr10040736
Sääskilahti E, Syrjänen S, Loimaranta V, Louvanto K. Hinokitiol Dysregulates Metabolism of Carcinoma Cell Lines and Induces Downregulation of HPV16E6 and E7 Oncogenes and p21 Upregulation in HPV Positive Cell Lines. Processes. 2022; 10(4):736. https://doi.org/10.3390/pr10040736
Chicago/Turabian StyleSääskilahti, Erika, Stina Syrjänen, Vuokko Loimaranta, and Karolina Louvanto. 2022. "Hinokitiol Dysregulates Metabolism of Carcinoma Cell Lines and Induces Downregulation of HPV16E6 and E7 Oncogenes and p21 Upregulation in HPV Positive Cell Lines" Processes 10, no. 4: 736. https://doi.org/10.3390/pr10040736
APA StyleSääskilahti, E., Syrjänen, S., Loimaranta, V., & Louvanto, K. (2022). Hinokitiol Dysregulates Metabolism of Carcinoma Cell Lines and Induces Downregulation of HPV16E6 and E7 Oncogenes and p21 Upregulation in HPV Positive Cell Lines. Processes, 10(4), 736. https://doi.org/10.3390/pr10040736