
Transgelin Contributes to a Poor Response of Metastatic Renal Cell Carcinoma to Sunitinib Treatment

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Patients
2.2. Proteomics
2.2.1. Lysis of Tissue Samples
2.2.2. Preparing Samples for Mass Spectrometry (MS) Analysis
2.2.3. Peptide Desalting Prior to LC-MS/MS
2.2.4. Liquid Chromatography (LC)-MS Analysis of Peptides
2.2.5. MS Data Analysis
2.2.6. Functional Analysis of Identified Proteins
2.3. Cellular Studies
2.3.1. Cell Cultures and Cultivation
2.3.2. Immunoblotting
2.3.3. CRISPR/Cas9 Gene Editing
2.3.4. Monoclonal Selection of CRISPR Modified Cells and Their Cultivation
2.3.5. siRNA Transfection, Cultivation of siRNA-Transfected Cells, and Scratch Assay
2.4. Statistical Analysis
3. Results
3.1. Spectral Library and Mass Spectrometry Data Matrix
3.2. Proteins Associated with Sunitinib Non-Responders
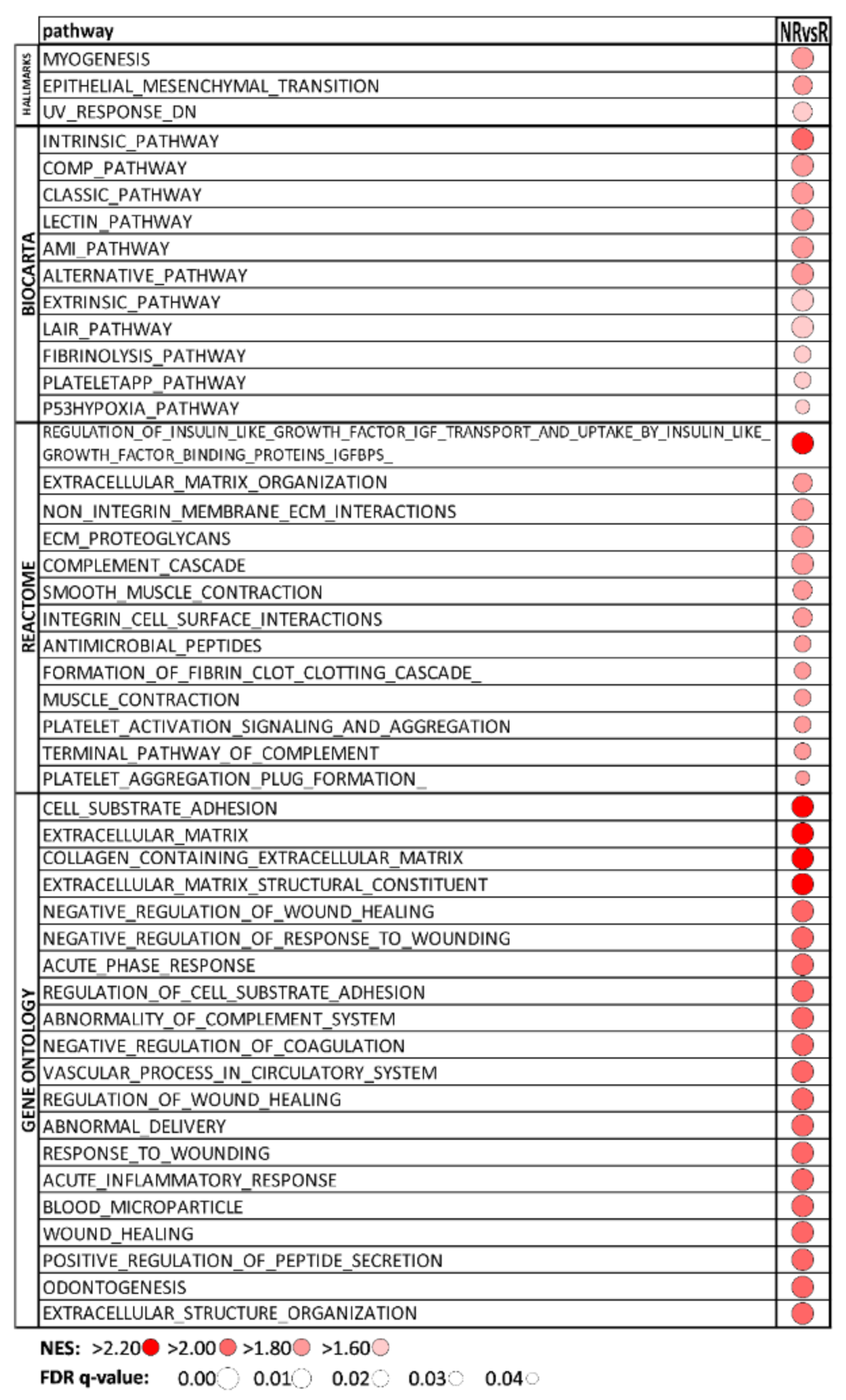
3.3. Hallmark and BIOCARTA Pathways, Reactome, and GO Associated with Sunitinib Non-Responders
3.4. Proteins Differentially Abundant in mccRCC Tumors vs. Adjacent Non-Cancerous Tissue
3.5. Pathways Enriched in mccRCC Tumors Compared to Non-Cancerous Tissues
3.6. CRISPR/Cas9 Knock-Down Shows That Transgelin Is Essential for the Proliferation of 786-0 RCC Cells
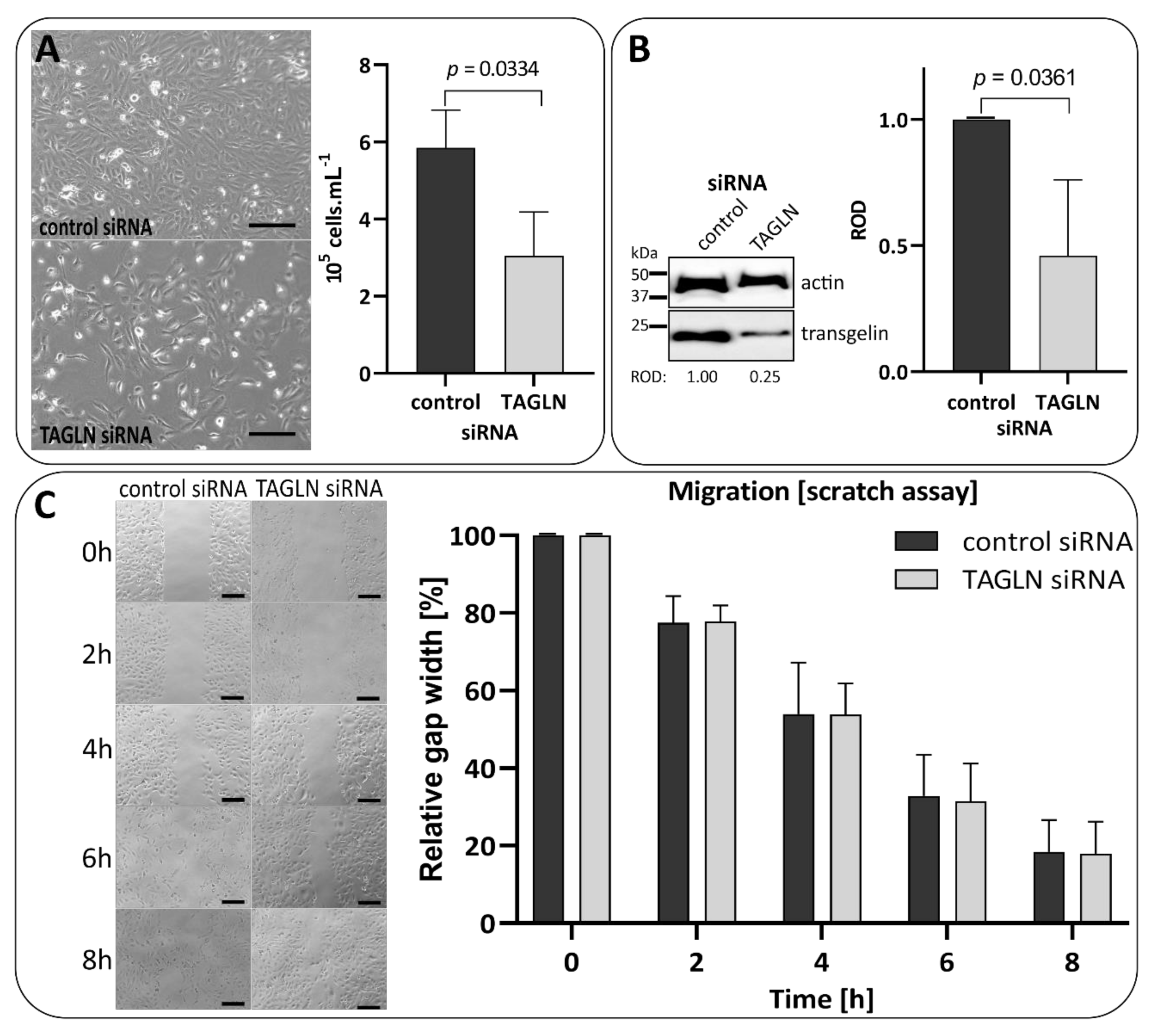
3.7. Transient Silencing of Transgelin Slows the Proliferation of 786-0 Cells Down
4. Discussion
4.1. Transgelin Is the Key Identified Protein in Intrinsic Sunitinib Resistance through Proliferation Support and EMT
4.2. Other Relevant Proteins in Sunitinib Non-Responding Tumors
4.3. Role of Enriched Signaling Pathways in Sunitinib Resistance
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [Green Version]
- Rini, B.I.; Campbell, S.C.; Escudier, B. Renal cell carcinoma. Lancet 2009, 373, 1119–1132. [Google Scholar] [CrossRef]
- Cohen, H.T.; McGovern, F.J. Renal-cell carcinoma. N. Engl. J. Med. 2005, 353, 2477–2490. [Google Scholar] [CrossRef] [Green Version]
- Doberstein, K.; Wieland, A.; Lee, S.B.; Blaheta, R.A.; Wedel, S.; Moch, H.; Schraml, P.; Pfeilschifter, J.; Kristiansen, G.; Gutwein, P. L1-CAM expression in ccRCC correlates with shorter patients survival times and confers chemoresistance in renal cell carcinoma cells. Carcinogenesis 2011, 32, 262–270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, S.; Zhang, H.; Chong, Y.; Guan, B.; Guo, P. YAP Promotes VEGFA Expression and Tumor Angiogenesis Though Gli2 in Human Renal Cell Carcinoma. Arch. Med. Res. 2019, 50, 225–233. [Google Scholar] [CrossRef]
- Goebell, P.J.; Ivanyi, P.; Bedke, J.; Bergmann, L.; Berthold, D.; Boegemann, M.; Busch, J.; Doehn, C.; Krege, S.; Retz, M.; et al. Consensus paper: Current state of first- and second-line therapy in advanced clear-cell renal cell carcinoma. Future Oncol. 2020, 16, 2307–2328. [Google Scholar] [CrossRef]
- Abrams, T.J.; Lee, L.B.; Murray, L.J.; Pryer, N.K.; Cherrington, J.M. SU11248 inhibits KIT and platelet-derived growth factor receptor beta in preclinical models of human small cell lung cancer. Mol. Cancer Ther. 2003, 2, 471–478. [Google Scholar] [PubMed]
- Mendel, D.B.; Laird, A.D.; Xin, X.; Louie, S.G.; Christensen, J.G.; Li, G.; Schreck, R.E.; Abrams, T.J.; Ngai, T.J.; Lee, L.B.; et al. In vivo antitumor activity of SU11248, a novel tyrosine kinase inhibitor targeting vascular endothelial growth factor and platelet-derived growth factor receptors: Determination of a pharmacokinetic/pharmacodynamic relationship. Clin. Cancer Res. 2003, 9, 327–337. [Google Scholar] [PubMed]
- Goodman, V.L.; Rock, E.P.; Dagher, R.; Ramchandani, R.P.; Abraham, S.; Gobburu, J.V.; Booth, B.P.; Verbois, S.L.; Morse, D.E.; Liang, C.Y.; et al. Approval Summary: Sunitinib for the Treatment of Imatinib Refractory or Intolerant Gastrointestinal Stromal Tumors and Advanced Renal Cell Carcinoma. Clin. Cancer Res. 2007, 13, 1367–1373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Motzer, R.J.; Hutson, T.; Tomczak, P.; Michaelson, D.; Bukowski, R.M.; Oudard, S.; Negrier, S.; Szczylik, C.; Pili, R.; Bjarnason, G.A.; et al. Overall Survival and Updated Results for Sunitinib Compared With Interferon Alfa in Patients With Metastatic Renal Cell Carcinoma. J. Clin. Oncol. 2009, 27, 3584–3590. [Google Scholar] [CrossRef] [PubMed]
- Motzer, R.J.; Hutson, T.; Tomczak, P.; Michaelson, D.; Bukowski, R.M.; Rixe, O.; Oudard, S.; Negrier, S.; Szczylik, C.; Kim, S.T.; et al. Sunitinib versus Interferon Alfa in Metastatic Renal-Cell Carcinoma. N. Engl. J. Med. 2007, 356, 115–124. [Google Scholar] [CrossRef]
- Morais, C. Sunitinib resistance in renal cell carcinoma. J. Kidney Cancer VHL 2014, 1, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Buczek, M.; Escudier, B.; Bartnik, E.; Szczylik, C.; Czarnecka, A. Resistance to tyrosine kinase inhibitors in clear cell renal cell carcinoma: From the patient’s bed to molecular mechanisms. Biochim. Biophys. Acta (BBA)—Rev. Cancer 2014, 1845, 31–41. [Google Scholar] [CrossRef] [PubMed]
- Busch, J.; Seidel, C.; Weikert, S.; Wolff, I.; Kempkensteffen, C.; Weinkauf, L.; Hinz, S.; Magheli, A.; Miller, K.; Grünwald, V. Intrinsic resistance to tyrosine kinase inhibitors is associated with poor clinical outcome in metastatic renal cell carcinoma. BMC Cancer 2011, 11, 295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, J.L.; Rak, J.W.; Coomber, B.L.; Hicklin, D.J.; Kerbel, R.S. Effect of p53 Status on Tumor Response to Antiangiogenic Therapy. Science 2002, 295, 1526–1528. [Google Scholar] [CrossRef]
- Loges, S.; Mazzone, M.; Hohensinner, P.; Carmeliet, P. Silencing or Fueling Metastasis with VEGF Inhibitors: Antiangiogenesis Revisited. Cancer Cell 2009, 15, 167–170. [Google Scholar] [CrossRef] [Green Version]
- Minardi, D.; Quaresima, L.; Santoni, M.; Bianconi, M.; Scartozzi, M.; Cascinu, S.; Muzzonigro, G. Recent Aspects of Sunitinib Therapy in Patients with Metastatic Clear-Cell Renal Cell Carcinoma: A Systematic Review of the Literature. Curr. Urol. Rep. 2015, 16, 3. [Google Scholar] [CrossRef] [PubMed]
- Gotink, K.J.; Broxterman, H.J.; Labots, M.; De Haas, R.R.; Dekker, H.; Honeywell, R.J.; Rudek, M.A.; Beerepoot, L.V.; Musters, R.J.; Jansen, G.; et al. Lysosomal Sequestration of Sunitinib: A Novel Mechanism of Drug Resistance. Clin. Cancer Res. 2011, 17, 7337–7346. [Google Scholar] [CrossRef] [Green Version]
- Harada, K.-I.; Miyake, H.; Kusuda, Y.; Fujisawa, M. Expression of epithelial-mesenchymal transition markers in renal cell carcinoma: Impact on prognostic outcomes in patients undergoing radical nephrectomy. BJU Int. 2012, 110, E1131–E1137. [Google Scholar] [CrossRef]
- Huang, D.; Ding, Y.; Zhou, M.; Rini, B.I.; Petillo, D.; Qian, C.-N.; Kahnoski, R.; Futreal, P.A.; Furge, K.A.; Teh, B.T. Interleukin-8 Mediates Resistance to Antiangiogenic Agent Sunitinib in Renal Cell Carcinoma. Cancer Res. 2010, 70, 1063–1071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sato, M.; Nakai, Y.; Nakata, W.; Yoshida, T.; Hatano, K.; Kawashima, A.; Fujita, K.; Uemura, M.; Takayama, H.; Nonomura, N. EMMPRIN Promotes Angiogenesis, Proliferation, Invasion and Resistance to Sunitinib in Renal Cell Carcinoma, and Its Level Predicts Patient Outcome. PLoS ONE 2013, 8, e74313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bielecka, Z.; Czarnecka, A.; Solarek, W.; Kornakiewicz, A.; Szczylik, C. Mechanisms of Acquired Resistance to Tyrosine Kinase Inhibitors in Clear-Cell Renal Cell Carcinoma (ccRCC). Curr. Signal Transduct. Ther. 2014, 8, 219–228. [Google Scholar] [CrossRef] [PubMed]
- Zama, I.N.; Hutson, T.E.; Elson, P.; Cleary, J.M.; Choueiri, T.K.; Heng, D.Y.; Ramaiya, N.; Michaelson, D.; Garcia, J.A.; Knox, J.J.; et al. Sunitinib rechallenge in metastatic renal cell carcinoma patients. Cancer 2010, 116, 5400–5406. [Google Scholar] [CrossRef] [PubMed]
- Bouchal, P.; Schubert, O.T.; Faktor, J.; Capkova, L.; Imrichova, H.; Zoufalova, K.; Paralova, V.; Hrstka, R.; Liu, Y.; Ebhardt, H.A.; et al. Breast Cancer Classification Based on Proteotypes Obtained by SWATH Mass Spectrometry. Cell Rep. 2019, 28, 832–843.e7. [Google Scholar] [CrossRef]
- Janacova, L.; Faktor, J.; Capkova, L.; Paralova, V.; Pospisilova, A.; Podhorec, J.; Ebhardt, H.A.; Hrstka, R.; Nenutil, R.; Aebersold, R.; et al. SWATH-MS Analysis of FFPE Tissues Identifies Stathmin as a Potential Marker of Endometrial Cancer in Patients Exposed to Tamoxifen. J. Proteome Res. 2020, 19, 2617–2630. [Google Scholar] [CrossRef] [PubMed]
- Wiśniewski, J.R.; Zougman, A.; Nagaraj, N.; Mann, M. Universal sample preparation method for proteome analysis. Nat. Methods 2009, 6, 359–362. [Google Scholar] [CrossRef]
- Maryáš, J.; Faktor, J.; Čápková, L.; Müller, P.; Skládal, P.; Bouchal, P. Pull-down Assay on Streptavidin Beads and Surface Plasmon Resonance Chips for SWATH-MS-based Interactomics. Cancer Genom. Proteom. 2018, 15, 395–404. [Google Scholar] [CrossRef] [Green Version]
- GeneArt Genomic Cleavage Detection Kit Manual. 10 May 2018. Available online: https://www.thermofisher.com/document-connect/document-connect.html?url=https%3A%2F%2Fassets.thermofisher.com%2FTFS-Assets%2FLSG%2Fmanuals%2FA24372_GeneArt_GenomicCleavage_Detect_Kit_man.pdf&title=R2VuZUFydCZyZWc7IEdlbm9taWMgQ2xlYXZhZ2UgRGV0ZWN0aW9uIEtpdCAtIFVzZXIgR3VpZGU= (accessed on 26 July 2021).
- Neely, B.A.; Wilkins, C.E.; Marlow, L.A.; Malyarenko, D.; Kim, Y.; Ignatchenko, A.; Sasinowska, H.; Sasinowski, M.; Nyalwidhe, J.O.; Kislinger, T.; et al. Proteotranscriptomic Analysis Reveals Stage Specific Changes in the Molecular Landscape of Clear-Cell Renal Cell Carcinoma. PLoS ONE 2016, 11, e0154074. [Google Scholar] [CrossRef]
- Song, Y.; Zhong, L.; Zhou, J.; Lu, M.; Xing, T.; Ma, L.; Shen, J. Data-Independent Acquisition-Based Quantitative Proteomic Analysis Reveals Potential Biomarkers of Kidney Cancer. Proteom.—Clin. Appl. 2017, 11, 1700066. [Google Scholar] [CrossRef]
- Sanjana, N.; Shalem, O.; Zhang, F. Improved vectors and genome-wide libraries for CRISPR screening. Nat. Methods 2014, 11, 783–784. [Google Scholar] [CrossRef] [Green Version]
- Shalem, O.; Sanjana, N.E.; Hartenian, E.; Shi, X.; Scott, D.A.; Mikkelsen, T.S.; Heckl, D.; Ebert, B.L.; Root, D.E.; Doench, J.; et al. Genome-Scale CRISPR-Cas9 Knockout Screening in Human Cells. Science 2013, 343, 84–87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dvorakova, M.; Nenutil, R.; Bouchal, P. Transgelins, cytoskeletal proteins implicated in different aspects of cancer development. Expert Rev. Proteom. 2014, 11, 149–165. [Google Scholar] [CrossRef] [PubMed]
- Gerolymos, M.; Karagianni, F.; Papasotiriou, M.; Kalliakmani, P.; Sotsiou, F.; Charonis, A.; Goumenos, D. Expression of Transgelin in Human Glomerulonephritis of Various Etiology. Nephron Clin. Pract. 2011, 119, c74–c82. [Google Scholar] [CrossRef] [PubMed]
- Klade, C.S.; Voss, T.; Krystek, E.; Ahorn, H.; Zatloukal, K.; Pummer, K.; Adolf, G.R. Identification of tumor antigens in renal cell carcinoma by serological proteome analysis. Proteomics 2001, 1, 890–898. [Google Scholar] [CrossRef]
- Finne, K.; Marti, H.-P.; Leh, S.; Skogstrand, T.; Vethe, H.; Tenstad, O.; Berven, F.S.; Scherer, A.; Vikse, B.E. Proteomic Analysis of Minimally Damaged Renal Tubular Tissue from Two-Kidney-One-Clip Hypertensive Rats Demonstrates Extensive Changes Compared to Tissue from Controls. Nephron 2016, 132, 70–80. [Google Scholar] [CrossRef]
- Marshall, C.B.; Krofft, R.D.; Blonski, M.J.; Kowalewska, J.; Logar, C.M.; Pippin, J.W.; Kim, F.; Feil, R.; Alpers, C.E.; Shankland, S.J. Role of smooth muscle protein SM22α in glomerular epithelial cell injury. Am. J. Physiol. Renal Physiol. 2011, 300, F1026–F1042. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elsafadi, M.; Manikandan, M.; Almalki, S.; Mahmood, A.; Shinwari, T.; Vishnubalaji, R.; Mobarak, M.; Alfayez, M.; Aldahmash, A.; Kassem, M.; et al. Transgelin is a poor prognostic factor associated with advanced colorectal cancer (CRC) stage promoting tumor growth and migration in a TGFbeta-dependent manner. Cell Death Dis. 2020, 11, 341. [Google Scholar] [CrossRef]
- Zhou, H.; Zhang, Y.; Chen, Q.; Lin, Y. AKT and JNK Signaling Pathways Increase the Metastatic Potential of Colorectal Cancer Cells by Altering Transgelin Expression. Dig. Dis. Sci. 2015, 61, 1091–1097. [Google Scholar] [CrossRef]
- Dvořáková, M.; Jeřábková, J.; Procházková, I.; Lenčo, J.; Nenutil, R.; Bouchal, P. Transgelin is upregulated in stromal cells of lymph node positive breast cancer. J. Proteom. 2016, 132, 103–111. [Google Scholar] [CrossRef]
- Chen, Z.; He, S.; Zhan, Y.; He, A.; Fang, D.; Gong, Y.; Li, X.; Zhou, L. TGF-beta-induced transgelin promotes bladder cancer metastasis by regulating epithelial-mesenchymal transition and invadopodia formation. EBioMedicine 2019, 47, 208–220. [Google Scholar] [CrossRef] [Green Version]
- Zhou, L.; Zhang, R.; Zhang, L.; Sun, Y.; Yao, W.; Zhao, A.; Li, J.; Yuan, Y. Upregulation of transgelin is an independent factor predictive of poor prognosis in patients with advanced pancreatic cancer. Cancer Sci. 2013, 104, 423–430. [Google Scholar] [CrossRef] [Green Version]
- Wu, X.; Dong, L.; Zhang, R.; Ying, K.; Shen, H. Transgelin overexpression in lung adenocarcinoma is associated with tumor progression. Int. J. Mol. Med. 2014, 34, 585–591. [Google Scholar] [CrossRef] [Green Version]
- Fu, J.; Wang, X.; Yue, Q. Functional loss of TAGLN inhibits tumor growth and increases chemosensitivity of non-small cell lung cancer. Biochem. Biophys. Res. Commun. 2020, 529, 1086–1093. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Zhang, Y.; Wu, L.; Xie, W.; Li, L.; Yuan, Y.; Chen, Y.; Lin, Y.; He, X. Elevated transgelin/TNS1 expression is a potential biomarker in human colorectal cancer. Oncotarget 2017, 9, 1107–1113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aikins, A.R.; Kim, M.; Raymundo, B.; Kim, C.-W. Featured Article: Downregulation of transgelin blocks interleukin-8 utilization and suppresses vasculogenic mimicry in breast cancer cells. Exp. Biol. Med. 2017, 242, 573–583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, H.; Konigshoff, M.; Jayachandran, A.; Handley, D.; Seeger, W.; Kaminski, N.; Eickelberg, O. Transgelin is a direct target of TGF-beta/Smad3-dependent epithelial cell migration in lung fibrosis. FASEB J. 2008, 22, 1778–1789. [Google Scholar] [CrossRef] [PubMed]
- Zhong, W.; Hou, H.; Liu, T.; Su, S.; Xi, X.; Liao, Y.; Xie, R.; Jin, G.; Liu, X.; Zhu, L.; et al. Cartilage Oligomeric Matrix Protein promotes epithelial-mesenchymal transition by interacting with Transgelin in Colorectal Cancer. Theranostics 2020, 10, 8790–8806. [Google Scholar] [CrossRef]
- Zhou, H.; Li, L.; Xie, W.; Wu, L.; Lin, Y.; He, X. TAGLN and High-mobility Group AT-Hook 2 (HMGA2) Complex Regulates TGF-beta-induced Colorectal Cancer Metastasis. OncoTargets Ther. 2020, 13, 10489–10498. [Google Scholar] [CrossRef]
- Chiu, I.-J.; Hsu, Y.-H.; Chang, J.-S.; Yang, J.-C.; Chiu, H.-W.; Lin, Y.-F. Lactotransferrin Downregulation Drives the Metastatic Progression in Clear Cell Renal Cell Carcinoma. Cancers 2020, 12, 847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ni, L.; Yuan, C.; Zhang, C.; Xiang, Y.; Wu, J.; Wang, X.; Wu, X. Co-Expression Network Analysis Identified LTF in Association with Metastasis Risk and Prognosis in Clear Cell Renal Cell Carcinoma. OncoTargets Ther. 2020, 13, 6975–6986. [Google Scholar] [CrossRef]
- Harada, K.; Miyake, H.; Kusuda, Y.; Fujisawa, M. Characterization of mechanism involved in acquired resistance to sorafenib in a mouse renal cell cancer RenCa model. Clin. Transl. Oncol. 2013, 16, 801–806. [Google Scholar] [CrossRef]
- Liu, Y.; Men, C.; Xu, Y.; Zhao, K.; Luo, L.; Dong, D.; Yu, Q. Clusterin promotes growth and invasion of clear cell renal carcinoma cell by upregulation of S100A4 expression. Cancer Biomark. 2018, 21, 915–923. [Google Scholar] [CrossRef]
- Nishi, K.; Ono, T.; Nakamura, T.; Fukunaga, N.; Izumi, M.; Watanabe, H.; Suenaga, A.; Maruyama, T.; Yamagata, Y.; Curry, S.; et al. Structural insights into differences in drug-binding selectivity between two forms of human alpha1-acid glycoprotein genetic variants, the A and F1*S forms. J. Biol. Chem. 2011, 286, 14427–14434. [Google Scholar] [CrossRef] [Green Version]
- Toyama, Y.; Ueyama, J.; Nomura, H.; Tsukiyama, I.; Saito, H.; Hisada, T.; Matsuura, K.; Hasegawa, T. Contribution of plasma proteins, albumin and alpha 1-acid glycoprotein, to pharmacokinetics of a multi-targeted receptor tyrosine kinase inhibitor, sunitinib, in analbuminemic rats. Anticancer Res. 2014, 34, 2283–2289. [Google Scholar] [PubMed]
- Koh, H.M.; An, H.J.; Ko, G.H.; Lee, J.H.; Lee, J.S.; Kim, D.C.; Song, D.H. Prognostic role of S100A9 expression in patients with clear cell renal cell carcinoma. Medicine 2019, 98, e17188. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.L.; Wu, Z.Z.; Xu, Y.; Wang, J.G.; Wang, Y.Q.; Cao, M.Q.; Wang, C.H. Saliva proteomic analysis reveals possible biomarkers of renal cell carcinoma. Open Chem. 2020, 18, 918–926. [Google Scholar] [CrossRef]
- Luo, T.; Chen, X.; Zeng, S.; Guan, B.; Hu, B.; Meng, Y.; Liu, F.; Wong, T.; Lu, Y.; Yun, C.; et al. Bioinformatic identification of key genes and analysis of prognostic values in clear cell renal cell carcinoma. Oncol. Lett. 2018, 16, 1747–1757. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Wei, X.; Feng, H.; Hu, B.; Liu, B.; Luan, Y.; Ruan, Y.; Liu, X.; Liu, Z.; Liu, J.; et al. Integrated Analysis to Identify a Redox-Related Prognostic Signature for Clear Cell Renal Cell Carcinoma. Oxidative Med. Cell. Longev. 2021, 2021, 6648093. [Google Scholar] [CrossRef]
- Maruschke, M.; Koczan, D.; Reuter, D.; Ziems, B.; Nizze, H.; Hakenberg, O.; Thiesen, H.-J. Putative Biomarker Genes for Grading Clear Cell Renal Cell Carcinoma. Urol. Int. 2011, 87, 205–217. [Google Scholar] [CrossRef]
- Brooks, S.A.; Brannon, A.R.; Parker, J.S.; Fisher, J.C.; Sen, O.; Kattan, M.; Hakimi, A.A.; Hsieh, J.J.; Choueiri, T.K.; Tamboli, P.; et al. ClearCode34: A Prognostic Risk Predictor for Localized Clear Cell Renal Cell Carcinoma. Eur. Urol. 2014, 66, 77–84. [Google Scholar] [CrossRef] [Green Version]
- Thibodeau, B.J.; Fulton, M.; Fortier, L.E.; Geddes, T.J.; Pruetz, B.L.; Ahmed, S.; Banes-Berceli, A.; Zhang, P.L.; Wilson, G.; Hafron, J. Characterization of clear cell renal cell carcinoma by gene expression profiling. Urol. Oncol. Semin. Orig. Investig. 2015, 34, 168.e1–168.e9. [Google Scholar] [CrossRef]
- Sandim, V.; Pereira, D.D.A.; Kalume, D.E.; Oliveira-Carvalho, A.L.; Ornellas, A.A.; Soares, M.R.; Alves, G.; Zingali, R.B. Proteomic analysis reveals differentially secreted proteins in the urine from patients with clear cell renal cell carcinoma. Urol. Oncol. Semin. Orig. Investig. 2016, 34, 5.e11–5.e25. [Google Scholar] [CrossRef]
- Landolt, L.; Eikrem, Ø.; Strauss, P.; Scherer, A.; Lovett, D.H.; Beisland, C.; Finne, K.; Osman, T.; Ibrahim, M.M.; Gausdal, G.; et al. Clear Cell Renal Cell Carcinoma is linked to Epithelial-to-Mesenchymal Transition and to Fibrosis. Physiol. Rep. 2017, 5, e13305. [Google Scholar] [CrossRef] [Green Version]
- Piva, F.; Giulietti, M.; Santoni, M.; Occhipinti, G.; Scarpelli, M.; López-Beltrán, A.; Cheng, L.; Principato, G.; Montironi, R. Epithelial to Mesenchymal Transition in Renal Cell Carcinoma: Implications for Cancer Therapy. Mol. Diagn. Ther. 2016, 20, 111–117. [Google Scholar] [CrossRef]
- Medici, D.; Nawshad, A. Type I collagen promotes epithelial-mesenchymal transition through ILK-dependent activation of NF-kappaB and LEF-1. Matrix Biol. 2010, 29, 161–165. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.H.; Albig, A.R.; Regner, M.; Schiemann, B.J.; Schiemann, W.P. Fibulin-5 initiates epithelial-mesenchymal transition (EMT) and enhances EMT induced by TGF-beta in mammary epithelial cells via a MMP-dependent mechanism. Carcinogenesis 2008, 29, 2243–2251. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Buckhaults, P.J.; Lee, J.R.; Xiong, H.; Farrell, C.; Podolsky, R.H.; Schade, R.R.; Dynan, W. Association of the Actin-Binding Protein Transgelin with Lymph Node Metastasis in Human Colorectal Cancer. Neoplasia 2009, 11, 864–873. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Genovese, F.; Manresa, A.A.; Leeming, D.J.; Karsdal, M.A.; Boor, P. The extracellular matrix in the kidney: A source of novel non-invasive biomarkers of kidney fibrosis? Fibrogenesis Tissue Repair 2014, 7, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, H.; Li, W.; Lv, Y.; Fan, Q.; Mao, X.; Long, T.; Xie, L.; Dong, C.; Yang, R.; Zhang, H. Exploring the mechanism of clear cell renal cell carcinoma metastasis and key genes based on multi-tool joint analysis. Gene 2019, 720, 144103. [Google Scholar] [CrossRef] [PubMed]
- Beuselinck, B.; Job, S.; Becht, E.; Karadimou, A.; Verkarre, V.; Couchy, G.; Giraldo, N.; Rioux-Leclercq, N.; Molinié, V.; Sibony, M.; et al. Molecular Subtypes of Clear Cell Renal Cell Carcinoma Are Associated with Sunitinib Response in the Metastatic Setting. Clin. Cancer Res. 2015, 21, 1329–1339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beuselinck, B.; Jean-Baptiste, J.; Couchy, G.; Job, S.; De Reynies, A.; Wolter, P.; Theodore, C.; Gravis, G.; Rousseau, B.; Albiges, L.; et al. RANK/OPG ratio of expression in primary clear-cell renal cell carcinoma is associated with bone metastasis and prognosis in patients treated with anti-VEGFR-TKIs. Br. J. Cancer 2015, 113, 1313–1322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beuselinck, B.; Verbiest, A.; Couchy, G.; Job, S.; de Reynies, A.; Meiller, C.; Albersen, M.; Verkarre, V.; Lerut, E.; Méjean, A.; et al. Pro-angiogenic gene expression is associated with better outcome on sunitinib in metastatic clear-cell renal cell carcinoma. Acta Oncol. 2017, 57, 498–508. [Google Scholar] [CrossRef] [Green Version]
- Dumond, A.; Brachet, E.; Durivault, J.; Vial, V.; Puszko, A.K.; Lepelletier, Y.; Montemagno, C.; Pagnuzzi-Boncompagni, M.; Hermine, O.; Garbay, C.; et al. Neuropilin 1 and Neuropilin 2 gene invalidation or pharmacological inhibition reveals their relevance for the treatment of metastatic renal cell carcinoma. J. Exp. Clin. Cancer Res. 2021, 40, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Shin, S.-J.; Jeon, Y.K.; Cho, Y.M.; Lee, J.; Chung, D.H.; Park, J.Y.; Go, H. The Association Between PD-L1 Expression and the Clinical Outcomes to Vascular Endothelial Growth Factor-Targeted Therapy in Patients With Metastatic Clear Cell Renal Cell Carcinoma. Oncologist 2015, 20, 1253–1260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Furukawa, J.; Miyake, H.; Fujisawa, M. GLI2 expression levels in radical nephrectomy specimens as a predictor of disease progression in patients with metastatic clear cell renal cell carcinoma following treatment with sunitinib. Mol. Clin. Oncol. 2016, 5, 186–192. [Google Scholar] [CrossRef] [Green Version]
- Stewart, G.; O’Mahony, F.C.; Laird, A.; Rashid, S.; Martin, S.A.; Eöry, L.; Lubbock, A.; Nanda, J.; O’Donnell, M.; Mackay, A.; et al. Carbonic Anhydrase 9 Expression Increases with Vascular Endothelial Growth Factor–Targeted Therapy and Is Predictive of Outcome in Metastatic Clear Cell Renal Cancer. Eur. Urol. 2014, 66, 956–963. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Categories/Groups | Patients (n = 16) | Responders (R, n = 8) | Non-Responders (NR, n = 8) | |
|---|---|---|---|---|
| sex | M | 10 | 5 | 5 |
| F | 6 | 3 | 3 | |
| age at diagnosis, mean (yrs) | 63.7 ± 9.5 | 69.0 ± 5.5 | 58.4 ± 10.3 | |
| Fuhrman grade | 1 | 1 | 1 | 0 |
| 2 | 3 | 2 | 1 | |
| 3 | 8 | 4 | 4 | |
| 4 | 4 | 1 | 3 | |
| pT | 1a | 1 | 1 | 0 |
| 1b | 3 | 3 | 0 | |
| 2a | 1 | 1 | 0 | |
| 2b | 1 | 0 | 1 | |
| 3a | 5 | 2 | 3 | |
| 3b | 1 | 0 | 1 | |
| 4 | 4 | 1 | 3 | |
| pN | 0 | 12 | 7 | 5 |
| 1 | 2 | 0 | 2 | |
| 2 | 2 | 1 | 1 | |
| mestastases at diagnosis | 0 | 4 | 4 | 0 |
| 1 | 12 | 4 | 8 | |
| relapse after surgery | yes | 4 | 4 | 0 |
| no | 12 | 4 | 8 | |
| sunitinib response | CR | 1 | 1 | 0 |
| PR | 7 | 7 | 0 | |
| PD | 8 | 0 | 8 |
| P vs. R | P vs. N | R vs. N | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Genes | Protein | Uniprot ID | AVG Log2 Ratio | q Value | AVG Log2 Ratio | q Value | AVG Log2 Ratio | q Value | |
| 1 | LTF | Lactotransferrin | P02788 | 2.445 | 0.004 | 2.237 | 0.001 | −0.208 | 0.531 |
| 2 | TAGLN | Transgelin | Q01995 | 2.407 | 0.007 | 2.667 | 0.021 | 0.260 | 0.328 |
| 3 | CLU | Clusterin | P10909 | 2.235 | 0.029 | 2.781 | 0.041 | 0.546 | 0.242 |
| 4 | ORM2 | Alpha-1-acid glycoprotein 2 | P19652 | 1.623 | 0.020 | 2.850 | 0.008 | 1.227 | 0.516 |
| 5 | S100A9 | Protein S100-A9 | P06702 | 1.553 | 0.035 | 2.659 | 0.006 | 1.106 | 0.397 |
| 6 | CFH | Complement factor H | P08603 | 1.542 | 0.019 | 2.936 | 0.000 | 1.395 | 0.228 |
| 7 | SERPINA3 | Alpha-1-antichymotrypsin | P01011 | 1.491 | 0.002 | 2.906 | 0.000 | 1.415 | 0.162 |
| 8 | HPX | Hemopexin | P02790 | 1.400 | 0.000 | 2.616 | 0.000 | 1.216 | 0.055 |
| 9 | PLG | Plasminogen | P00747 | 1.345 | 0.001 | 3.213 | 0.000 | 1.868 | 0.201 |
| 10 | SERPINC1 | Antithrombin-III | P01008 | 1.309 | 0.024 | 1.831 | 0.015 | 0.521 | 0.517 |
| 11 | KNG1 | Kininogen-1 | P01042 | 1.180 | 0.007 | 2.403 | 0.000 | 1.223 | 0.111 |
| 12 | SERPINA1 | Alpha-1-antitrypsin | P01009 | 1.150 | 0.000 | 2.420 | 0.000 | 1.270 | 0.475 |
| 13 | MYH11 | Myosin-11 | P35749 | 1.133 | 0.000 | 2.197 | 0.000 | 1.064 | 0.130 |
| 14 | CP | Ceruloplasmin | P00450 | 1.108 | 0.003 | 2.660 | 0.000 | 1.553 | 0.060 |
| 15 | ITIH4 | Inter-alpha-trypsin inhibitor heavy chain H4 | Q14624 | 0.888 | 0.029 | 1.865 | 0.011 | 0.977 | 0.396 |
| 16 | IGHM | Immunoglobulin heavy constant mu | P01871 | 0.826 | 0.025 | 2.562 | 0.000 | 1.735 | 0.181 |
| 17 | CLTC | Clathrin heavy chain 1 | Q00610 | 0.784 | 0.001 | 1.075 | 0.007 | 0.291 | 0.373 |
| 18 | ERP29 | Endoplasmic reticulum resident protein 29 | P30040 | −0.610 | 0.039 | −1.019 | 0.004 | −0.409 | 0.209 |
| 19 | NDUFB11 | NADH-ubiquinone oxidoreductase ESSS subunit | Q9NX14 | −0.617 | 0.046 | −1.765 | 0.010 | −1.148 | 0.129 |
| 20 | HSD17B4 | Peroxisomal multifunctional enzyme type 2 | P51659 | −0.618 | 0.003 | −0.841 | 0.000 | −0.223 | 0.148 |
| 21 | DNPH1 | 2′-deoxynucleoside 5′-phosphate N-hydrolase 1 | O43598 | −0.785 | 0.036 | −0.751 | 0.025 | 0.034 | 0.355 |
| 22 | CTSC | Dipeptidyl peptidase 1 | P53634 | −0.804 | 0.019 | −0.969 | 0.001 | −0.165 | 0.406 |
| 23 | HSPA12A | Heat shock 70 kDa protein 12A | O43301 | −0.806 | 0.025 | −0.856 | 0.002 | −0.050 | 0.408 |
| 24 | DYNLL2 | Dynein light chain 2, cytoplasmic | Q96FJ2 | −0.858 | 0.039 | −1.066 | 0.001 | −0.208 | 0.222 |
| 25 | PPME1 | Protein phosphatase methylesterase 1 | Q9Y570 | −0.889 | 0.022 | −3.817 | 0.048 | −2.928 | 0.260 |
| 26 | ACSS1 | Acetyl-coenzyme A synthetase 2-like, mitochondrial | Q9NUB1 | −1.666 | 0.041 | −1.816 | 0.021 | −0.150 | 0.503 |
| 27 | GSTA2 | Glutathione S-transferase A2 | P09210 | −2.219 | 0.000 | −1.776 | 0.000 | 0.443 | 0.389 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bouchalova, P.; Beranek, J.; Lapcik, P.; Potesil, D.; Podhorec, J.; Poprach, A.; Bouchal, P. Transgelin Contributes to a Poor Response of Metastatic Renal Cell Carcinoma to Sunitinib Treatment. Biomedicines 2021, 9, 1145. https://doi.org/10.3390/biomedicines9091145
Bouchalova P, Beranek J, Lapcik P, Potesil D, Podhorec J, Poprach A, Bouchal P. Transgelin Contributes to a Poor Response of Metastatic Renal Cell Carcinoma to Sunitinib Treatment. Biomedicines. 2021; 9(9):1145. https://doi.org/10.3390/biomedicines9091145
Chicago/Turabian StyleBouchalova, Pavla, Jindrich Beranek, Petr Lapcik, David Potesil, Jan Podhorec, Alexandr Poprach, and Pavel Bouchal. 2021. "Transgelin Contributes to a Poor Response of Metastatic Renal Cell Carcinoma to Sunitinib Treatment" Biomedicines 9, no. 9: 1145. https://doi.org/10.3390/biomedicines9091145