
Development of New Meridianin/Leucettine-Derived Hybrid Small Molecules as Nanomolar Multi-Kinase Inhibitors with Antitumor Activity

 , , ,
, , ,  ,
,
Abstract
:1. Introduction
2. Materials and Methods
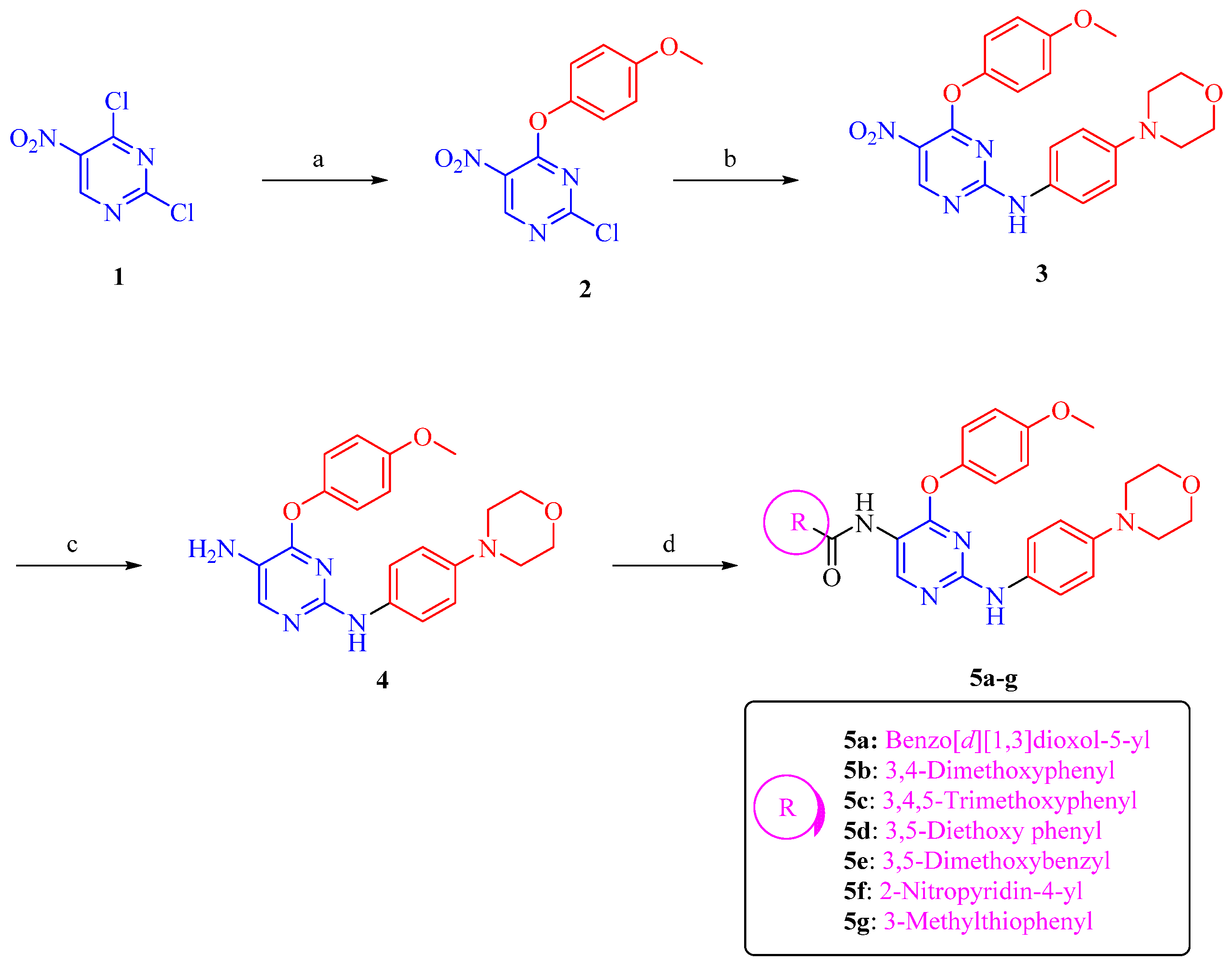
2.1. Chemistry
General
2.2. Biological Evaluation
2.2.1. In Vitro Kinase Inhibition Assay
2.2.2. In Vitro Antitumor Activity towards 60 Cancer Cell Lines
2.2.3. Molecular Modeling Study
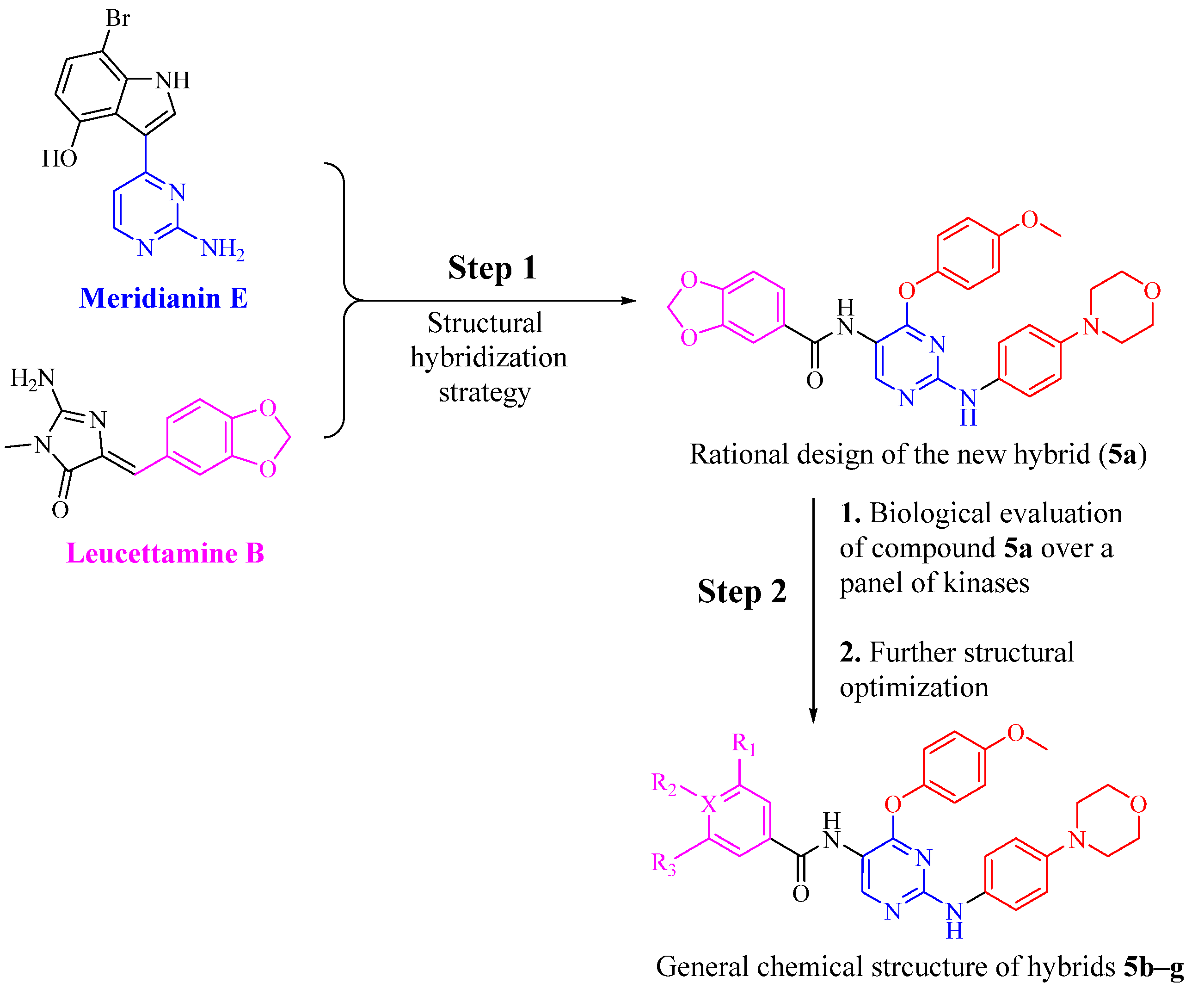
3. Results and Discussion
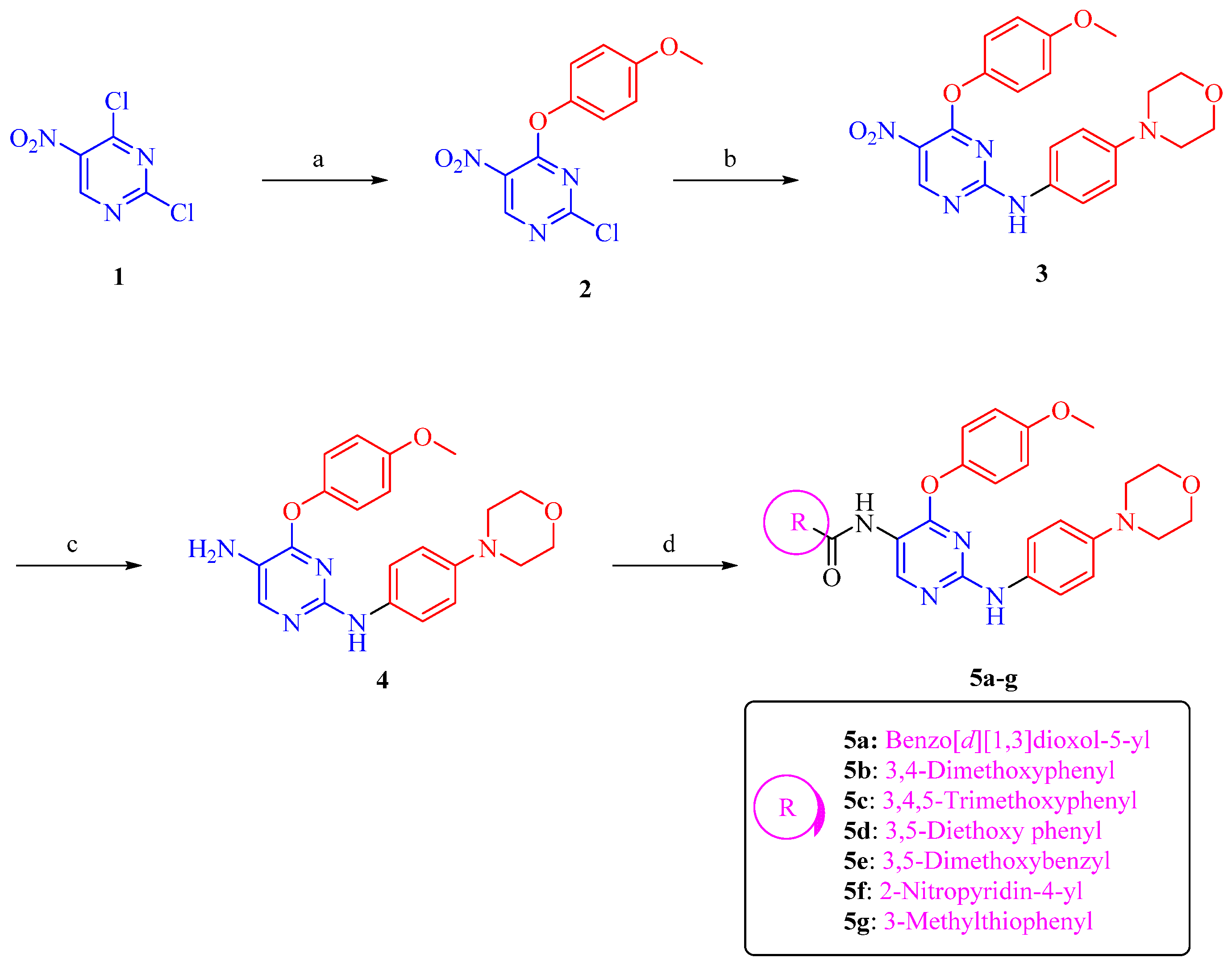
3.1. Chemical Synthesis
3.2. Biological Evaluation
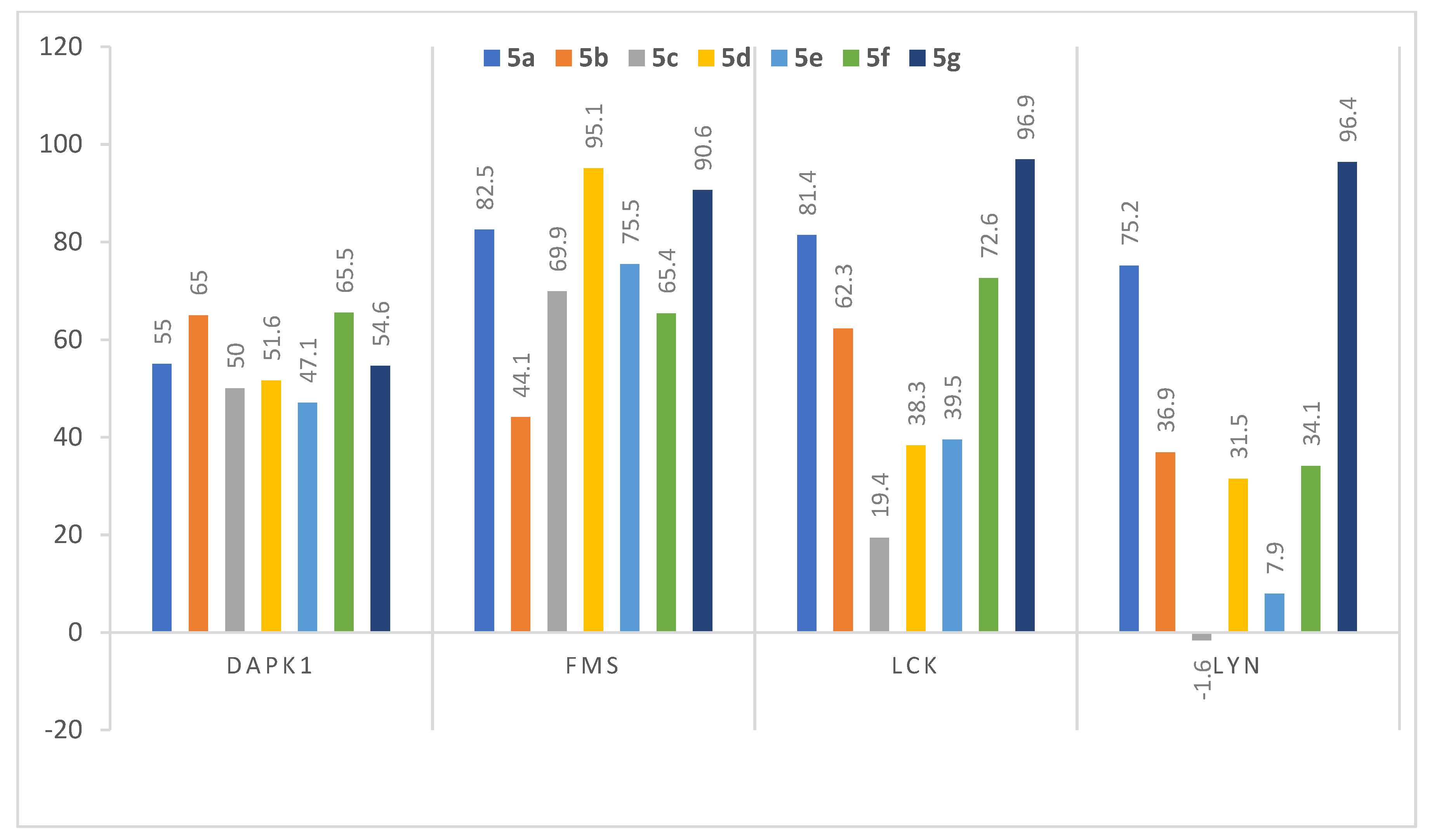
3.2.1. Assessment of Kinase Inhibitory Activity of Compound 5a against a Panel of Kinases
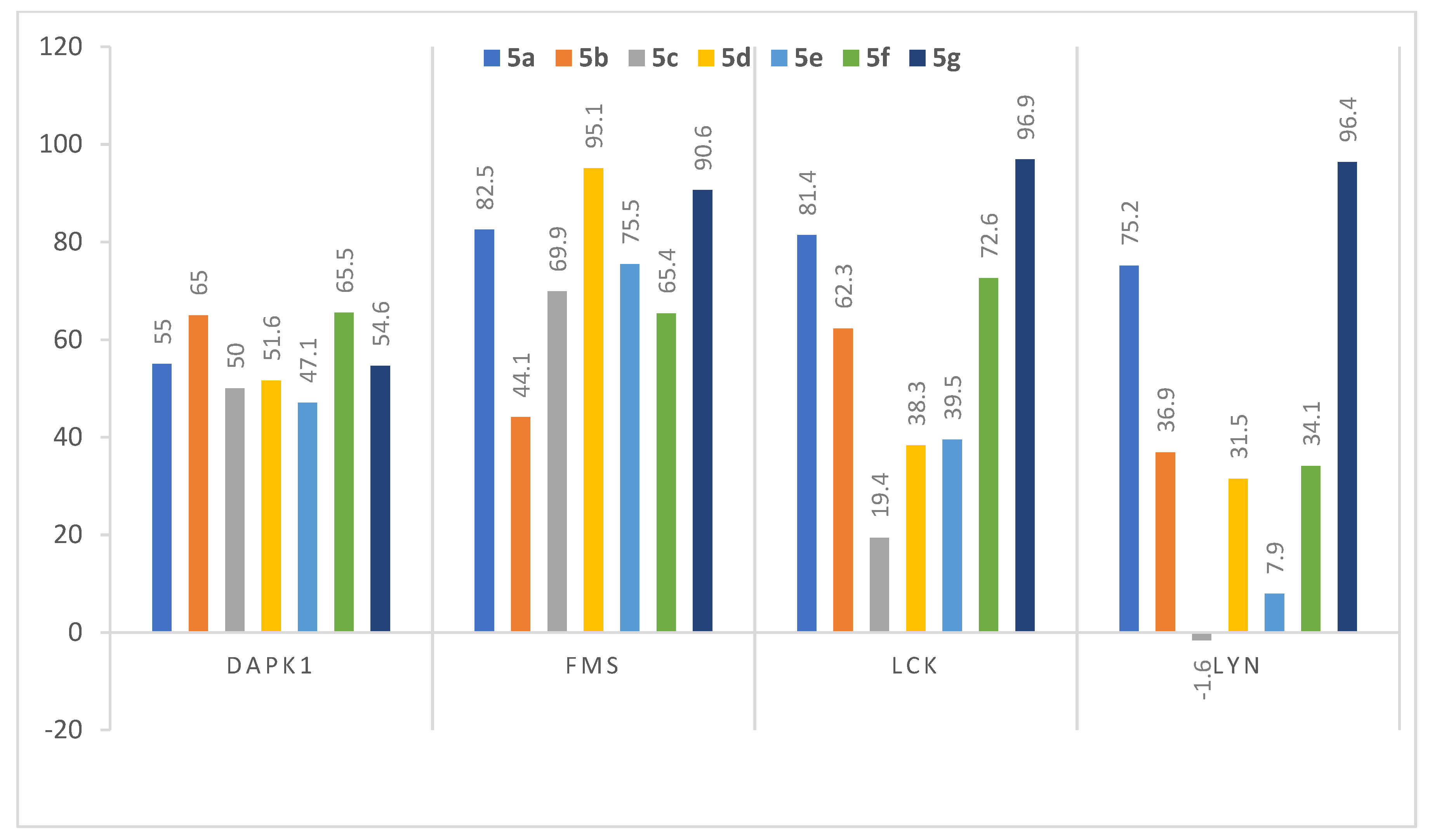
3.2.2. Assessment of Kinase Inhibitory Activity of Compounds 5b–5g against FMS, LCK, LYN, and DAPK1 Kinases
3.2.3. Dose-Dependent Assay of the Most Active Analogs 5d and 5g over FMS, LCK, and LYN Kinases
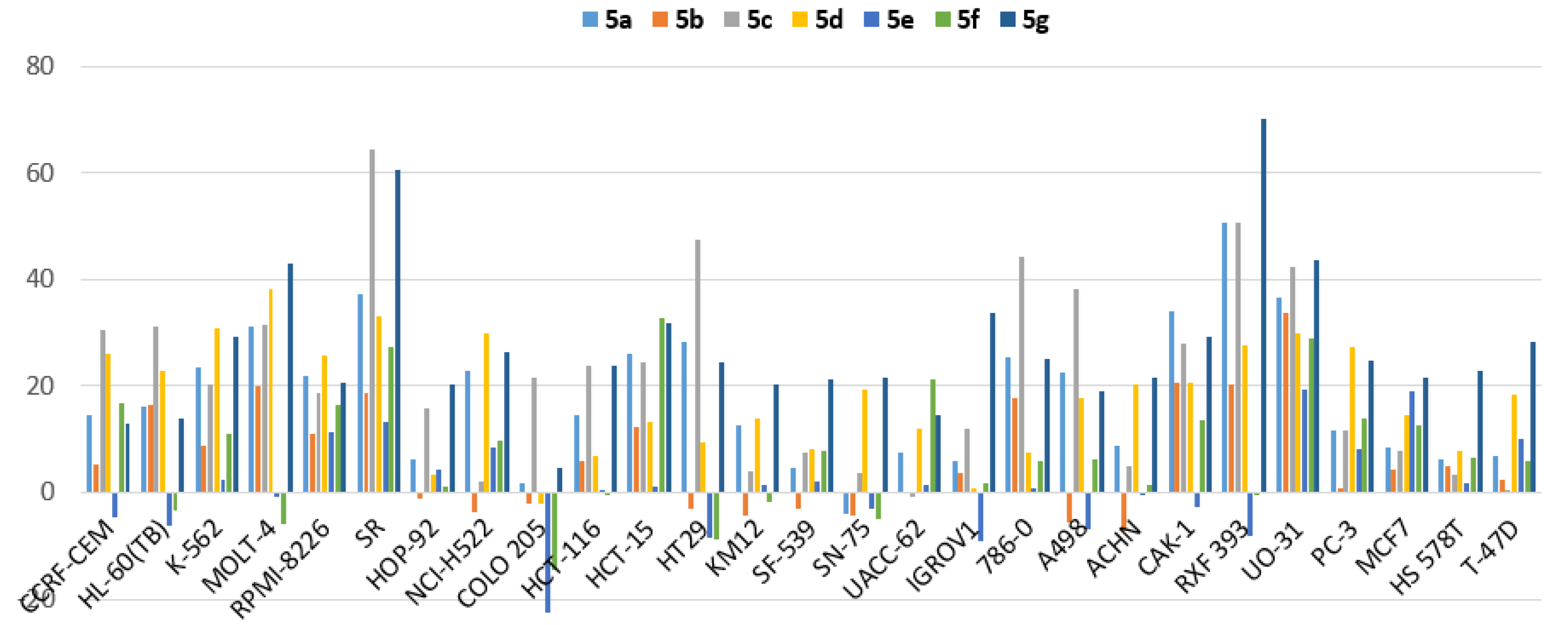
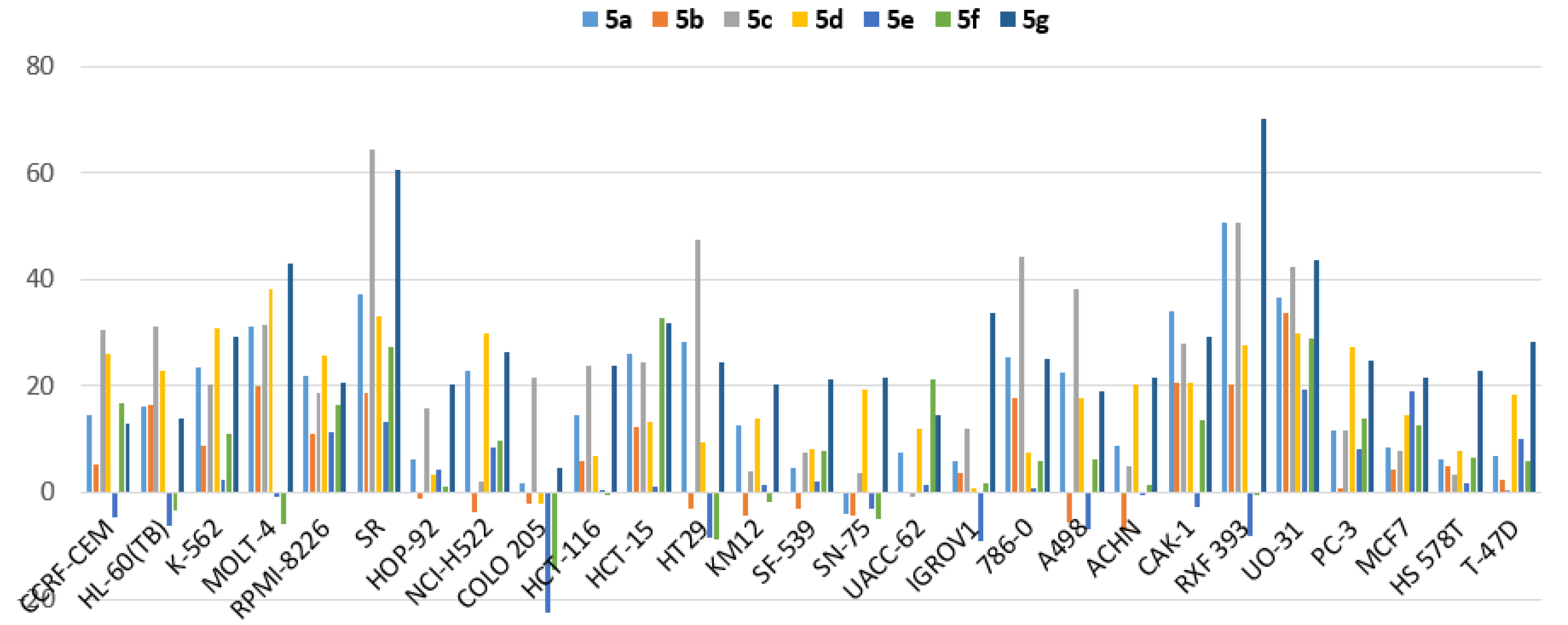
3.2.4. Efficacy and Spectrum against Diverse Cancer Cells in Growth Inhibition (GI) Assays
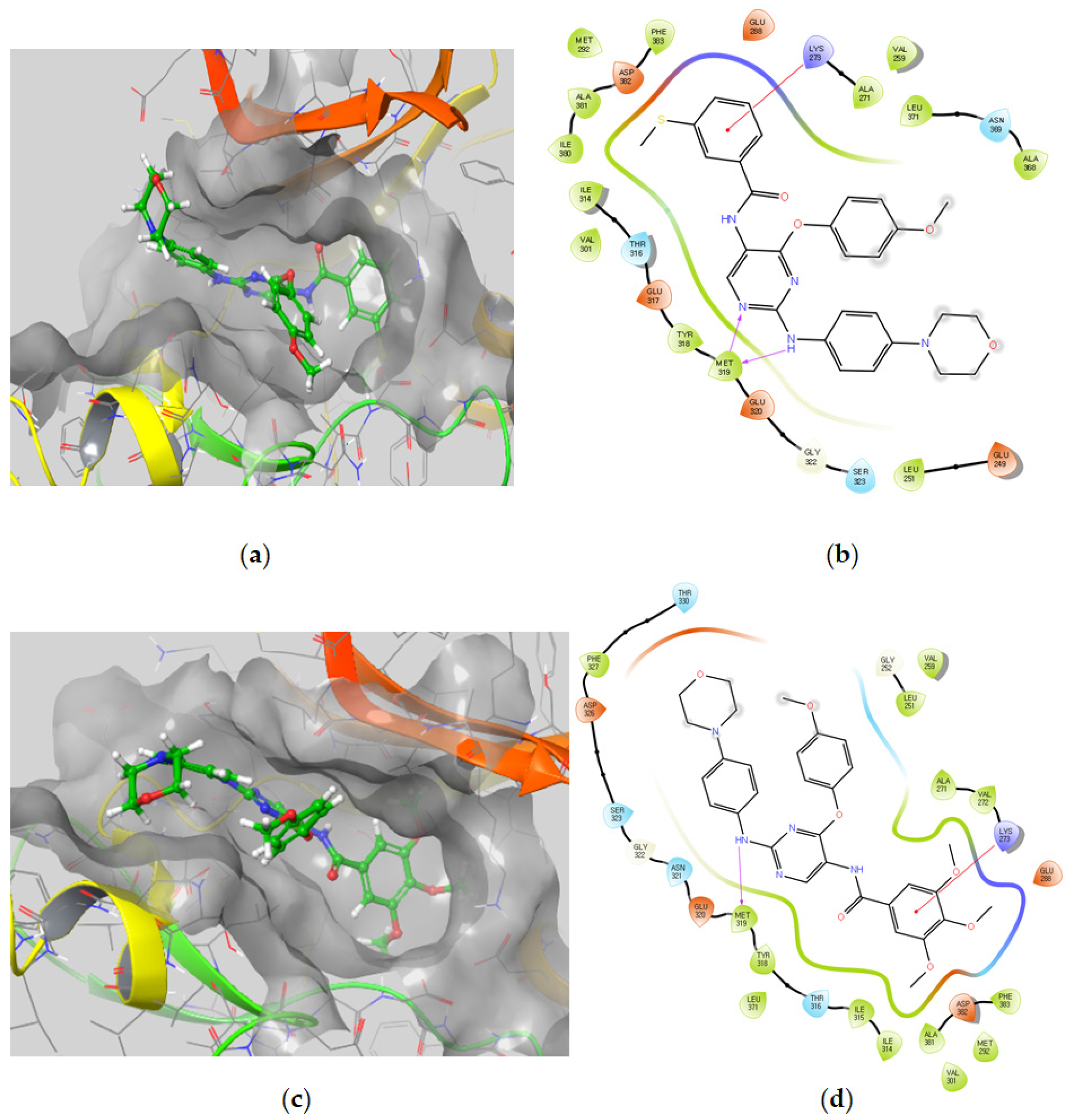
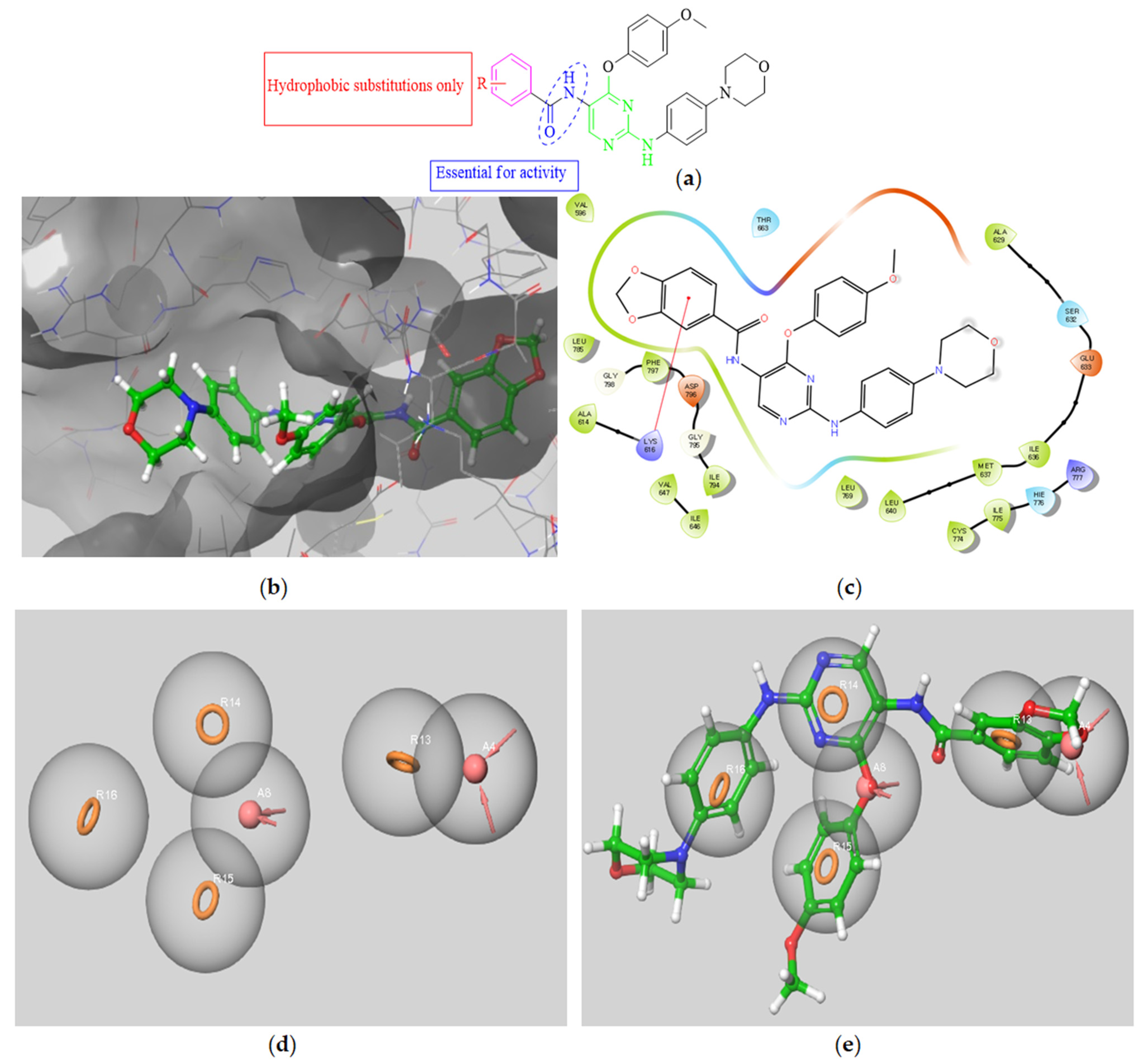
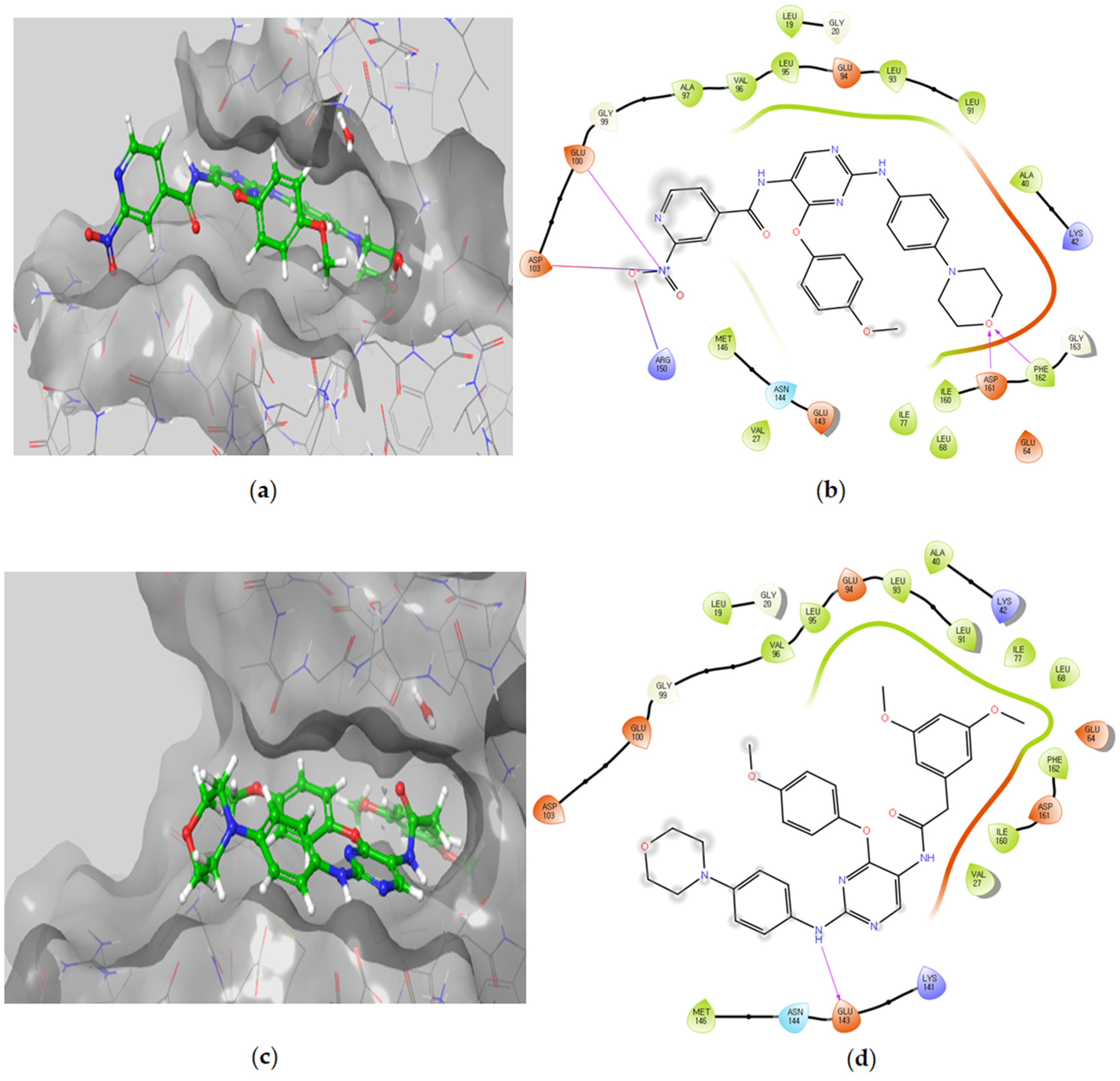
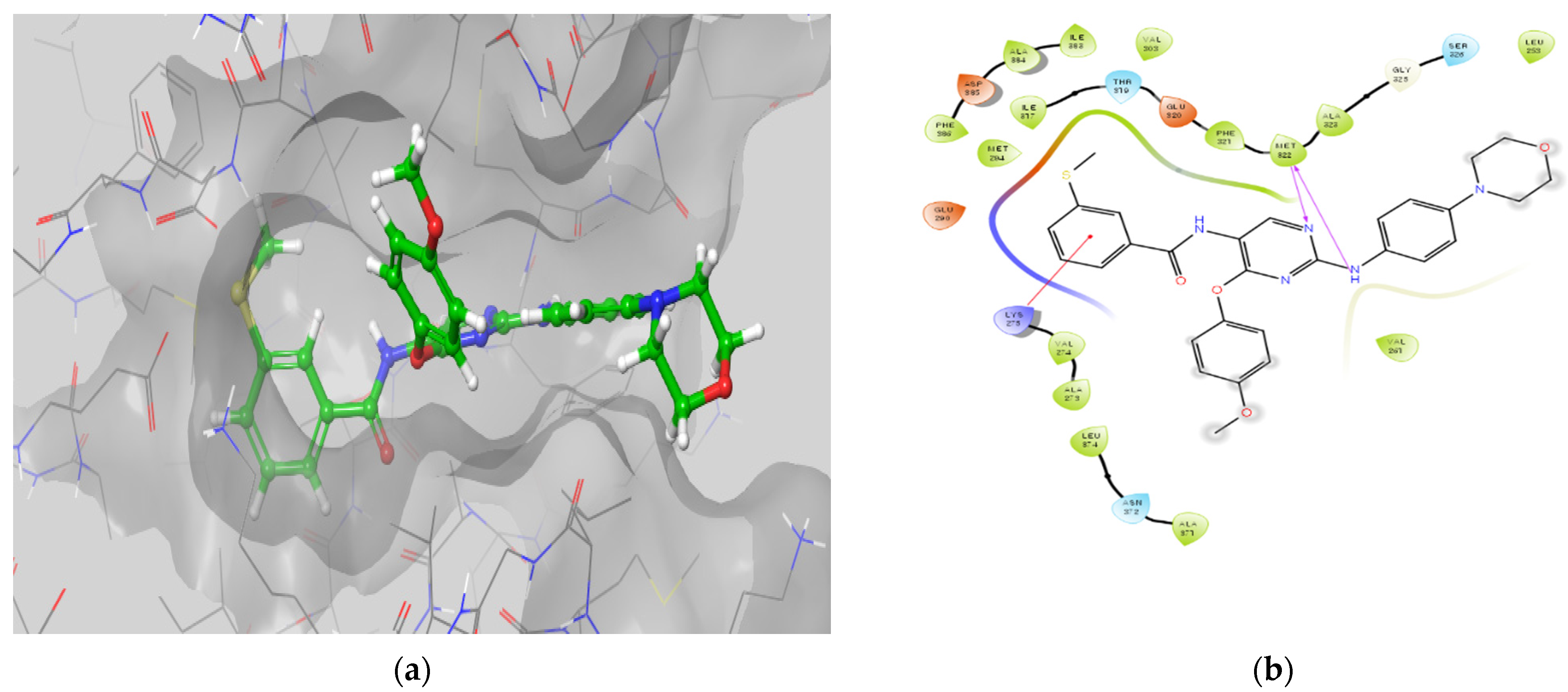
3.2.5. Molecular Docking Studies
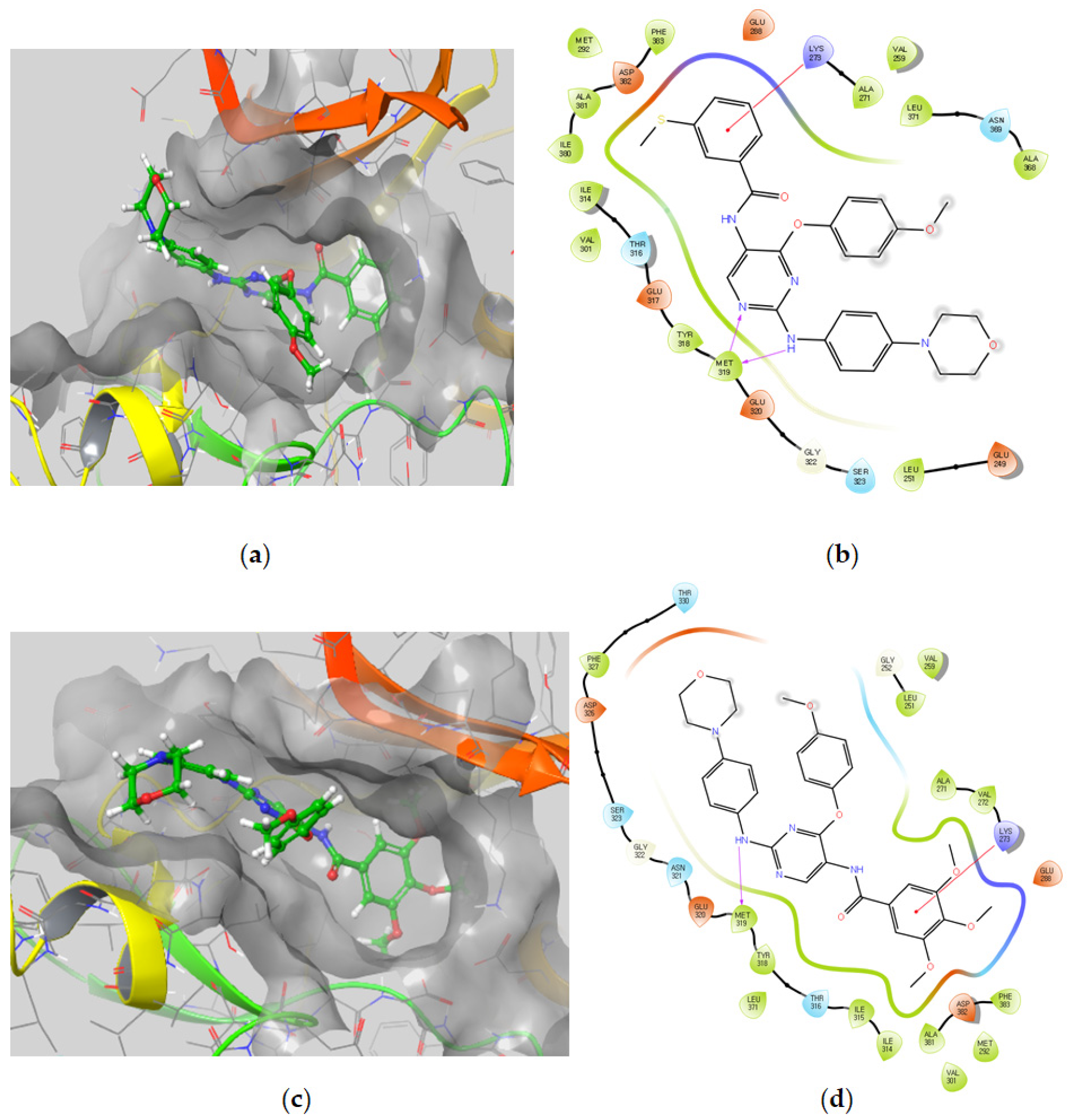
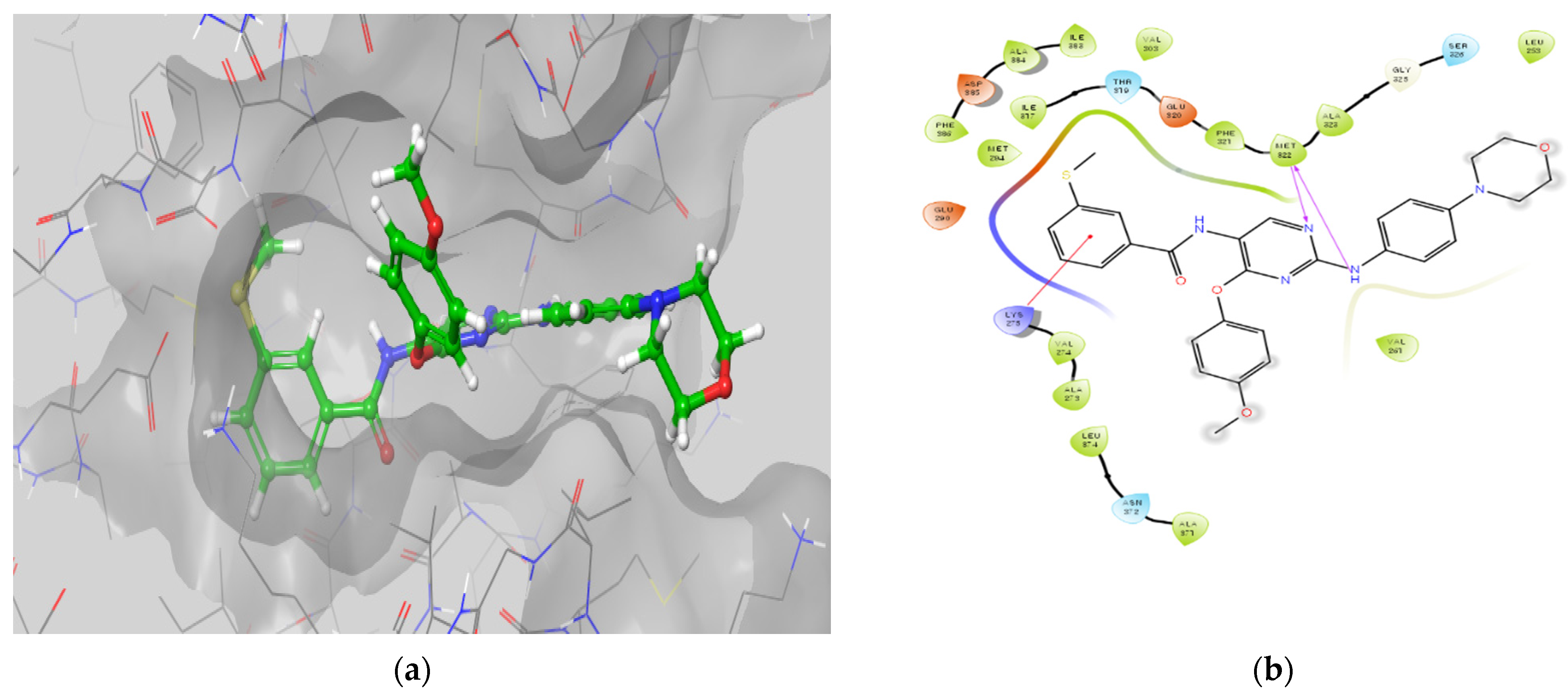
Molecular Docking Models within the LCK Binding Site
Molecular Docking Models within the FMS Binding Site
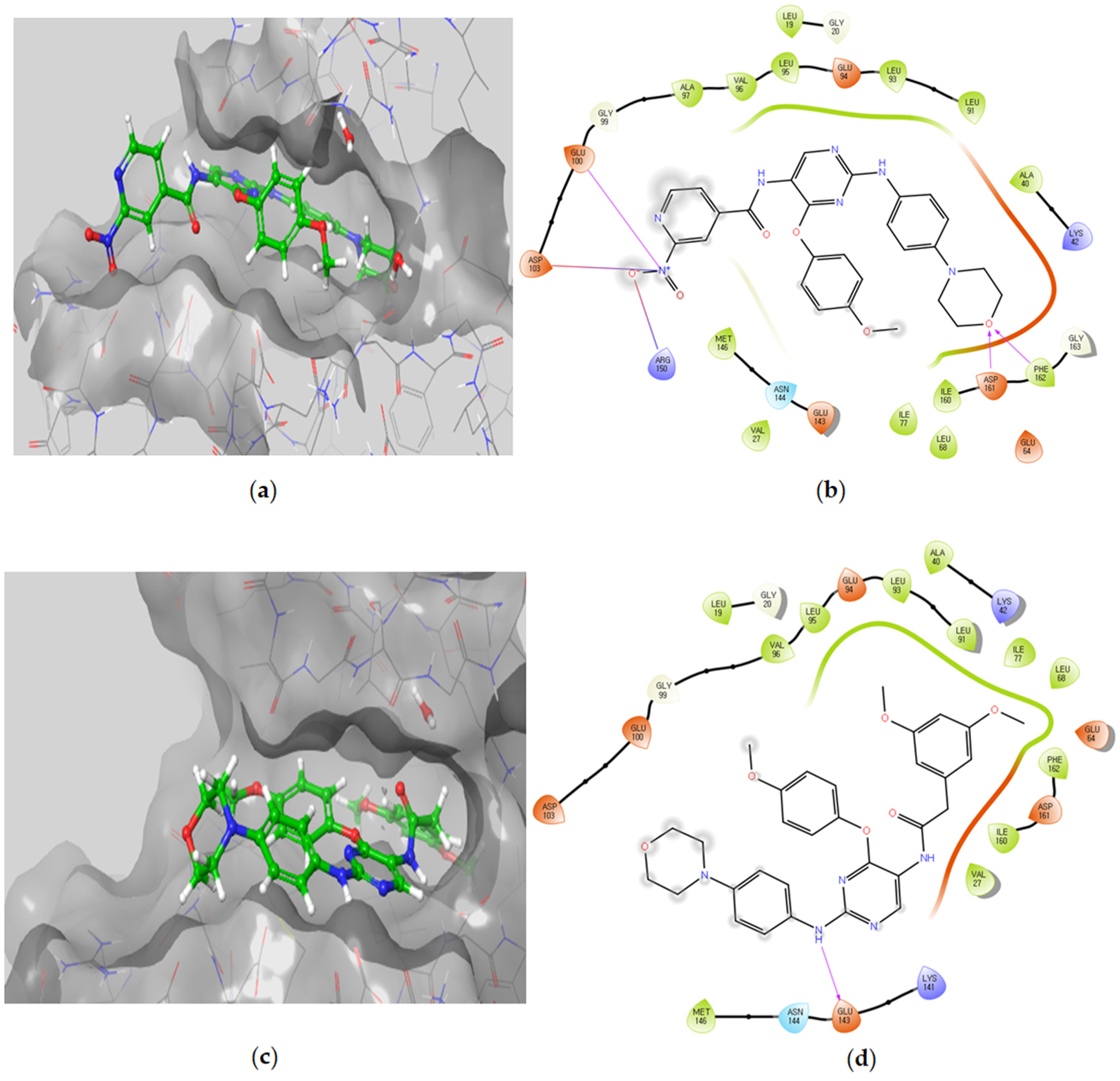
Molecular Docking Models within the DAPK1 Binding Site
Molecular Docking Models within the LYN Binding Site
3.2.6. In Silico Pharmacokinetic Study
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Bharate, S.B.; Sawant, S.D.; Singh, P.P.; Vishwakarma, R.A. Kinase Inhibitors of Marine Origin. Chem. Rev. 2013, 113, 6761–6815. [Google Scholar] [CrossRef]
- Bharate, S.B.; Yadav, R.R.; Battula, S.; Vishwakarma, R.A. Meridianins: Marine-derived potent kinase inhibitors. Mini Rev. Med. Chem. 2012, 12, 618–631. [Google Scholar] [CrossRef] [PubMed]
- Eldehna, W.M.; Hassan, G.S.; Al-Rashood, S.T.; Alkahtani, H.M.; Almehizia, A.A.; Al-Ansary, G.H. Marine-Inspired Bis-indoles Possessing Antiproliferative Activity against Breast Cancer; Design, Synthesis, and Biological Evaluation. Mar. Drugs 2020, 18, 190. [Google Scholar] [CrossRef] [Green Version]
- Gerwick, W.H.; Fenner, A.M. Drug discovery from marine microbes. Microb. Ecol. 2013, 65, 800–806. [Google Scholar] [CrossRef] [PubMed]
- Wang, E.; Sorolla, M.A.; Krishnan, P.D.G.; Sorolla, A. From Seabed to Bedside: A Review on Promising Marine Anticancer Compounds. Biomolecules 2020, 10, 248. [Google Scholar] [CrossRef] [Green Version]
- Pereira, F. Have marine natural product drug discovery efforts been productive and how can we improve their efficiency? Expert Opin. Drug Discov. 2019, 14, 717–722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Song, K.; Li, L.; Chen, L. Structure-Based Drug Design Strategies and Challenges. Curr. Top. Med. Chem. 2018, 18, 998–1006. [Google Scholar] [CrossRef] [PubMed]
- Nagasaka, M.; Gadgeel, S.M. Role of chemotherapy and targeted therapy in early-stage non-small cell lung cancer. Expert Rev. Anticancer. Ther. 2018, 18, 63–70. [Google Scholar] [CrossRef]
- Hughes, J.P.; Rees, S.; Kalindjian, S.B.; Philpott, K.L. Principles of early drug discovery. Br. J. Pharmacol. 2011, 162, 1239–1249. [Google Scholar] [CrossRef] [Green Version]
- Hoelder, S.; Clarke, P.A.; Workman, P. Discovery of small molecule cancer drugs: Successes, challenges and opportunities. Mol. Oncol. 2012, 6, 155–176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elkamhawy, A.; Viswanath, A.N.I.; Pae, A.N.; Kim, H.Y.; Heo, J.C.; Park, W.K.; Lee, C.O.; Yang, H.; Kim, K.H.; Nam, D.H.; et al. Discovery of potent and selective cytotoxic activity of new quinazoline-ureas against TMZ-resistant glioblastoma multiforme (GBM). Eur. J. Med. Chem. 2015, 103, 210–222. [Google Scholar] [CrossRef]
- Bhullar, K.S.; Lagarón, N.O.; McGowan, E.M.; Parmar, I.; Jha, A.; Hubbard, B.P.; Rupasinghe, H.P.V. Kinase-targeted cancer therapies: Progress, challenges and future directions. Mol. Cancer 2018, 17, 48. [Google Scholar] [CrossRef]
- Madhusudan, S.; Ganesan, T.S. Tyrosine kinase inhibitors in cancer therapy. Clin. Biochem. 2004, 37, 618–635. [Google Scholar] [CrossRef]
- Cicenas, J.; Cicenas, E. Multi-kinase inhibitors, AURKs and cancer. Med. Oncol. 2016, 33, 43. [Google Scholar] [CrossRef]
- Nada, H.; Elkamhawy, A.; Lee, K. Structure Activity Relationship of Key Heterocyclic Anti-Angiogenic Leads of Promising Potential in the Fight against Cancer. Molecules 2021, 26, 553. [Google Scholar] [CrossRef] [PubMed]
- Elkamhawy, A.; Farag, A.K.; Viswanath, A.N.I.; Bedair, T.M.; Leem, D.G.; Lee, K.-T.; Pae, A.N.; Roh, E.J. Targeting EGFR/HER2 tyrosine kinases with a new potent series of 6-substituted 4-anilinoquinazoline hybrids: Design, synthesis, kinase assay, cell-based assay, and molecular docking. Bioorg. Med. Chem. Lett. 2015, 25, 5147–5154. [Google Scholar] [CrossRef] [PubMed]
- Elkamhawy, A.; Park, J.-e.; Cho, N.-C.; Sim, T.; Pae, A.N.; Roh, E.J. Discovery of a broad spectrum antiproliferative agent with selectivity for DDR1 kinase: Cell line-based assay, kinase panel, molecular docking, and toxicity studies. J. Enzym. Inhib. Med. Chem. 2016, 31, 158–166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elkamhawy, A.; Paik, S.; Hassan, A.H.E.; Lee, Y.S.; Roh, E.J. Hit discovery of 4-amino-N-(4-(3-(trifluoromethyl)phenoxy)pyrimidin-5-yl)benzamide: A novel EGFR inhibitor from a designed small library. Bioorganic Chem. 2017, 75, 393–405. [Google Scholar] [CrossRef] [PubMed]
- Elkamhawy, A.; Kim, N.y.; Hassan, A.H.E.; Park, J.-e.; Yang, J.-E.; Oh, K.-S.; Lee, B.H.; Lee, M.Y.; Shin, K.J.; Lee, K.-T.; et al. Design, synthesis and biological evaluation of novel thiazolidinedione derivatives as irreversible allosteric IKK-β modulators. Eur. J. Med. Chem. 2018, 157, 691–704. [Google Scholar] [CrossRef] [PubMed]
- Elkamhawy, A.; youn Kim, N.; Hassan, A.H.E.; Park, J.-e.; Yang, J.-E.; Elsherbeny, M.H.; Paik, S.; Oh, K.-S.; Lee, B.H.; Lee, M.Y.; et al. Optimization study towards more potent thiazolidine-2,4-dione IKK-β modulator: Synthesis, biological evaluation and in silico docking simulation. Bioorg. Chem. 2019, 92, 103261. [Google Scholar] [CrossRef] [PubMed]
- Al-Sanea, M.M.; Elkamhawy, A.; Paik, S.; Lee, K.; El Kerdawy, A.M.; Syed Nasir Abbas, B.; Joo Roh, E.; Eldehna, W.M.; Elshemy, H.A.H.; Bakr, R.B.; et al. Sulfonamide-based 4-anilinoquinoline derivatives as novel dual Aurora kinase (AURKA/B) inhibitors: Synthesis, biological evaluation and in silico insights. Bioorg. Med. Chem. 2020, 28, 115525. [Google Scholar] [CrossRef]
- Zhao, J.; Huang, Y.; Ma, G.; Lin, L.; Feng, P. One-Pot Protocol To Synthesize 2-Aminophenols from Anilines via Palladium-Catalyzed C–H Acetoxylation. Organometallics 2019, 38, 2084–2091. [Google Scholar] [CrossRef]
- Ellis, H.; Ma, C.X. PI3K Inhibitors in Breast Cancer Therapy. Curr. Oncol. Rep. 2019, 21, 110. [Google Scholar] [CrossRef]
- Wang, M.; Wang, T.; Zhang, X.; Wu, X.; Jiang, S. Cyclin-dependent kinase 7 inhibitors in cancer therapy. Future Med. Chem. 2020, 12, 813–833. [Google Scholar] [CrossRef]
- Otto, T.; Sicinski, P. Cell cycle proteins as promising targets in cancer therapy. Nat. Rev. Cancer 2017, 17, 93–115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Degirmenci, U.; Wang, M.; Hu, J. Targeting Aberrant RAS/RAF/MEK/ERK Signaling for Cancer Therapy. Cells 2020, 9, 198. [Google Scholar] [CrossRef] [Green Version]
- Wang, P.F.; Qiu, H.Y.; He, Y.; Zhu, H.L. Cyclin-dependent kinase 4/6 inhibitors for cancer therapy: A patent review (2015–2019). Expert Opin. Ther. Pat. 2020, 30, 795–805. [Google Scholar] [CrossRef] [PubMed]
- Roskoski, R., Jr. Small molecule inhibitors targeting the EGFR/ErbB family of protein-tyrosine kinases in human cancers. Pharmacol. Res. 2019, 139, 395–411. [Google Scholar] [CrossRef]
- Zhong, L.; Li, Y.; Xiong, L.; Wang, W.; Wu, M.; Yuan, T.; Yang, W.; Tian, C.; Miao, Z.; Wang, T.; et al. Small molecules in targeted cancer therapy: Advances, challenges, and future perspectives. Signal Transduct. Target. Ther. 2021, 6, 201. [Google Scholar] [CrossRef]
- Li, T.; Wang, N.; Zhang, T.; Zhang, B.; Sajeevan, T.P.; Joseph, V.; Armstrong, L.; He, S.; Yan, X.; Naman, C.B. A Systematic Review of Recently Reported Marine Derived Natural Product Kinase Inhibitors. Mar. Drugs 2019, 17, 493. [Google Scholar] [CrossRef] [Green Version]
- Franco, L.H.; Joffé, E.B.d.K.; Puricelli, L.; Tatian, M.; Seldes, A.M.; Palermo, J.A. Indole Alkaloids from the Tunicate Aplidium meridianum. J. Nat. Prod. 1998, 61, 1130–1132. [Google Scholar] [CrossRef]
- Gompel, M.; Leost, M.; De Kier Joffe, E.B.; Puricelli, L.; Franco, L.H.; Palermo, J.; Meijer, L. Meridianins, a new family of protein kinase inhibitors isolated from the ascidian Aplidium meridianum. Bioorg. Med. Chem. Lett. 2004, 14, 1703–1707. [Google Scholar] [CrossRef]
- Hsu, M.-H.; Hsieh, C.-Y.; Kapoor, M.; Chang, J.-H.; Chu, H.-L.; Cheng, T.-M.; Hsu, K.-C.; Lin, T.E.; Tsai, F.-Y.; Horng, J.-C. Leucettamine B analogs and their carborane derivative as potential anti-cancer agents: Design, synthesis, and biological evaluation. Bioorg. Chem. 2020, 98, 103729. [Google Scholar] [CrossRef]
- Chan, G.W.; Mong, S.; Hemling, M.E.; Freyer, A.J.; Offen, P.H.; DeBrosse, C.W.; Sarau, H.M.; Westley, J.W. New Leukotriene B4 Receptor Antagonist: Leucettamine A and Related Imidazole Alkaloids from the Marine Sponge Leucetta microraphis. J. Nat. Prod. 1993, 56, 116–121. [Google Scholar] [CrossRef] [PubMed]
- Debdab, M.; Carreaux, F.; Renault, S.; Soundararajan, M.; Fedorov, O.; Filippakopoulos, P.; Lozach, O.; Babault, L.; Tahtouh, T.; Baratte, B.; et al. Leucettines, a Class of Potent Inhibitors of cdc2-Like Kinases and Dual Specificity, Tyrosine Phosphorylation Regulated Kinases Derived from the Marine Sponge Leucettamine B: Modulation of Alternative Pre-RNA Splicing. J. Med. Chem. 2011, 54, 4172–4186. [Google Scholar] [CrossRef]
- Debdab, M.; Renault, S.; Lozach, O.; Meijer, L.; Paquin, L.; Carreaux, F.; Bazureau, J.-P. Synthesis and preliminary biological evaluation of new derivatives of the marine alkaloid leucettamine B as kinase inhibitors. Eur. J. Med. Chem. 2010, 45, 805–810. [Google Scholar] [CrossRef] [PubMed]
- Tahtouh, T.; Elkins, J.M.; Filippakopoulos, P.; Soundararajan, M.; Burgy, G.; Durieu, E.; Cochet, C.; Schmid, R.S.; Lo, D.C.; Delhommel, F.; et al. Selectivity, Cocrystal Structures, and Neuroprotective Properties of Leucettines, a Family of Protein Kinase Inhibitors Derived from the Marine Sponge Alkaloid Leucettamine B. J. Med. Chem. 2012, 55, 9312–9330. [Google Scholar] [CrossRef] [PubMed]
- Park, J.-e.; Elkamhawy, A.; Hassan, A.H.E.; Pae, A.N.; Lee, J.; Paik, S.; Park, B.-G.; Roh, E.J. Synthesis and evaluation of new pyridyl/pyrazinyl thiourea derivatives: Neuroprotection against amyloid-β-induced toxicity. Eur. J. Med. Chem. 2017, 141, 322–334. [Google Scholar] [CrossRef] [PubMed]
- Elkamhawy, A.; Hassan, A.H.E.; Paik, S.; Sup Lee, Y.; Lee, H.H.; Shin, J.S.; Lee, K.T.; Roh, E.J. EGFR inhibitors from cancer to inflammation: Discovery of 4-fluoro-N-(4-(3-(trifluoromethyl)phenoxy)pyrimidin-5-yl)benzamide as a novel anti-inflammatory EGFR inhibitor. Bioorg. Chem. 2019, 86, 112–118. [Google Scholar] [CrossRef]
- Farag, A.K.; Elkamhawy, A.; Londhe, A.M.; Lee, K.T.; Pae, A.N.; Roh, E.J. Novel LCK/FMS inhibitors based on phenoxypyrimidine scaffold as potential treatment for inflammatory disorders. Eur. J. Med. Chem. 2017, 141, 657–675. [Google Scholar] [CrossRef]
- Skehan, P.; Storeng, R.; Scudiero, D.; Monks, A.; McMahon, J.; Vistica, D.; Warren, J.T.; Bokesch, H.; Kenney, S.; Boyd, M.R. New colorimetric cytotoxicity assay for anticancer-drug screening. JNCI J. Natl. Cancer Inst. 1990, 82, 1107–1112. [Google Scholar] [CrossRef] [PubMed]
- Elkamhawy, A.; Al-Sanea, M.M.; Song, C.; Sim, T.; Roh, E.J. Design and Synthesis of New [1,2,3]Triazolo[4,5-d]pyrimidine Derivatives as Potential Antiproliferative Agents. Bull. Korean Chem. Soc. 2015, 36, 1863–1873. [Google Scholar] [CrossRef]
- Al-Sanea, M.M.; Elkamhawy, A.; Zakaria, A.; Park, B.S.; Kwon, Y.; Lee, S.H.; Lee, S.W.; Kim, I.T. Synthesis and in Vitro Screening of Phenylbipyridinylpyrazole Derivatives as Potential Antiproliferative Agents. Molecules 2015, 20, 1031–1045. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El-Gamal, M.I.; Anbar, H.S.; Yoo, K.H.; Oh, C.H. FMS Kinase Inhibitors: Current Status and Future Prospects. Med. Res. Rev. 2013, 33, 599–636. [Google Scholar] [CrossRef]
- Sun, Y.; Yang, Y.; Zhao, Y.; Li, X.; Zhang, Y.; Liu, Z. The role of the tyrosine kinase Lyn in allergy and cancer. Mol. Immunol. 2021, 131, 121–126. [Google Scholar] [CrossRef]
- Chen, D.; Zhou, X.Z.; Lee, T.H. Death-Associated Protein Kinase 1 as a Promising Drug Target in Cancer and Alzheimer’s Disease. Recent Pat. Anti-Cancer Drug Discov. 2019, 14, 144–157. [Google Scholar] [CrossRef]
- Singh, P.; Ravanan, P.; Talwar, P. Death Associated Protein Kinase 1 (DAPK1): A Regulator of Apoptosis and Autophagy. Front. Mol. Neurosci. 2016, 9, 46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agodi, A.; Barchitta, M.; Quattrocchi, A.; Maugeri, A.; Vinciguerra, M. DAPK1 Promoter Methylation and Cervical Cancer Risk: A Systematic Review and a Meta-Analysis. PLoS ONE 2015, 10, e0135078. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elkamhawy, A.; Ali, E.M.H.; Lee, K. New horizons in drug discovery of lymphocyte-specific protein tyrosine kinase (Lck) inhibitors: A decade review (2011–2021) focussing on structure-activity relationship (SAR) and docking insights. J. Enzym. Inhib. Med. Chem. 2021, 36, 1574–1602. [Google Scholar] [CrossRef] [PubMed]
- Kitagawa, D.; Yokota, K.; Gouda, M.; Narumi, Y.; Ohmoto, H.; Nishiwaki, E.; Akita, K.; Kirii, Y. Activity-based kinase profiling of approved tyrosine kinase inhibitors. Genes Cells 2013, 18, 110–122. [Google Scholar] [CrossRef]
- Teicher, B.A. Anticancer Drug Development Guide: Preclinical Screening, Clinical Trials, and Approval; Springer Science & Business Media: Berlin/Heidelberg, Germany, 2013. [Google Scholar]
- Di, L.; Kerns, E.H. Drug-Like Properties: Concepts, Structure Design and Methods from ADME to Toxicity Optimization; Academic Press: Cambridge, MA, USA, 2015. [Google Scholar]
- Zhang, P.; Xu, S.; Zhu, Z.; Xu, J. Multi-target design strategies for the improved treatment of Alzheimer’s disease. Eur. J. Med. Chem. 2019, 176, 228–247. [Google Scholar] [CrossRef] [PubMed]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daina, A.; Zoete, V. A BOILED-Egg to Predict Gastrointestinal Absorption and Brain Penetration of Small Molecules. ChemMedChem 2016, 11, 1117–1121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alavijeh, M.S.; Chishty, M.; Qaiser, M.Z.; Palmer, A.M. Drug metabolism and pharmacokinetics, the blood-brain barrier, and central nervous system drug discovery. NeuroRx 2005, 2, 554–571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pajouhesh, H.; Lenz, G.R. Medicinal chemical properties of successful central nervous system drugs. NeuroRx 2005, 2, 541–553. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type of Kinase | Family | Kinase | Percent Inhibition |
|---|---|---|---|
| Receptor Tyrosine Kinases | TAM family | c-MER | 4.4 ± 0.1 |
| EGF receptor family | EGFR | 26.99 ± 0.9 | |
| PVR family | FMS | 82.5 ± 0.6 | |
| PDGFRα | 24.0 ± 0.4 | ||
| FLT1/VEGFR1 | −2.4 ± 1.7 | ||
| KDR/VEGFR2 | 7.9 ± 1.4 | ||
| HGF receptor | c-MET | 12.0 ± 13.7 | |
| Non-Receptor Tyrosine Kinases | SRC-B family | LCK | 81.4 ± 0.6 |
| LYN | 75.2 ± 0.0 | ||
| JAK family | JAK3 | 11.2 ± 1.2 | |
| Tyrosine Kinase-Like kinases | RAF family | BRAF | 4.1 ± 10.5 |
| Calcium/Calmodulin-dependent kinases (CAMKs) | DAPK family | DAPK1 | 55 ± 1.1 |
| CMGC serine/threonine kinases | Cyclin-dependent kinase family | CDK2/cyclin A | 20.1 ± 0.1 |
| P21-activated serine/threonine kinases | PAK Family | PAK1 | −29.1 ± 3.4 |
| Cpd | Chemical Structure | Percent Inhibition a | |||
|---|---|---|---|---|---|
| DAPK1 | FMS | LCK | LYN | ||
| 5a | 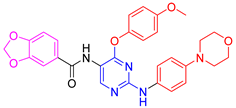 | 55 ± 1.1 | 82.5 ± 0.6 | 81.4 ± 0.6 | 75.2 ± 0.0 |
| 5b | 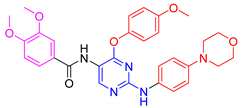 | 65 ± 1.2 | 44.1 ± 0.2 | 62.3 ± 0.8 | 36.9 ± 4.5 |
| 5c | 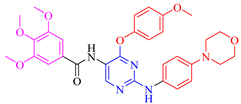 | 50 ± 0.1 | 69.9 ± 0.4 | 19.4 ± 0.9 | −1.6 ± 0.3 |
| 5d | 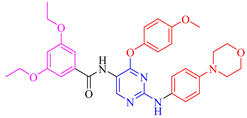 | 51.6 ± 0.5 | 95.1 ± 0.3 | 38.3 ± 4.2 | 31.5 ± 5.8 |
| 5e | 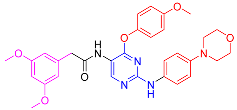 | 47.1 ± 0.7 | 75.5 ± 0.8 | 39.5 ± 0.4 | 7.9 ± 0.4 |
| 5f | 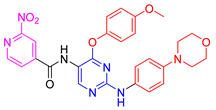 | 65.5 ± 1.4 | 65.4 ± 0.1 | 72.6 ± 0.6 | 34.1 ± 2.2 |
| 5g |  | 54.6 ± 0.8 | 90.6 ± 0.8 | 96.9 ± 0.3 | 96.4 ± 0.1 |
| Compound | FMS IC50 (nM) | LCK IC50 (nM) | LYN IC50 (nM) |
|---|---|---|---|
| 5d | 213 ± 1 | NT | NT |
| 5g | 110 ± 8 | 87.7 ± 8.3 | 169 ± 31 |
| Imatinib | 1000 | 160 | 190 |
| Cancer Type | Cell Line | Percent Growth Inhibition (GI) | ||||||
|---|---|---|---|---|---|---|---|---|
| 5a | 5b | 5c | 5d | 5e | 5f | 5g | ||
| Leukemia | CCRF-CEM | 14.46 | 5.37 | 30.5 | 26.1 | −4.73 | 16.66 | 12.96 |
| HL-60(TB) | 16.16 | 16.41 | 31.0 | 22.8 | −6.34 | −3.19 | 13.94 | |
| K-562 | 23.4 | 8.76 | 20.4 | 30.7 | 2.31 | 10.89 | 29.1 | |
| MOLT-4 | 31.1 | 19.9 | 31.6 | 38.2 | −0.81 | −5.98 | 43.1 | |
| RPMI-8226 | 21.8 | 11.12 | 18.76 | 25.6 | 11.31 | 16.32 | 20.5 | |
| SR | 37.1 | 18.75 | 64.4 | 33.2 | 13.21 | 27.4 | 60.5 | |
| Non-Small Cell Lung Cancer | HOP-92 | 6.16 | −1.19 | 15.76 | 3.34 | 4.46 | 1.12 | 20.2 |
| NCI-H522 | 22.7 | −3.5 | 2.25 | 29.8 | 8.4 | 9.77 | 26.4 | |
| Colon Cancer | COLO 205 | 1.75 | −2.21 | 21.6 | −2.11 | −22.42 | −14.56 | 4.7 |
| HCT-116 | 14.57 | 6.01 | 23.9 | 6.73 | 0.34 | −0.19 | 23.7 | |
| HCT-15 | 26.0 | 12.32 | 34.4 | 13.24 | 1.13 | 32.8 | 31.7 | |
| HT29 | 28.3 | −2.93 | 47.4 | 9.54 | −8.47 | −8.86 | 24.3 | |
| KM12 | 12.72 | −4.34 | 3.99 | 14 | 1.32 | −1.85 | 20.2 | |
| CNS Cancer | SF-539 | 4.73 | −3.17 | 7.64 | 8.01 | 2.09 | 7.72 | 21.1 |
| SNB-75 | −3.98 | −4.29 | 3.59 | 19.2 | −3.1 | −4.975 | 21.5 | |
| Melanoma | UACC-62 | 7.38 | 0.1 | −0.63 | 11.99 | 1.34 | 21.4 | 14.69 |
| Ovarian Cancer | IGROV1 | 5.92 | 3.6 | 11.87 | 0.72 | −9.02 | 1.91 | 33.9 |
| Renal Cancer | 786-0 | 25.3 | 17.64 | 44.2 | 7.41 | 0.75 | 5.78 | 25.0 |
| A498 | 22.4 | −5.56 | 38.3 | 17.86 | −6.85 | 6.39 | 18.93 | |
| ACHN | 8.91 | −7.28 | 4.83 | 20.4 | −0.26 | 1.61 | 21.4 | |
| CAKI-1 | 33.9 | 20.6 | 28.1 | 20.6 | −2.58 | 13.69 | 29.4 | |
| RXF 393 | 50.5 | 20.4 | 50.6 | 27.5 | −8.16 | −0.36 | 70.1 | |
| UO-31 | 36.7 | 33.6 | 42.3 | 29.9 | 19.36 | 29 | 43.7 | |
| Prostate Cancer | PC-3 | 11.71 | 0.75 | 11.58 | 27.3 | 8.05 | 13.85 | 24.9 |
| Breast Cancer | MCF7 | 8.51 | 4.24 | 7.94 | 14.43 | 18.87 | 12.54 | 21.5 |
| HS 578T | 6.26 | 5.09 | 3.23 | 7.96 | 1.64 | 6.7 | 22.7 | |
| T-47D | 6.78 | 2.41 | 0.52 | 18.4 | 10.16 | 5.93 | 28.2 | |
| Compound | Docking Score | Ligand Atoms | Receptor Atoms | Interaction Type | Percent Inhibition |
|---|---|---|---|---|---|
| 5a | −9.75 | N3 N7 O38 | Met319 Met319 Asp382 | HBA HBD HBA | 81.4 ± 0.6 |
| 5b | −8.15 | N3 N7 | Met319 Met319 | HBA HBD | 62.3 ± 0.8 |
| 5c | −7.39 | N7 Pyridine ring | Met319 Lys273 | HBD π-π stacking | 19.4 ± 1.0 |
| 5d | −6.84 | O31 O27 | Ser329 Asp382 | HBA HBA | 38.3 ± 4.2 |
| 5e | −8.96 | O31 O27 | Met319 SER323 | HBA HBA | 37.5 ± 0.3 |
| 5f | −9.62 | N3 N7 Pyridine ring N38 O39 O40 | Met319 Met319 Lys273 Glu288 Phe383 Asp382 | HBA HBD π-π stacking Salt bridge HBA HBA | 72.6 ± 0.6 |
| 5g | −9.32 | N3 N7 Pyridine ring | Met319 Met319 Lys273 | HBA HBD π-π stacking | 96.9 ± 0.3 |
| Compound | TPSA | Solubility in Water | BBB Permeability | Intestinal Absorption |
|---|---|---|---|---|
| 5a | 116.3 | Moderately soluble | no | high |
| 5b | 116.3 | Moderately soluble | no | high |
| 5c | 125.5 | Moderately soluble | no | low |
| 5d | 116.3 | Poorly soluble | no | low |
| 5e | 116.3 | Moderately soluble | no | high |
| 5f | 156.5 | Moderately soluble | no | low |
| 5g | 123.1 | Poorly soluble | no | low |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Elsherbeny, M.H.; Elkamhawy, A.; Nada, H.; Abdellattif, M.H.; Lee, K.; Roh, E.J. Development of New Meridianin/Leucettine-Derived Hybrid Small Molecules as Nanomolar Multi-Kinase Inhibitors with Antitumor Activity. Biomedicines 2021, 9, 1131. https://doi.org/10.3390/biomedicines9091131
Elsherbeny MH, Elkamhawy A, Nada H, Abdellattif MH, Lee K, Roh EJ. Development of New Meridianin/Leucettine-Derived Hybrid Small Molecules as Nanomolar Multi-Kinase Inhibitors with Antitumor Activity. Biomedicines. 2021; 9(9):1131. https://doi.org/10.3390/biomedicines9091131
Chicago/Turabian StyleElsherbeny, Mohamed H., Ahmed Elkamhawy, Hossam Nada, Magda H. Abdellattif, Kyeong Lee, and Eun Joo Roh. 2021. "Development of New Meridianin/Leucettine-Derived Hybrid Small Molecules as Nanomolar Multi-Kinase Inhibitors with Antitumor Activity" Biomedicines 9, no. 9: 1131. https://doi.org/10.3390/biomedicines9091131
APA StyleElsherbeny, M. H., Elkamhawy, A., Nada, H., Abdellattif, M. H., Lee, K., & Roh, E. J. (2021). Development of New Meridianin/Leucettine-Derived Hybrid Small Molecules as Nanomolar Multi-Kinase Inhibitors with Antitumor Activity. Biomedicines, 9(9), 1131. https://doi.org/10.3390/biomedicines9091131