
Albumin Fusion at the N-Terminus or C-Terminus of HM-3 Leads to Improved Pharmacokinetics and Bioactivities

 ,
,
Abstract
:
1. Introduction
2. Materials and Methods
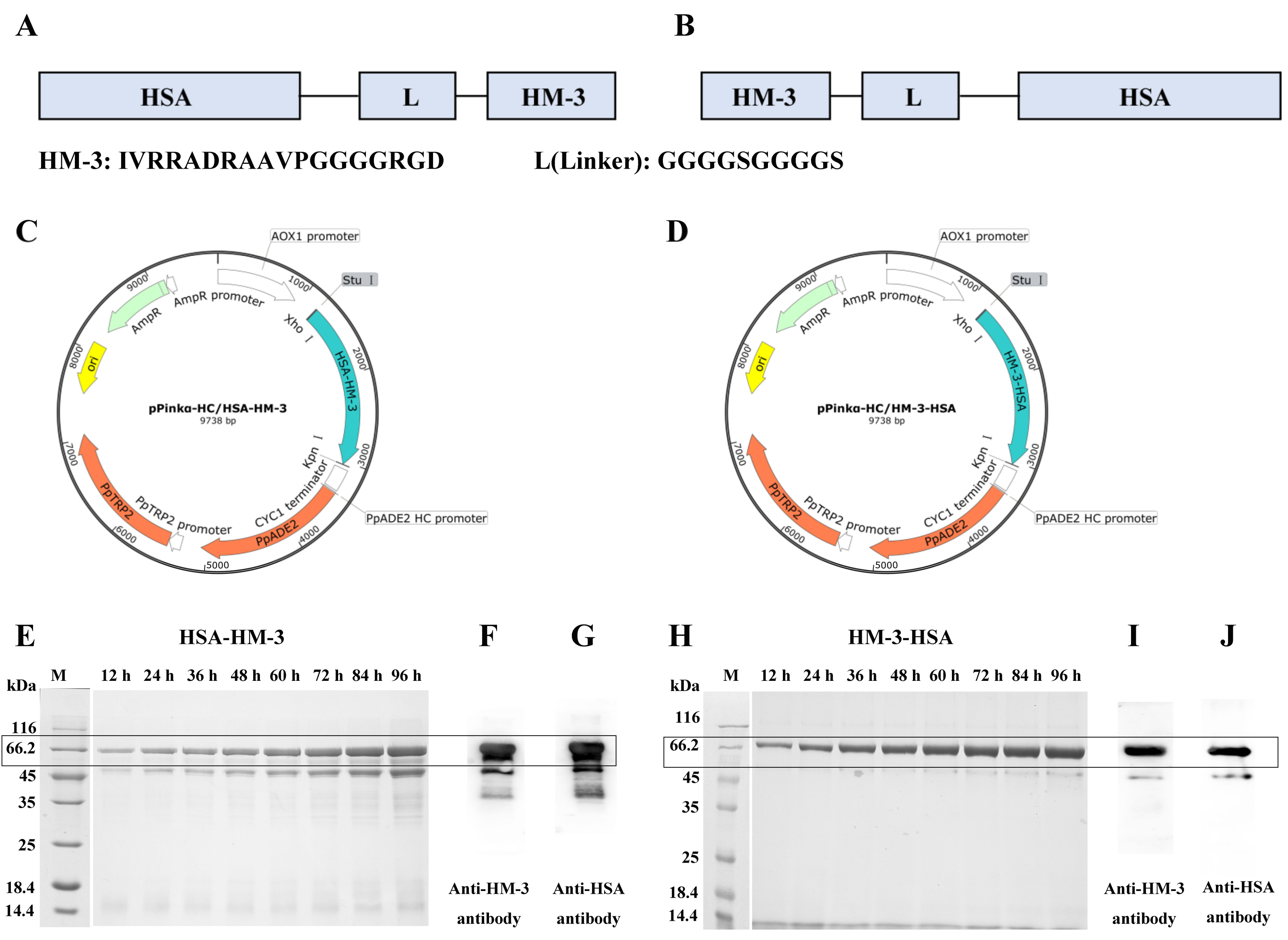
2.1. Construction of the Expression Plasmid
2.2. Expression and Purification
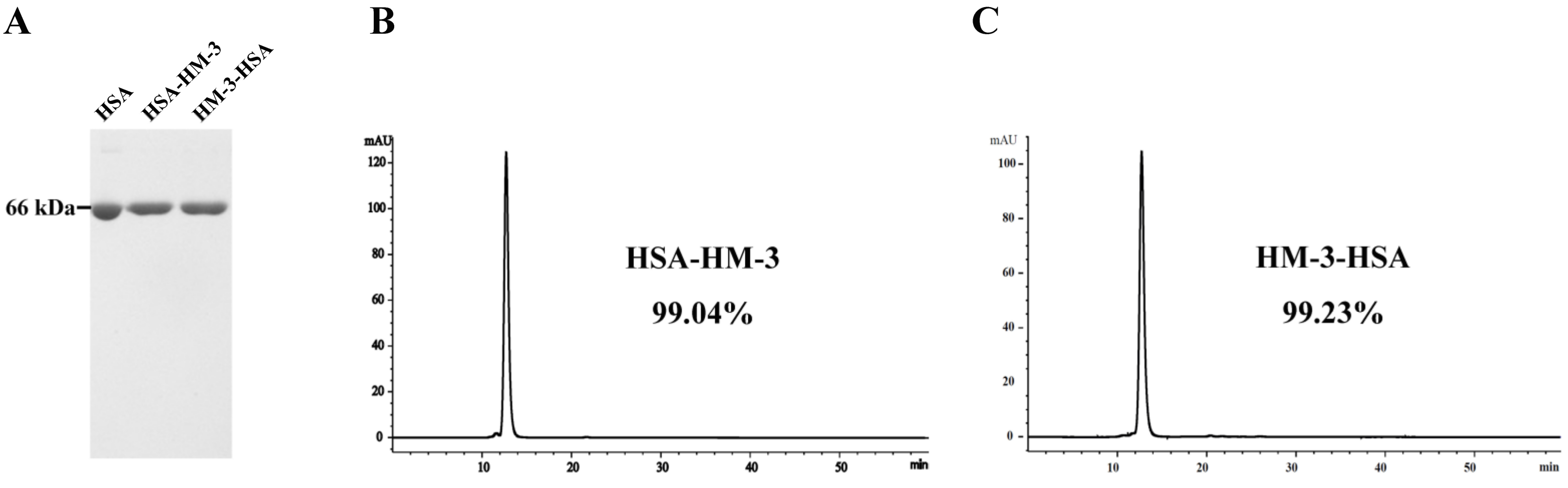
2.3. HPLC Analysis
2.4. Stability of Fusion Proteins Test
2.5. Circular Dichroism Spectrometry
2.6. Cell Lines and Animals
2.7. Pharmacokinetics Analysis
2.8. MTT Assay
2.9. Colony Formation Assay
2.10. Cell Cycle Analysis
2.11. Migration Assay
2.12. Invasion Assay
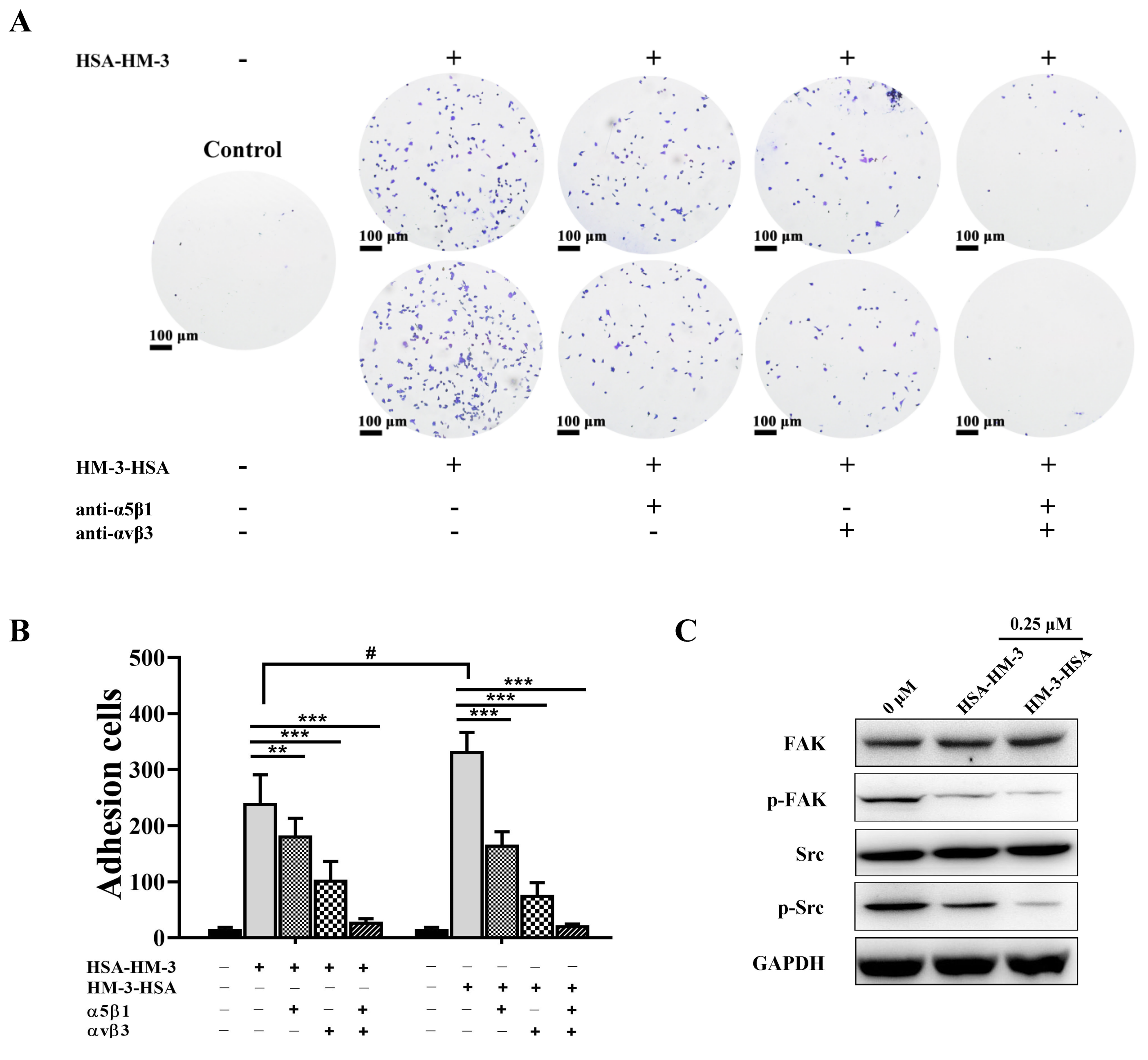
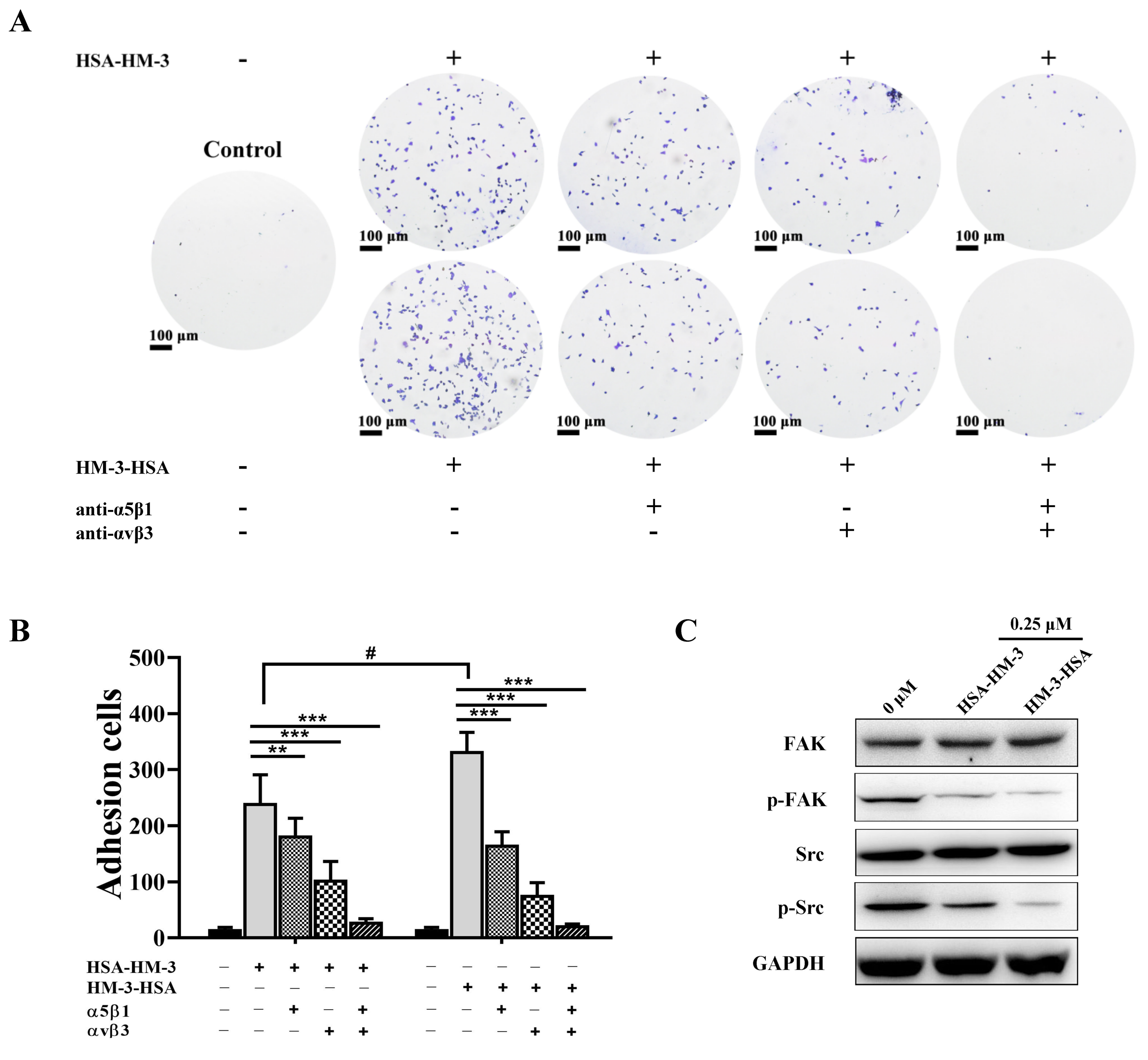
2.13. Integrin Antibody-Blocking Assay
2.14. Western Blot Assay
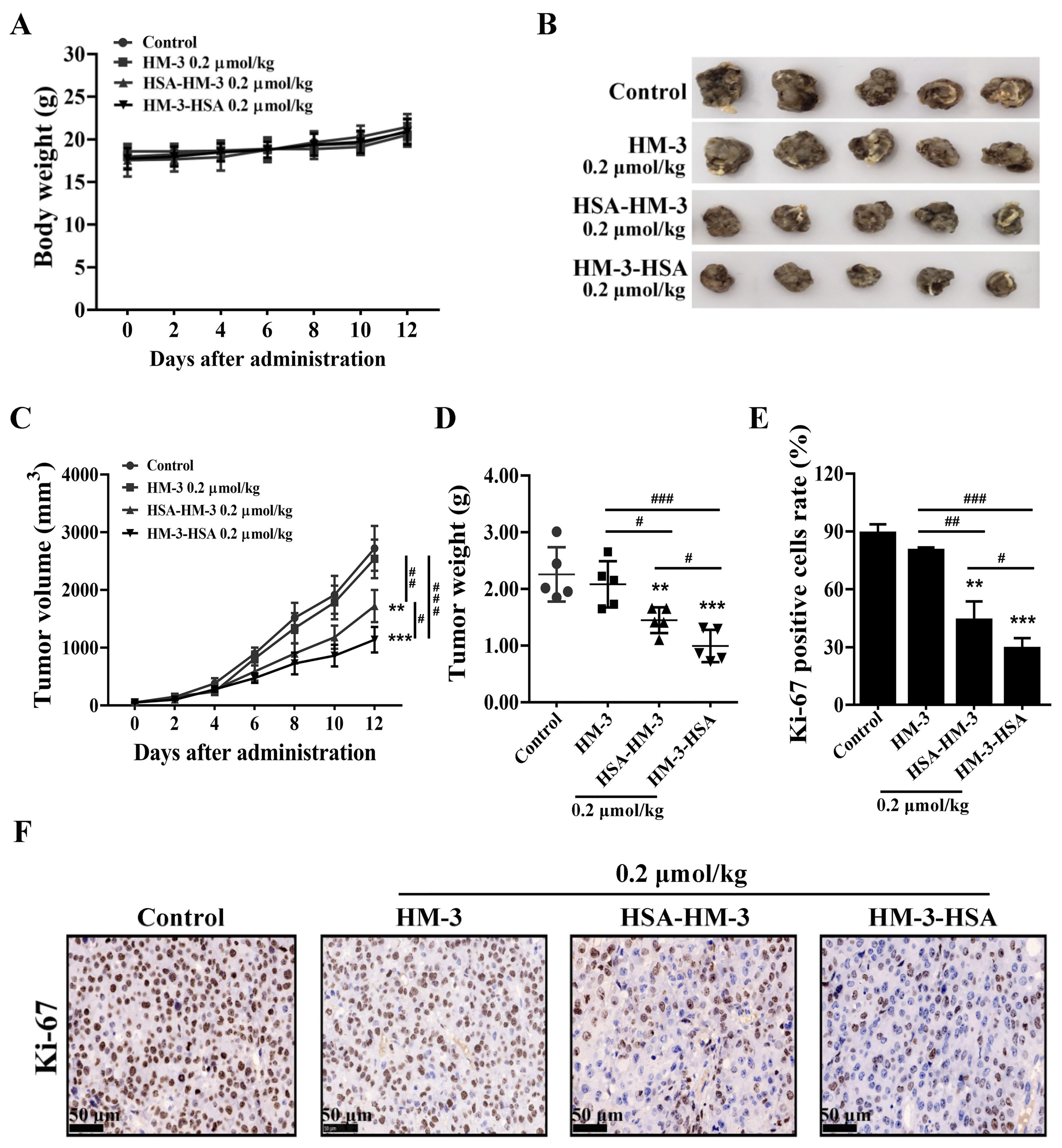
2.15. Tumor Implantation and Treatment
2.16. Immunohistochemistry
2.17. Analytic Methods
3. Results
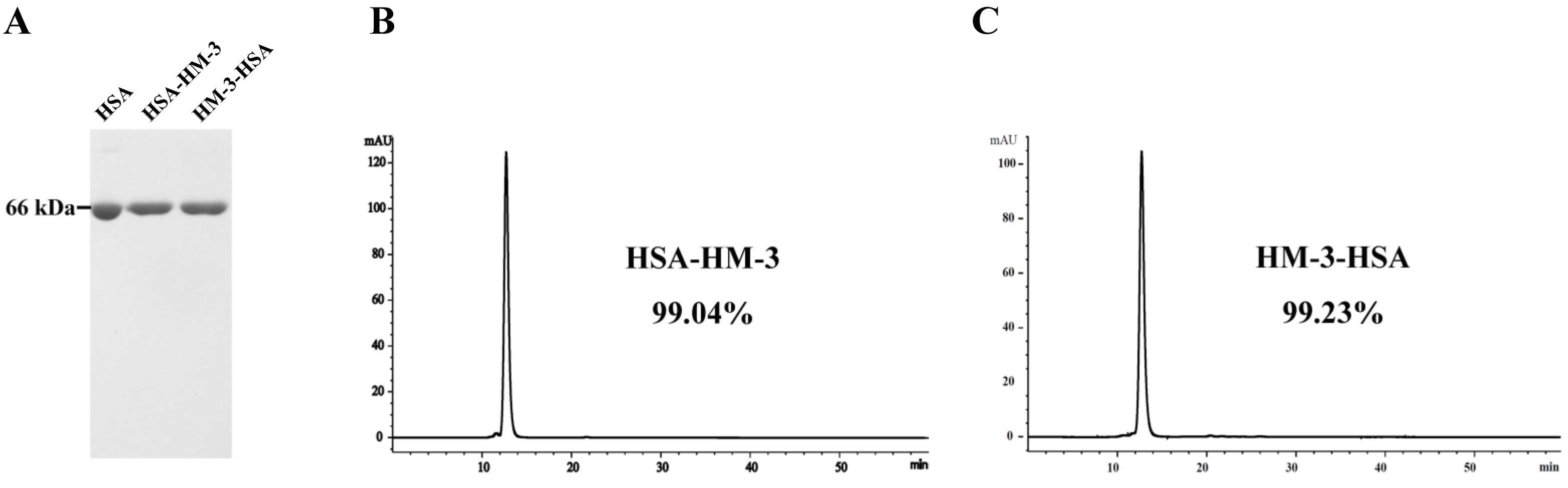
3.1. Expression and WB Identification of the Fusion Proteins
3.2. Purification and HPLC Analysis of the Fusion Proteins
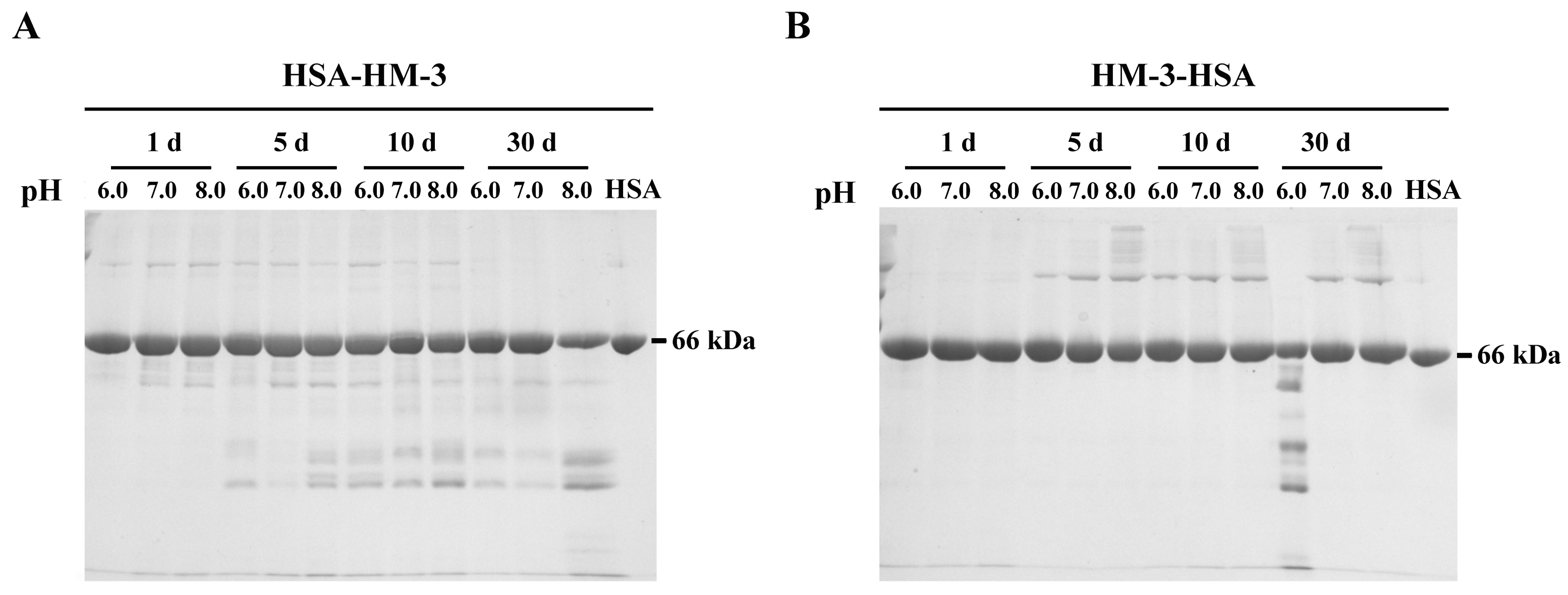
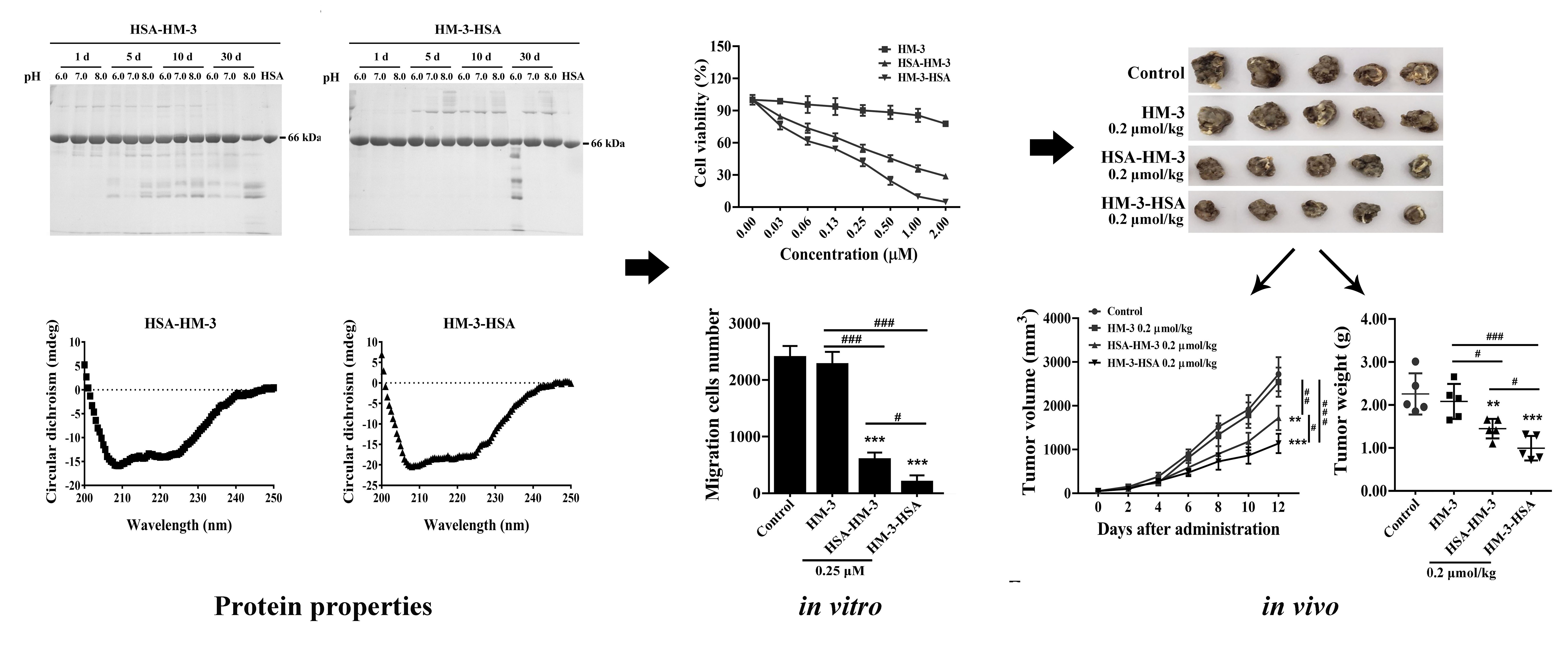
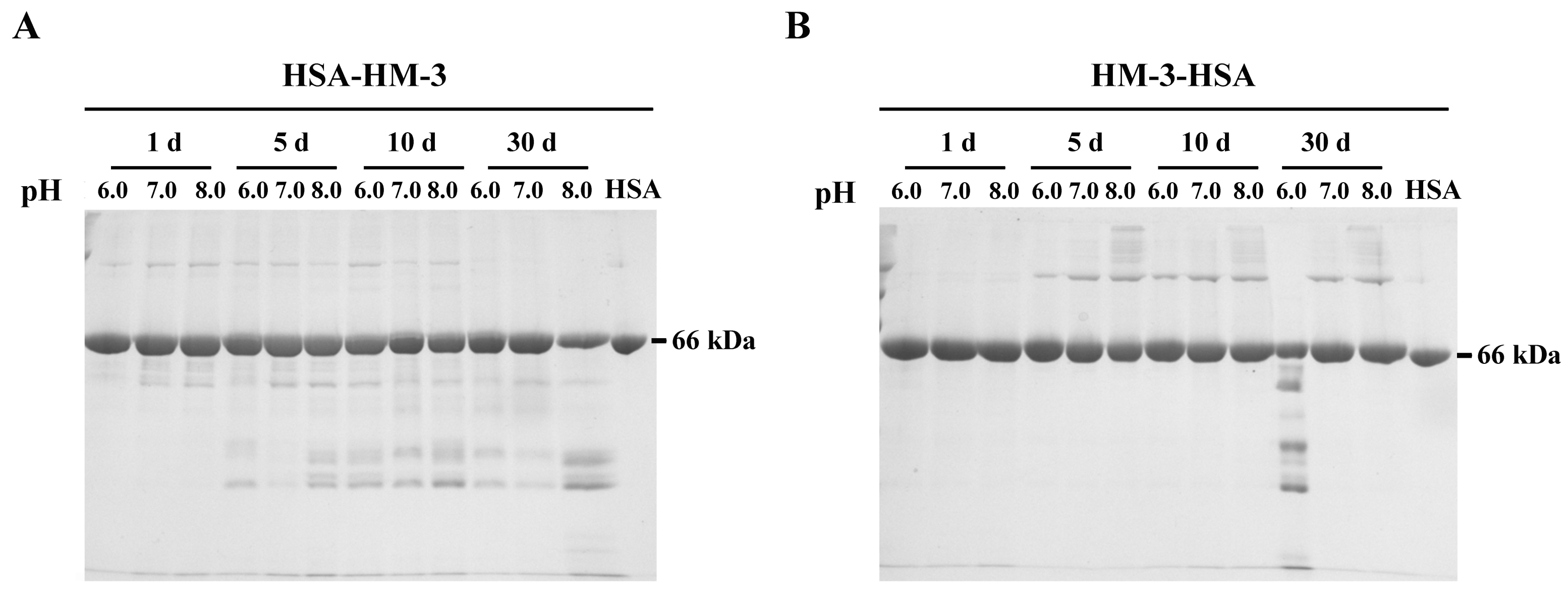
3.3. Stability of Fusion Proteins
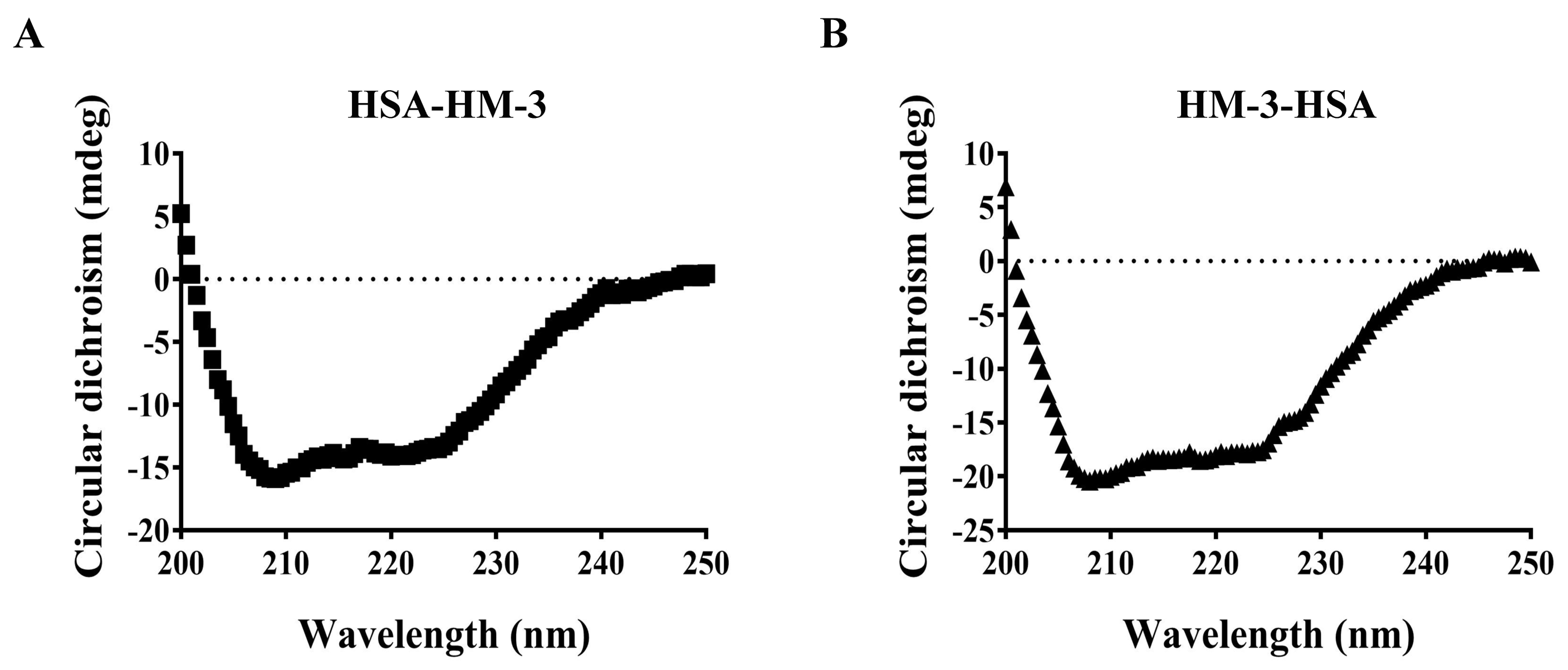
3.4. Circular Dichroism Spectral Analysis
3.5. Pharmacokinetic Analysis
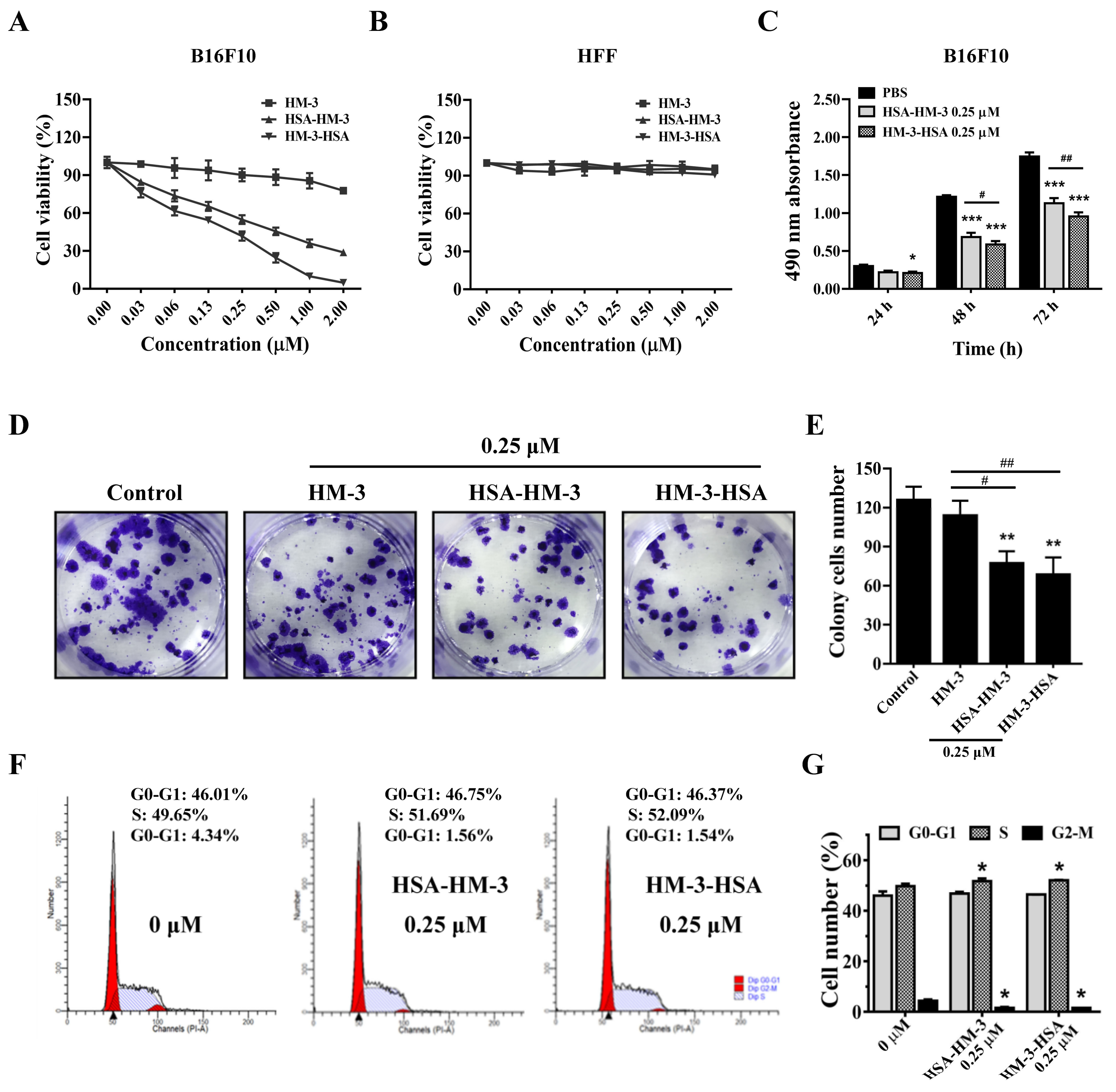
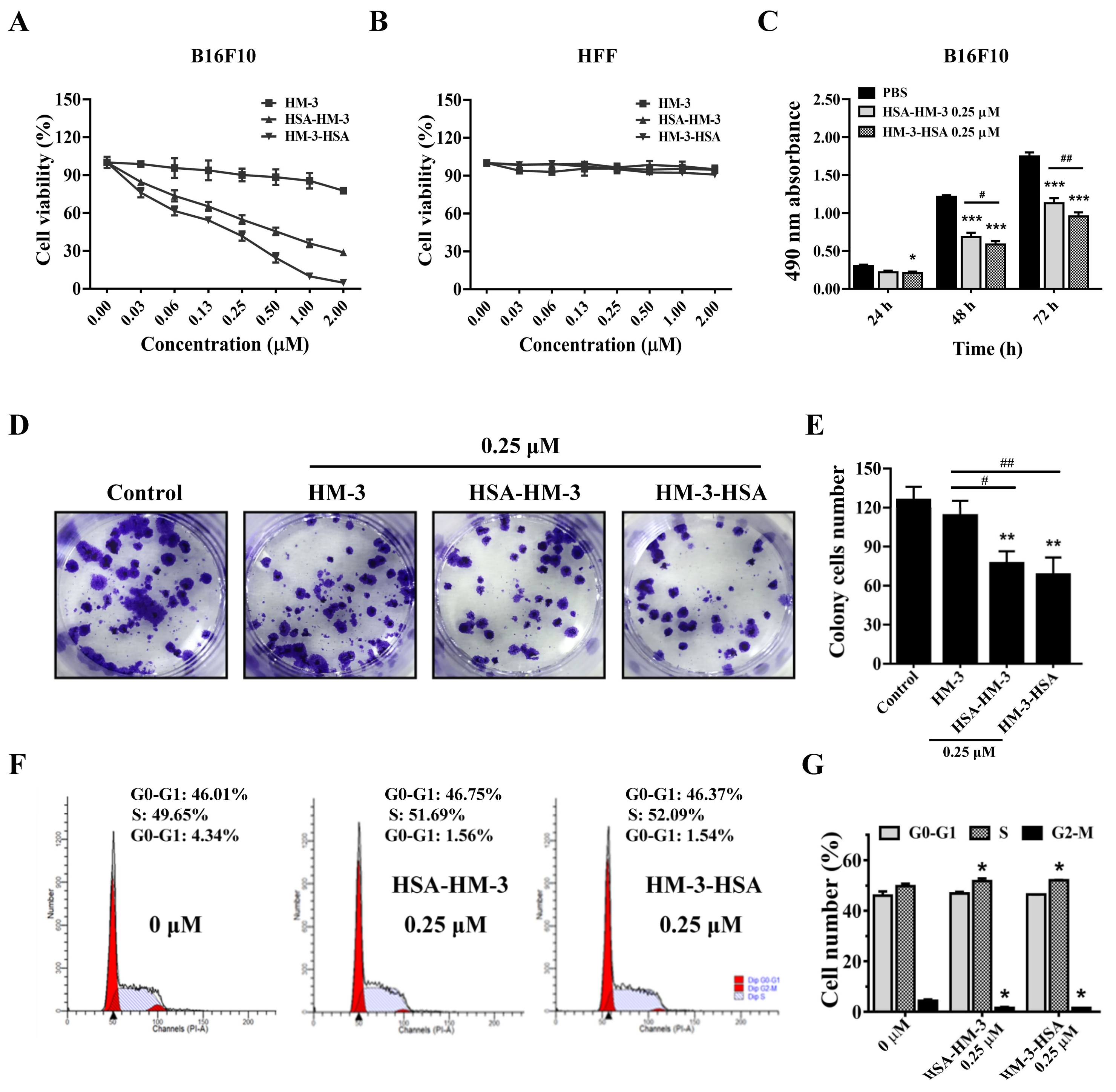
3.6. HM-3/HSA Inhibited B16F10 Cells’ Viability
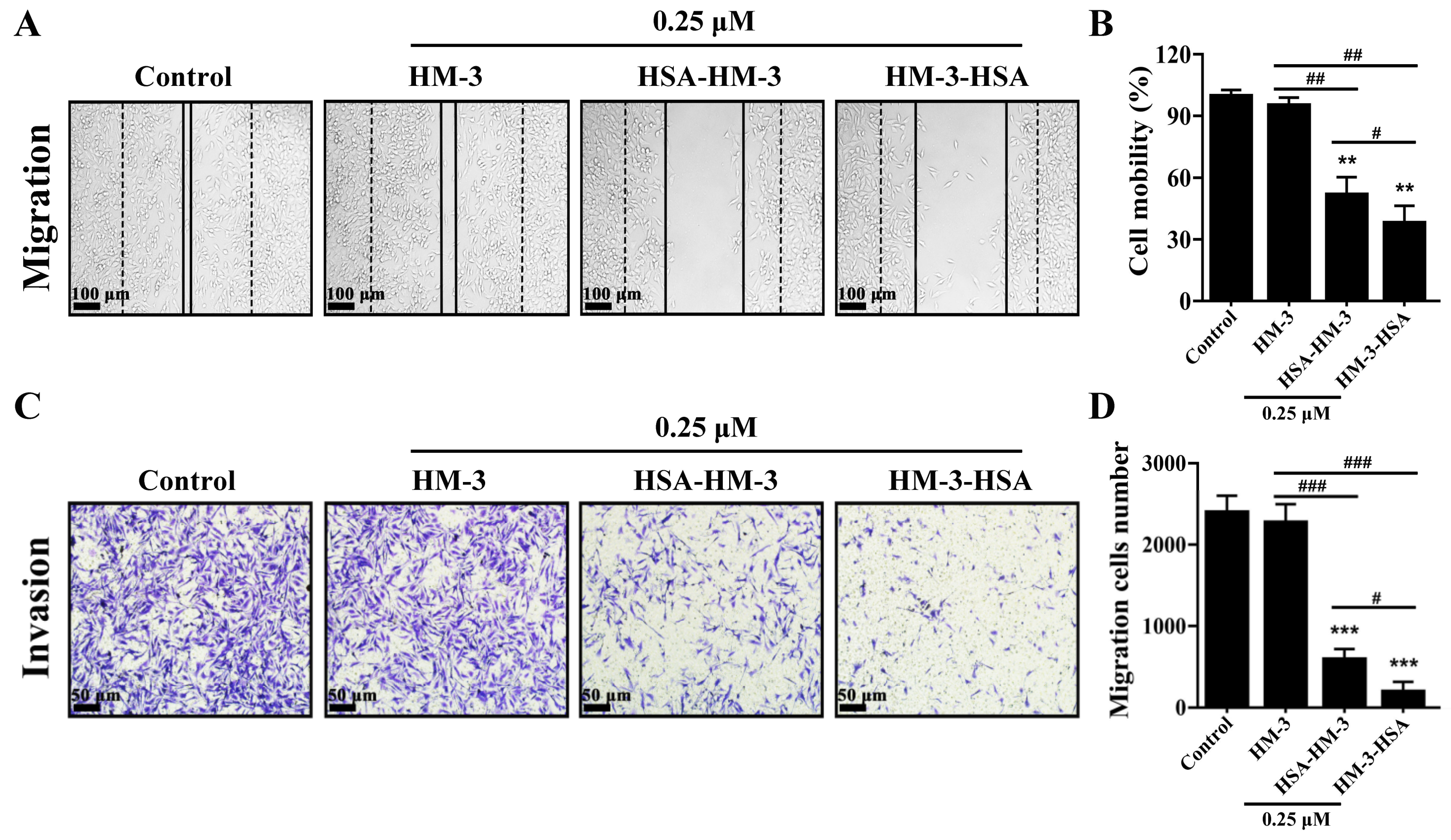
3.7. HM-3/HSA Inhibited B16F10 Cells’ Migration and Invasion
3.8. HM-3/HSA Bind to Integrin and Inhibited Integrin Signaling Pathways
3.9. HM-3/HSA Inhibited the Growth of the B16F10 Tumor
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| DMSO | dimethyl sulfoxide |
| FBS | fetal bovine serum |
| PBS | phosphate buffer saline |
| HSA | human serum albumin |
| MTT | 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide |
| PCR | polymerase chain reaction |
| CD | circular dichroism |
References
- Xu, H.M.; Yin, R.; Chen, L.; Siraj, S.; Huang, X.; Wang, M.; Fang, H.; Wang, Y. An RGD-modified endostatin-derived synthetic peptide shows antitumor activity in vivo. Bioconjug. Chem. 2008, 19, 1980–1986. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Pan, L.; Ren, Y.; Yang, Y.; Huang, X.; Liu, Z. RGD-modified angiogenesis inhibitor HM-3 dose: Dual function during cancer treatment. Bioconjug. Chem. 2011, 22, 1386–1393. [Google Scholar] [CrossRef] [PubMed]
- Wickström, S.A.; Alitalo, K.; Keski-Oja, J. An endostatin-derived peptide interacts with integrins and regulates actin cytoskeleton and migration of endothelial cells. J. Biol. Chem. 2004, 279, 20178–20185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, J.; Wang, W.; Liu, C.; Li, M.; Nice, E.; Xu, H. Receptor tyrosine kinase inhibitor Sunitinib and integrin antagonist peptide HM-3 show similar lipid raft dependent biphasic regulation of tumor angiogenesis and metastasis. J. Exp. Clin. Cancer Res. 2019, 38, 381. [Google Scholar] [CrossRef] [Green Version]
- Huang, R.; Li, J.; Wang, Y.; Zhang, L.; Ma, X.; Wang, H.; Li, W.; Cao, X.; Xu, H.; Hu, J. The Protective Effect of a Long-Acting and Multi-Target HM-3-Fc Fusion Protein in Rheumatoid Arthritis. Int. J. Mol. Sci. 2018, 19, 2683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, K.; Zheng, X.; Xu, H.-M.; Zhang, J.; Chen, Y.; Xi, T.; Feng, T. Studies of poly(ethylene glycol) modification of HM-3 polypeptides. Bioconjug. Chem. 2009, 20, 932–936. [Google Scholar] [CrossRef]
- Zhu, B.; Xu, H.-M.; Zhao, L.; Huang, X.; Zhang, F. Site-specific modification of anti-angiogenesis peptide HM-3 by polyethylene glycol molecular weight of 20 kDa. J. Biochem. 2010, 148, 341–347. [Google Scholar] [CrossRef]
- Rabbani, G.; Ahn, S.N. Structure, enzymatic activities, glycation and therapeutic potential of human serum albumin: A natural cargo. Int. J. Biol. Macromol. 2019, 123, 979–990. [Google Scholar] [CrossRef]
- Kenanova, V.E.; Olafsen, T.; Salazar, F.B.; Williams, L.E.; Knowles, S.; Wu, A.M. Tuning the serum persistence of human serum albumin domain III:diabody fusion proteins. Protein Eng. Des. Sel. 2010, 23, 789–798. [Google Scholar] [CrossRef] [Green Version]
- Tan, H.; Su, W.; Zhang, W.; Wang, P.; Sattler, M.; Zou, P. Recent Advances in Half-life Extension Strategies for Therapeutic Peptides and Proteins. Curr. Pharm. Des. 2018, 24, 4932–4946. [Google Scholar] [CrossRef] [PubMed]
- Kinoshita, R.; Ishima, Y.; Chuang, V.T.G.; Nakamura, H.; Fang, J.; Watanabe, H.; Shimizu, T.; Okuhira, K.; Ishida, T.; Maeda, H.; et al. Improved anticancer effects of albumin-bound paclitaxel nanoparticle via augmentation of EPR effect and albumin-protein interactions using S-nitrosated human serum albumin dimer. Biomaterials 2017, 140, 162–169. [Google Scholar] [CrossRef]
- Li, R.; Yang, H.; Jia, D.; Nie, Q.; Cai, H.; Fan, Q.; Wan, L.; Li, L.; Lu, X. Fusion to an albumin-binding domain with a high affinity for albumin extends the circulatory half-life and enhances the in vivo antitumor effects of human TRAIL. J. Control. Release 2016, 228, 96–106. [Google Scholar] [CrossRef]
- Roscoe, I.; Parker, M.; Dong, D.; Li, X.; Li, Z. Human Serum Albumin and the p53-Derived Peptide Fusion Protein Promotes Cytotoxicity Irrespective of p53 Status in Cancer Cells. Mol. Pharm. 2018, 15, 5046–5057. [Google Scholar] [CrossRef]
- Ueda, K.; Shimizu, M.; Ohashi, A.; Murata, D.; Suzuki, T.; Kobayashi, N.; Baba, J.; Takeuchi, T.; Shiga, Y.; Nakamura, M.; et al. Albumin fusion at the N-terminus or C-terminus of human lactoferrin leads to improved pharmacokinetics and anti-proliferative effects on cancer cell lines. Eur. J. Pharm. Sci. 2020, 155, 105551. [Google Scholar] [CrossRef]
- Dou, W.F.; Lei, J.Y.; Zhang, L.F.; Xu, Z.H.; Chen, Y.; Jin, J. Expression, purification, and characterization of recombinant human serum albumin fusion protein with two human glucagon-like peptide-1 mutants in Pichia pastoris. Protein Expr. Purif. 2008, 61, 45–49. [Google Scholar] [CrossRef] [PubMed]
- Hoogenboezem, E.N.; Duvall, C.L. Harnessing albumin as a carrier for cancer therapies. Adv. Drug Deliv. Rev. 2018, 130, 73–89. [Google Scholar] [CrossRef] [PubMed]
- Ru, Y.; Zhi, D.; Guo, D.; Wang, Y.; Li, Y.; Wang, M.; Wei, S.; Wang, H.; Wang, N.; Che, J.; et al. Expression and bioactivity of recombinant human serum albumin and dTMP fusion proteins in CHO cells. Appl. Microbiol. Biotechnol. 2016, 100, 7565–7575. [Google Scholar] [CrossRef] [PubMed]
- Shiga, Y.; Murata, D.; Sugimoto, A.; Oshima, Y.; Tada, M.; Ishii-Watabe, A.; Imai, K.; Tomii, K.; Takeuchi, T.; Kagaya, S.; et al. Hinge-Deficient IgG1 Fc Fusion: Application to Human Lactoferrin. Mol. Pharm. 2017, 14, 3025–3035. [Google Scholar] [CrossRef] [PubMed]
- Valiyari, S.; Salimi, M.; Bouzari, S. Novel fusion protein NGR-sIL-24 for targetedly suppressing cancer cell growth via apoptosis. Cell Biol. Toxicol. 2020, 36, 179–193. [Google Scholar] [CrossRef]
- Wu, T.; Jiang, X.; Xu, B.; Wang, Y. Ponatinib inhibits growth of patient-derived xenograft of cholangiocarcinoma expressing FGFR2-CCDC6 fusion protein in nude mice. Nan Fang Yi Ke Da Xue Xue Bao 2020, 40, 1448–1456. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Ren, Y.; Pan, L.; Xu, H.M. In vivo anti-tumor activity of polypeptide HM-3 modified by different polyethylene glycols (PEG). Int. J. Mol. Sci. 2011, 12, 2650–2663. [Google Scholar] [CrossRef]
- Eskew, M.W.; Koslen, M.M.; Benight, A.S. Ligand binding to natural and modified human serum albumin. Anal. Biochem. 2021, 612, 113843. [Google Scholar] [CrossRef] [PubMed]
- Hseu, Y.C.; Tsou, H.T.; Kumar, K.J.; Lin, K.Y.; Chang, H.W.; Yang, H.L. The Antitumor Activity of Antrodia camphorata in Melanoma Cells: Modulation of Wnt/β-Catenin Signaling Pathways. Evid. Based Complement. Altern. Med. 2012, 2012, 197309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ratheesh, A.; Jain, M.; Gude, R.P. Antimetastatic Action of Pentoxifylline, a Methyl Xanthine Derivative, Through its Effect on PKC Mediated Integrin Transport in B16F10 Melanoma Cells. World J. Oncol. 2010, 1, 194–203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dayan, A.; Fleminger, G.; Ashur-Fabian, O. Targeting the Achilles’ heel of cancer cells via integrin-mediated delivery of ROS-generating dihydrolipoamide dehydrogenase. Oncogene 2019, 38, 5050–5061. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Jin, H.; Hu, J.; Li, X.; Ruan, H.; Xu, H.; Wei, L.; Dong, W.; Teng, F.; Gu, J.; et al. COL4A1 promotes the growth and metastasis of hepatocellular carcinoma cells by activating FAK-Src signaling. J. Exp. Clin. Cancer Res. 2020, 39, 148. [Google Scholar] [CrossRef] [PubMed]
- Mitra, S.K.; Schlaepfer, D.D. Integrin-regulated FAK-Src signaling in normal and cancer cells. Curr. Opin. Cell Biol. 2006, 18, 516–523. [Google Scholar] [CrossRef]
- Liu, Z.; Li, W.; Xu, H.M.; Huang, X.; Pan, L.; Ren, Y.; Yang, Y.; Li, Y.; Pu, C.; Zhang, C. In vivo pharmacokinetics, immunogenicity and mechanism of PEGylated antitumor polypeptide. Curr. Pharm. Des. 2012, 18, 1655–1662. [Google Scholar] [CrossRef]
- Tian, S.; Li, Q.; Yao, W.; Xu, C. Construction and characterization of a potent, long-lasting recombinant human serum albumin-interferon α1 fusion protein expressed in Pichia pastoris. Protein Expr. Purif. 2013, 90, 124–128. [Google Scholar] [CrossRef]
- Huang, Y.S.; Chen, Z.; Yang, Z.Y.; Wang, T.Y.; Zhou, L.; Wu, J.B.; Zhou, L.F. Preparation and characterization of a potent, long-lasting recombinant human serum albumin-interferon-alpha2b fusion protein expressed in Pichia pastoris. Eur. J. Pharm. Biopharm. 2007, 67, 301–308. [Google Scholar] [CrossRef]
- Ding, Y.; Fan, J.; Li, W.; Peng, Y.; Yang, R.; Deng, L.; Fu, Q. The effect of albumin fusion structure on the production and bioactivity of the somatostatin-28 fusion protein in Pichia pastoris. J. Ind. Microbiol. Biotechnol. 2014, 41, 997–1006. [Google Scholar] [CrossRef]
- Ding, Y.; Fan, J.; Li, W.; Yang, R.; Peng, Y.; Deng, L.; Wu, Y.; Fu, Q. The effect of albumin fusion patterns on the production and bioactivity of the somatostatin-14 fusion protein in Pichia pastoris. Appl. Biochem. Biotechnol. 2013, 170, 1637–1648. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Zhi, D.; Wang, H.; Ru, Y.; Ren, H.; Wang, N.; Liu, Y.; Li, Y.; Li, H. TAT-HSA-α-MSH fusion protein with extended half-life inhibits tumor necrosis factor-α in brain inflammation of mice. Appl. Microbiol. Biotechnol. 2016, 100, 5353–5361. [Google Scholar] [CrossRef] [PubMed]
- Froehlich, E.; Mandeville, J.S.; Jennings, C.J.; Sedaghat-Herati, R.; Tajmir-Riahi, H.A. Dendrimers bind human serum albumin. J. Phys. Chem. B 2009, 113, 6986–6993. [Google Scholar] [CrossRef] [PubMed]
- Syed, S.; Schuyler, P.D.; Kulczycky, M.; Sheffield, W.P. Potent antithrombin activity and delayed clearance from the circulation characterize recombinant hirudin genetically fused to albumin. Blood 1997, 89, 3243–3252. [Google Scholar] [CrossRef] [Green Version]
- Zhan, J.; Chen, Y.; Yuan, H.Y.; Li, H.; Lu, H. Fusion of HSA influences TNF-α neutralizing activity of shTNFRs. Biotechnol. Lett. 2012, 34, 417–423. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Purification Step | HSA-HM-3 | HM-3-HSA | ||||
|---|---|---|---|---|---|---|
| Volume (mL) | Concentration (mg/L) | Recovery (%) | Volume (mL) | Concentration (mg/L) | Recovery (%) | |
| Culture supernatant | 1000 | 205.5 | 100 | 1000 | 218.6 | 100 |
| DEAE Sepharose 6 Fast Flow | 355 | 492.0 | 85.0 | 391 | 481.9 | 86.2 |
| Phenyl Sepharose Fast Flow | 151 | 618.9 | 53.5 | 149 | 651.3 | 51.5 |
| Parameter | HSA-HM-3 | HM-3-HSA |
|---|---|---|
| Alpha helix | 46.4 | 56.3 |
| Parallel | 10.1 | 8.1 |
| Beta-turn | 15.2 | 12.8 |
| Random coil | 28.3 | 22.8 |
| Parameter | HSA-HM-3 | HM-3-HSA |
|---|---|---|
| t1/2 (h) | 14.99 | 17.36 |
| CLz (L/h/kg) | 140.23 | 156.62 |
| AUC(0−t) (μg/L *h) | 127.99 | 110.43 |
| AUC (0−∞) (μg/L *h) | 142.62 | 127.70 |
| Vz (L/kg) | 3032.95 | 3923.60 |
| Cmax (μg/L) | 5.99 | 7.09 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, T.; Zhang, H.-Z.; Ge, G.-F.; Yue, Z.-R.; Wang, R.-Y.; Zhang, Q.; Gu, Y.; Song, M.-J.; Li, W.-B.; Ma, M.-Z.; et al. Albumin Fusion at the N-Terminus or C-Terminus of HM-3 Leads to Improved Pharmacokinetics and Bioactivities. Biomedicines 2021, 9, 1084. https://doi.org/10.3390/biomedicines9091084
Li T, Zhang H-Z, Ge G-F, Yue Z-R, Wang R-Y, Zhang Q, Gu Y, Song M-J, Li W-B, Ma M-Z, et al. Albumin Fusion at the N-Terminus or C-Terminus of HM-3 Leads to Improved Pharmacokinetics and Bioactivities. Biomedicines. 2021; 9(9):1084. https://doi.org/10.3390/biomedicines9091084
Chicago/Turabian StyleLi, Ting, Han-Zi Zhang, Guang-Fei Ge, Zhao-Rong Yue, Ru-Yue Wang, Qian Zhang, Yan Gu, Mei-Juan Song, Wen-Bo Li, Min-Zhi Ma, and et al. 2021. "Albumin Fusion at the N-Terminus or C-Terminus of HM-3 Leads to Improved Pharmacokinetics and Bioactivities" Biomedicines 9, no. 9: 1084. https://doi.org/10.3390/biomedicines9091084
APA StyleLi, T., Zhang, H.-Z., Ge, G.-F., Yue, Z.-R., Wang, R.-Y., Zhang, Q., Gu, Y., Song, M.-J., Li, W.-B., Ma, M.-Z., Wang, M.-Z., Yang, H., Li, Y., & Li, H.-Y. (2021). Albumin Fusion at the N-Terminus or C-Terminus of HM-3 Leads to Improved Pharmacokinetics and Bioactivities. Biomedicines, 9(9), 1084. https://doi.org/10.3390/biomedicines9091084