
The NF-κB Nucleolar Stress Response Pathway

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
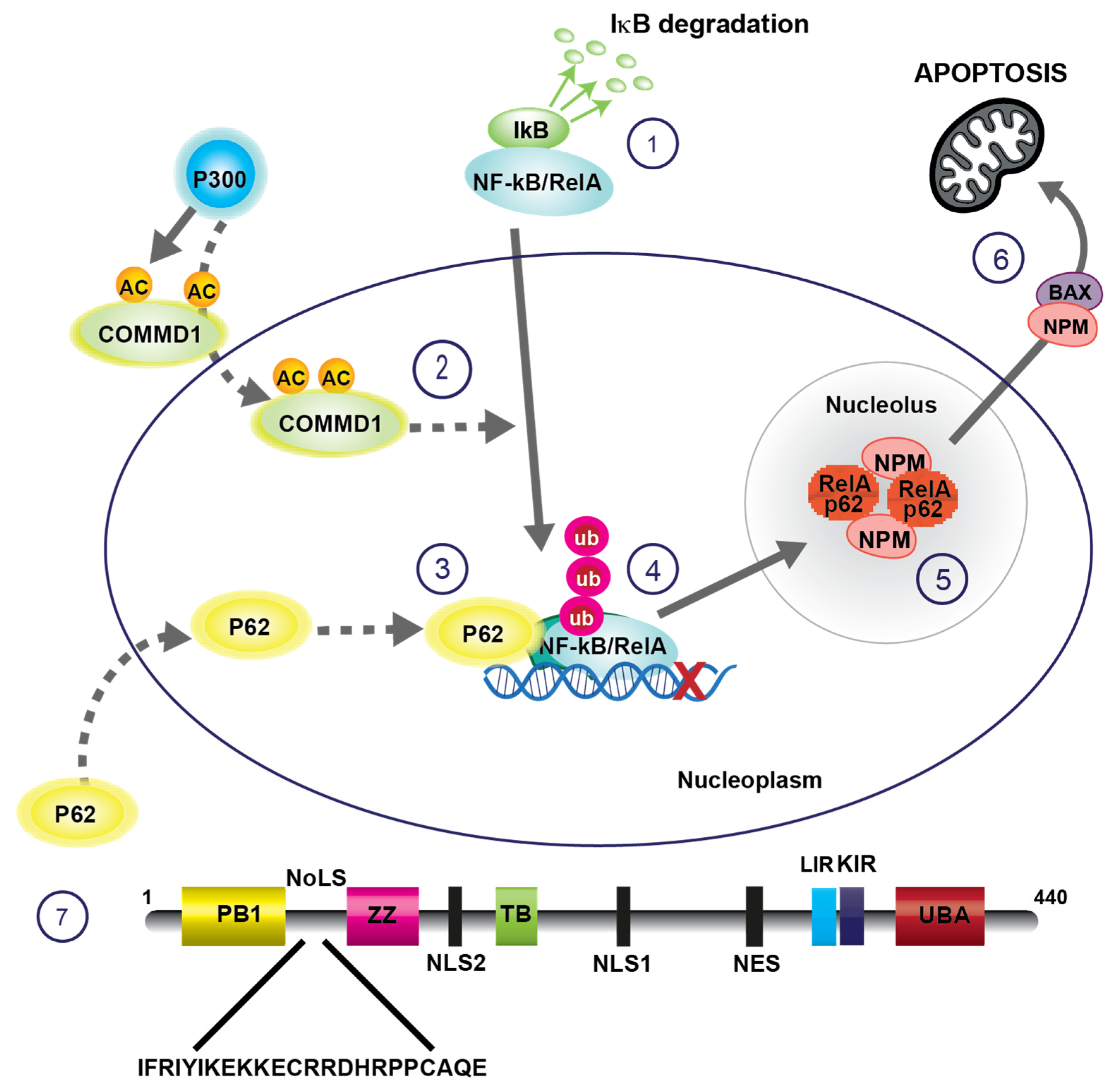
2. Regulation of Apoptosis by Nucleolar Sequestration of NF-κB/RelA
2.1. NF-κB and Disease
2.2. Sequestration in Nucleoli Represses NF-κB Activity and Induces Apoptosis
2.3. COMMD1-Mediated Ubiquitination of RelA Signals for Nucleolar Translocation
2.4. RelA Is Sequestered in Nucleolar Aggresomes
2.5. P62 Transports Ubiquitinated RelA to Nucleolar Aggresomes
2.6. RelA-Nucleophosmin Signalling
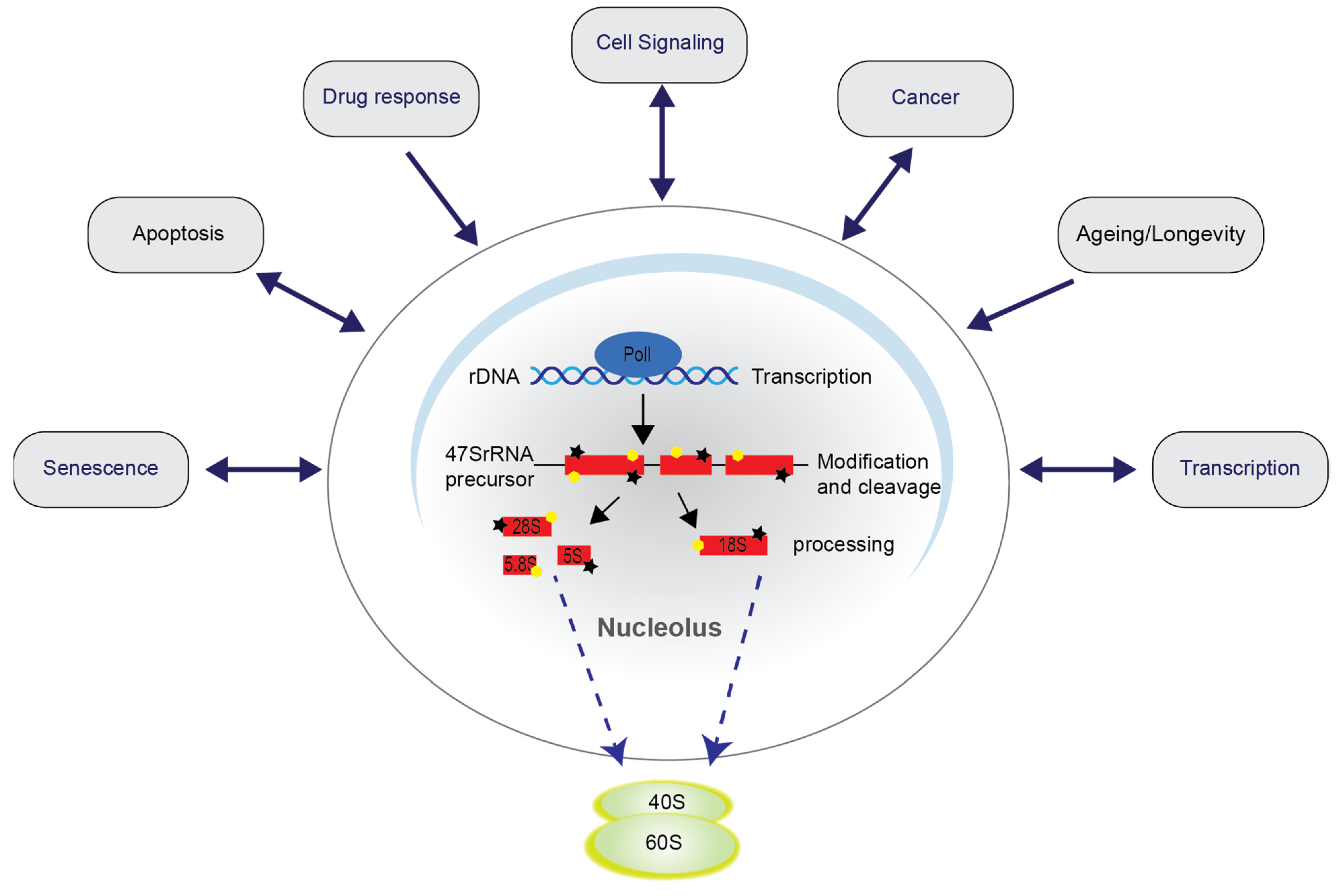
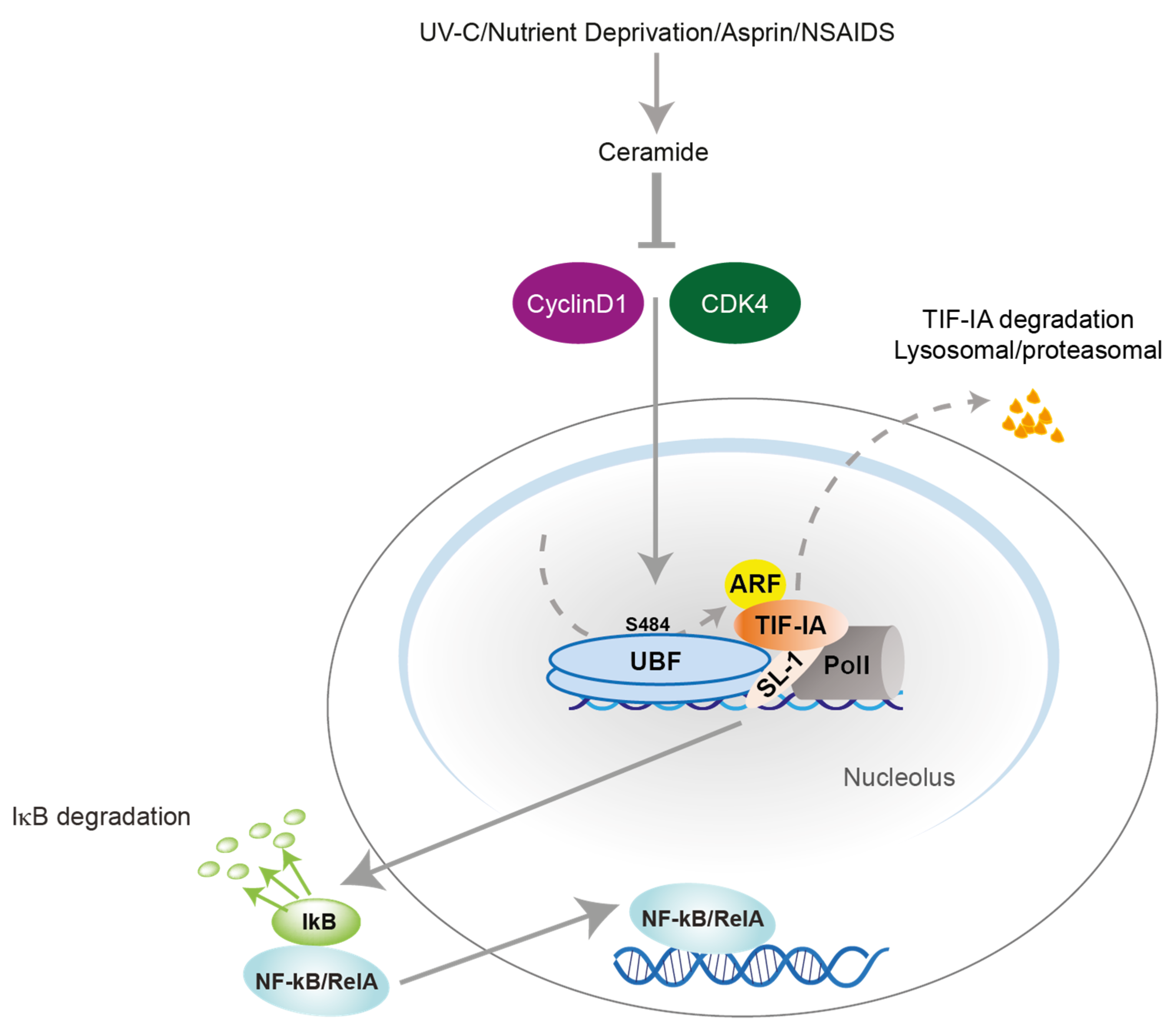
3. Nucleolar Disruption Lies Upstream of NF-κB Pathway Activation in Response to Stress
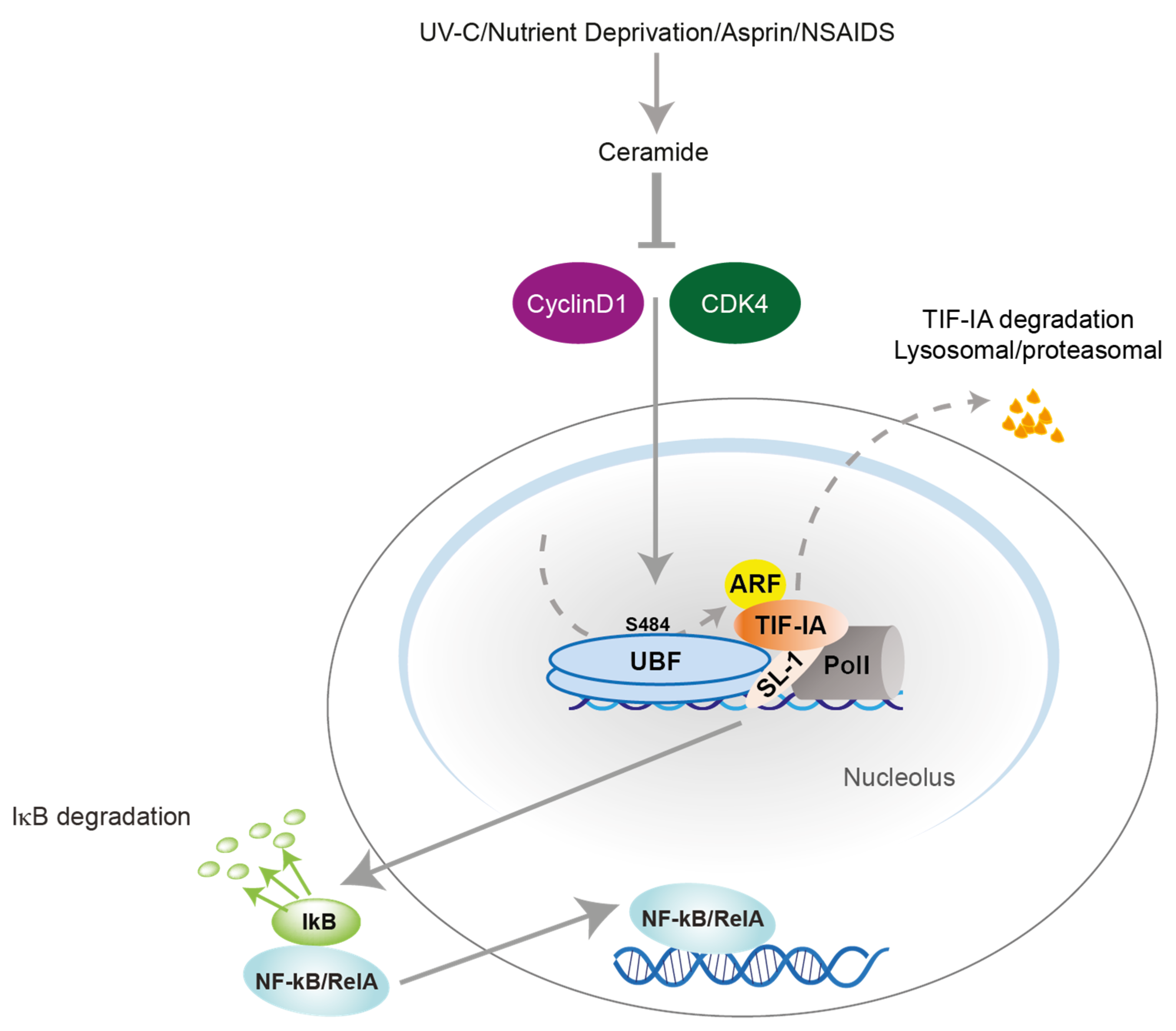
3.1. TIF-IA-NF-κB Nucleolar Stress Response
3.2. TIF-IA-NF-κB Nucleolar Stress Response in Senescence and Ageing
4. Aspirin Acts against Colon Cancer Cells by Targeting the NF-κB Nucleolar Stress Response Pathway
5. Summary
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Taniguchi, K.; Karin, M. NF-κB, inflammation, immunity and cancer: Coming of age. Nat. Rev. Immunol. 2018, 18, 309–324. [Google Scholar] [CrossRef] [PubMed]
- Prescott, J.A.; Cook, S.J. Targeting IKKbeta in Cancer: Challenges and Opportunities for the Therapeutic Utilisation of IKKbeta Inhibitors. Cells 2018, 7, 115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perkins, N.D. The diverse and complex roles of NF-κB subunits in cancer. Nat. Rev. Cancer 2012, 12, 121–132. [Google Scholar] [CrossRef]
- Chen, J.; Stark, L.A. Insights into the Relationship between Nucleolar Stress and the NF-κB Pathway. Trends Genet. 2019, 35, 768–780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.; Stark, L.A. Crosstalk between NF-κB and Nucleoli in the Regulation of Cellular Homeostasis. Cells 2018, 7, 157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lobb, I.T.; Morin, P.; Martin, K.; Thoms, H.C.; Wills, J.C.; Lleshi, X.; Olsen, K.C.F.; Duncan, R.R.; Stark, L.A. A Role for the Autophagic Receptor, SQSTM1/p62, in Trafficking NF-κB/RelA to Nucleolar Aggresomes. Mol. Cancer Res. 2021, 19, 274–287. [Google Scholar] [CrossRef]
- Boulon, S.; Westman, B.J.; Hutten, S.; Boisvert, F.M.; Lamond, A.I. The Nucleolus under Stress. Mol. Cell 2010, 40, 216–227. [Google Scholar] [CrossRef]
- Mangan, H.; Gailin, M.O.; McStay, B. Integrating the genomic architecture of human nucleolar organizer regions with the biophysical properties of nucleoli. FEBS J. 2017, 284, 3977–3985. [Google Scholar] [CrossRef]
- Nemeth, A.; Grummt, I. Dynamic regulation of nucleolar architecture. Curr Opin Cell Biol. 2018, 52, 105–111. [Google Scholar] [CrossRef]
- Chen, J.; Lobb, I.T.; Morin, P.; Novo, S.M.; Simpson, J.; Kennerknecht, K.; von Kriegsheim, A.; Batchelor, E.E.; Oakley, F.; Stark, L.A. Identification of a novel TIF-IA-NF-κB nucleolar stress response pathway. Nucleic Acids Res. 2018, 46, 6188–6205. [Google Scholar] [CrossRef] [Green Version]
- DiDonato, J.A.; Mercurio, F.; Karin, M. NF-κB and the link between inflammation and cancer. Immunol. Rev. 2012, 246, 379–400. [Google Scholar] [CrossRef]
- Gilmore, T.D. Introduction to NF-κB: Players, pathways, perspectives. Oncogene 2006, 25, 6680–6684. [Google Scholar] [CrossRef] [Green Version]
- Cartwright, T.; Perkins, N.D.; C, L.W. NFKB1: A suppressor of inflammation, ageing and cancer. FEBS J. 2016, 283, 1812–1822. [Google Scholar] [CrossRef] [Green Version]
- Pahl, H.L. Activators and target genes of Rel/NF-κB transcription factors. Oncogene 1999, 18, 6853–6866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ankers, J.M.; Awais, R.; Jones, N.A.; Boyd, J.; Ryan, S.; Adamson, A.D.; Harper, C.V.; Bridge, L.; Spiller, D.G.; Jackson, D.A.; et al. Dynamic NF-κB and E2F interactions control the priority and timing of inflammatory signalling and cell proliferation. Elife 2016, 5. [Google Scholar] [CrossRef]
- Tak, P.P.; Firestein, G.S. NF-κB: A key role in inflammatory diseases. J. Clin. Investig. 2001, 107, 7–11. [Google Scholar] [CrossRef] [PubMed]
- Mankan, A.K.; Lawless, M.W.; Gray, S.G.; Kelleher, D.; McManus, R. NF-κB regulation: The nuclear response. J. Cell Mol. Med. 2009, 13, 631–643. [Google Scholar] [CrossRef]
- Dolcet, X.; Llobet, D.; Pallares, J.; Matias-Guiu, X. NF-κB in development and progression of human cancer. Virchows Arch. 2005, 446, 475–482. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Hawke, N.; Baldwin, A.S. NF-κB and IKK as therapeutic targets in cancer. Cell Death Differ. 2006, 13, 738–747. [Google Scholar] [CrossRef] [Green Version]
- Hayden, M.S.; Ghosh, S. NF-κB, the first quarter-century: Remarkable progress and outstanding questions. Genes Dev. 2012, 26, 203–234. [Google Scholar] [CrossRef] [Green Version]
- Ashburner, B.P.; Westerheide, S.D.; Baldwin, A.S., Jr. The p65 (RelA) subunit of NF-κB interacts with the histone deacetylase (HDAC) corepressors HDAC1 and HDAC2 to negatively regulate gene expression. Mol. Cell Biol. 2001, 21, 7065–7077. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.F.; Greene, W.C. Shaping the nuclear action of NF-κB. Nat. Rev. Mol. Cell Biol. 2004, 5, 392–401. [Google Scholar] [CrossRef]
- Chen, L.F.; Williams, S.A.; Mu, Y.; Nakano, H.; Duerr, J.M.; Buckbinder, L.; Greene, W.C. NF-κB RelA phosphorylation regulates RelA acetylation. Mol. Cell Biol. 2005, 25, 7966–7975. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campbell, A.E.; Ferraz Franco, C.; Su, L.I.; Corbin, E.K.; Perkins, S.; Kalyuzhnyy, A.; Jones, A.R.; Brownridge, P.J.; Perkins, N.D.; Eyers, C.E. Temporal modulation of the NF-κB RelA network in response to different types of DNA damage. Biochem. J. 2021, 478, 533–551. [Google Scholar] [CrossRef]
- Webster, G.A.; Perkins, N.D. Transcriptional cross talk between NF-κB and p53. Mol. Cell Biol. 1999, 19, 3485–3495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stark, L.A.; Dunlop, M.G. Nucleolar sequestration of RelA (p65) regulates NF-κB-driven transcription and apoptosis. Mol. Cell Biol. 2005, 25, 5985–6004. [Google Scholar] [CrossRef] [Green Version]
- Olson, M.O.; Dundr, M.; Szebeni, A. The nucleolus: An old factory with unexpected capabilities. Trends Cell Biol. 2000, 10, 189–196. [Google Scholar] [CrossRef]
- Grummt, I. The nucleolus-guardian of cellular homeostasis and genome integrity. Chromosoma 2013, 122, 487–497. [Google Scholar] [CrossRef]
- Mayer, C.; Grummt, I. Cellular stress and nucleolar function. Cell Cycle 2005, 4, 1036–1038. [Google Scholar] [CrossRef] [Green Version]
- van Sluis, M.; McStay, B. Nucleolar reorganization in response to rDNA damage. Curr Opin Cell Biol. 2017, 46, 81–86. [Google Scholar] [CrossRef]
- Boisvert, F.M.; van Koningsbruggen, S.; Navascues, J.; Lamond, A.I. The multifunctional nucleolus. Nat. Rev. Mol. Cell Biol. 2007, 8, 574–585. [Google Scholar] [CrossRef]
- Moore, H.M.; Bai, B.; Boisvert, F.M.; Latonen, L.; Rantanen, V.; Simpson, J.C.; Pepperkok, R.; Lamond, A.I.; Laiho, M. Quantitative proteomics and dynamic imaging of the nucleolus reveal distinct responses to UV and ionizing radiation. Mol. Cell Proteomics 2011, 10, M111. [Google Scholar] [CrossRef] [Green Version]
- Andersen, J.S.; Lyon, C.E.; Fox, A.H.; Leung, A.K.; Lam, Y.W.; Steen, H.; Mann, M.; Lamond, A.I. Directed proteomic analysis of the human nucleolus. Curr. Biol. 2002, 12, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Visintin, R.; Hwang, E.S.; Amon, A. Cfi1 prevents premature exit from mitosis by anchoring Cdc14 phosphatase in the nucleolus. Nature 1999, 398, 818–823. [Google Scholar] [CrossRef] [PubMed]
- Latonen, L. Phase-to-Phase With Nucleoli-Stress Responses, Protein Aggregation and Novel Roles of RNA. Front. Cell Neurosci. 2019, 13, 151. [Google Scholar] [CrossRef]
- Frottin, F.; Schueder, F.; Tiwary, S.; Gupta, R.; Korner, R.; Schlichthaerle, T.; Cox, J.; Jungmann, R.; Hartl, F.U.; Hipp, M.S. The nucleolus functions as a phase-separated protein quality control compartment. Science 2019, 365, 342–347. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Bokros, M.; Theodoridis, P.R.; Lee, S. Nucleolar Sequestration: Remodeling Nucleoli Into Amyloid Bodies. Front. Genet. 2019, 10, 1179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brecht, R.M.; Liu, C.C.; Beilinson, H.A.; Khitun, A.; Slavoff, S.A.; Schatz, D.G. Nucleolar localization of RAG1 modulates V(D)J recombination activity. Proc. Natl. Acad. Sci. USA 2020, 117, 4300–4309. [Google Scholar] [CrossRef] [PubMed]
- Tao, W.; Levine, A.J. P19(ARF) stabilizes p53 by blocking nucleo-cytoplasmic shuttling of Mdm2. Proc. Natl. Acad. Sci. USA 1999, 96, 6937–6941. [Google Scholar] [CrossRef] [Green Version]
- Holmberg Olausson, K.; Nister, M.; Lindstrom, M.S. p53 -Dependent and -Independent Nucleolar Stress Responses. Cells 2012, 1, 774–798. [Google Scholar] [CrossRef] [Green Version]
- Mekhail, K.; Gunaratnam, L.; Bonicalzi, M.E.; Lee, S. HIF activation by pH-dependent nucleolar sequestration of VHL. Nat. Cell Biol. 2004, 6, 642–647. [Google Scholar] [CrossRef] [PubMed]
- Stark, L.A.; Din, F.V.N.; Zwacka, R.M.; Dunlop, M.G. Aspirin-induced activation of the NF-kB signalling pathway: A novel mechanism for aspirin-mediated apoptosis in colon cancer cells. FASEB J. 2001, 15, 1273–1275. [Google Scholar] [CrossRef]
- Loveridge, C.J.; Macdonald, A.D.; Thoms, H.C.; Dunlop, M.G.; Stark, L.A. The proapoptotic effects of sulindac, sulindac sulfone and indomethacin are mediated by nucleolar translocation of the RelA(p65) subunit of NF-κB. Oncogene 2008, 27, 2648–2655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parrondo, R.; de las Pozas, A.; Reiner, T.; Rai, P.; Perez-Stable, C. NF-κB activation enhances cell death by antimitotic drugs in human prostate cancer cells. Mol. Cancer 2010, 9, 182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sniderhan, L.F.; Garcia-Bates, T.M.; Burgart, M.; Bernstein, S.H.; Phipps, R.P.; Maggirwar, S.B. Neurotrophin signaling through tropomyosin receptor kinases contributes to survival and proliferation of non-Hodgkin lymphoma. Exp. Hematol. 2009, 37, 1295–1309. [Google Scholar] [CrossRef] [Green Version]
- Lee, D.H.; Forscher, C.; Di Vizio, D.; Koeffler, H.P. Induction of p53-independent apoptosis by ectopic expression of HOXA5 in human liposarcomas. Sci. Rep. 2015, 5, 12580. [Google Scholar] [CrossRef] [Green Version]
- Dadsetan, S.; Balzano, T.; Forteza, J.; Agusti, A.; Cabrera-Pastor, A.; Taoro-Gonzalez, L.; Hernandez-Rabaza, V.; Gomez-Gimenez, B.; ElMlili, N.; Llansola, M.; et al. Infliximab reduces peripheral inflammation, neuroinflammation, and extracellular GABA in the cerebellum and improves learning and motor coordination in rats with hepatic encephalopathy. J. Neuroinflamm. 2016, 13, 245. [Google Scholar] [CrossRef] [Green Version]
- Audas, T.E.; Jacob, M.D.; Lee, S. Immobilization of proteins in the nucleolus by ribosomal intergenic spacer noncoding RNA. Mol. Cell 2012, 45, 147–157. [Google Scholar] [CrossRef]
- Audas, T.E.; Jacob, M.D.; Lee, S. The nucleolar detention pathway: A cellular strategy for regulating molecular networks. Cell Cycle 2012, 11, 2059–2062. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arabi, A.; Rustum, C.; Hallberg, E.; Wright, A.P. Accumulation of c-Myc and proteasomes at the nucleoli of cells containing elevated c-Myc protein levels. J. Cell Sci. 2003, 116, 1707–1717. [Google Scholar] [CrossRef] [Green Version]
- Mattsson, K.; Pokrovskaja, K.; Kiss, C.; Klein, G.; Szekely, L. Proteins associated with the promyelocytic leukemia gene product (PML)-containing nuclear body move to the nucleolus upon inhibition of proteasome-dependent protein degradation. Proc. Natl. Acad. Sci. USA 2001, 98, 1012–1017. [Google Scholar] [CrossRef] [PubMed]
- Latonen, L. Nucleolar aggresomes as counterparts of cytoplasmic aggresomes in proteotoxic stress. Proteasome inhibitors induce nuclear ribonucleoprotein inclusions that accumulate several key factors of neurodegenerative diseases and cancer. Bioessays 2011, 33, 386–395. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, Y.; Yang, L.; Sun, F.; Li, S.; Wang, Y.; Zhang, G.A.; Dong, T.; Zhang, L.L.; Duan, W.; et al. The nucleolus functions as the compartment for histone H2B protein degradation. iScience 2021, 24, 102256. [Google Scholar] [CrossRef] [PubMed]
- Thoms, H.C.; Loveridge, C.J.; Simpson, J.; Clipson, A.; Reinhardt, K.; Dunlop, M.G.; Stark, L.A. Nucleolar targeting of RelA(p65) is regulated by COMMD1-dependent ubiquitination. Cancer Res. 2010, 70, 139–149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maine, G.N.; Mao, X.; Komarck, C.M.; Burstein, E. COMMD1 promotes the ubiquitination of NF-κB subunits through a cullin-containing ubiquitin ligase. EMBO J. 2007, 26, 436–447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burstein, E.; Hoberg, J.E.; Wilkinson, A.S.; Rumble, J.M.; Csomos, R.A.; Komarck, C.M.; Maine, G.N.; Wilkinson, J.C.; Mayo, M.W.; Duckett, C.S. COMMD proteins, a novel family of structural and functional homologs of MURR1. J. Biol. Chem. 2005, 280, 22222–22232. [Google Scholar] [CrossRef] [Green Version]
- Mao, X.; Gluck, N.; Li, D.; Maine, G.N.; Li, H.; Zaidi, I.W.; Repaka, A.; Mayo, M.W.; Burstein, E. GCN5 is a required cofactor for a ubiquitin ligase that targets NF-κB/RelA. Genes Dev. 2009, 23, 849–861. [Google Scholar] [CrossRef] [Green Version]
- Riera-Romo, M. COMMD1: A Multifunctional Regulatory Protein. J. Cell Biochem. 2018, 119, 34–51. [Google Scholar] [CrossRef] [PubMed]
- Bartuzi, P.; Hofker, M.H.; van de Sluis, B. Tuning NF-κB activity: A touch of COMMD proteins. Biochim. Biophys. Acta 2013, 1832, 2315–2321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Hara, A.; Simpson, J.; Morin, P.; Loveridge, C.J.; Williams, A.C.; Novo, S.M.; Stark, L.A. p300-mediated acetylation of COMMD1 regulates its stability, and the ubiquitylation and nucleolar translocation of the RelA NF-κB subunit. J. Cell Sci. 2014, 127, 3659–3665. [Google Scholar]
- Ehm, P.; Nalaskowski, M.M.; Wundenberg, T.; Jucker, M. The tumor suppressor SHIP1 colocalizes in nucleolar cavities with p53 and components of PML nuclear bodies. Nucleus 2015, 6, 154–164. [Google Scholar] [CrossRef] [Green Version]
- Vilotti, S.; Codrich, M.; Dal, F.M.; Pinto, M.; Ferrer, I.; Collavin, L.; Gustincich, S.; Zucchelli, S. Parkinson’s disease DJ-1 L166P alters rRNA biogenesis by exclusion of TTRAP from the nucleolus and sequestration into cytoplasmic aggregates via TRAF6. PLoS ONE 2012, 7, e35051. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Latonen, L.; Moore, H.M.; Bai, B.; Jaamaa, S.; Laiho, M. Proteasome inhibitors induce nucleolar aggregation of proteasome target proteins and polyadenylated RNA by altering ubiquitin availability. Oncogene 2011, 30, 790–805. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Souquere, S.; Weil, D.; Pierron, G. Comparative ultrastructure of CRM1-Nucleolar bodies (CNoBs), Intranucleolar bodies (INBs) and hybrid PML/p62 bodies uncovers new facets of nuclear body dynamic and diversity. Nucleus 2015, 6, 326–338. [Google Scholar] [CrossRef] [Green Version]
- Elson, E.L. Fluorescence correlation spectroscopy: Past, present, future. Biophys J. 2011, 101, 2855–2870. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanchez-Martin, P.; Saito, T.; Komatsu, M. p62/SQSTM1: ‘Jack of all trades’ in health and cancer. FEBS J. 2019, 286, 8–23. [Google Scholar] [CrossRef] [Green Version]
- Lamark, T.; Svenning, S.; Johansen, T. Regulation of selective autophagy: The p62/SQSTM1 paradigm. Essays Biochem. 2017, 61, 609–624. [Google Scholar] [CrossRef] [PubMed]
- Bjorkoy, G.; Lamark, T.; Brech, A.; Outzen, H.; Perander, M.; Overvatn, A.; Stenmark, H.; Johansen, T. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J. Cell Biol. 2005, 171, 603–614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pankiv, S.; Lamark, T.; Bruun, J.A.; Overvatn, A.; Bjorkoy, G.; Johansen, T. Nucleocytoplasmic shuttling of p62/SQSTM1 and its role in recruitment of nuclear polyubiquitinated proteins to promyelocytic leukemia bodies. J. Biol. Chem. 2010, 285, 5941–5953. [Google Scholar] [CrossRef] [Green Version]
- Salmina, K.; Huna, A.; Inashkina, I.; Belyayev, A.; Krigerts, J.; Pastova, L.; Vazquez-Martin, A.; Erenpreisa, J. Nucleolar aggresomes mediate release of pericentric heterochromatin and nuclear destruction of genotoxically treated cancer cells. Nucleus 2017, 8, 205–221. [Google Scholar] [CrossRef] [Green Version]
- Feng, Y.; Duan, T.; Du, Y.; Jin, S.; Wang, M.; Cui, J.; Wang, R.F. LRRC25 Functions as an Inhibitor of NF-κB Signaling Pathway by Promoting p65/RelA for Autophagic Degradation. Sci. Rep. 2017, 7, 13448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khandelwal, N.; Simpson, J.; Taylor, G.; Rafique, S.; Whitehouse, A.; Hiscox, J.; Stark, L.A. Nucleolar NF-κB/RelA mediates apoptosis by causing cytoplasmic relocalization of nucleophosmin. Cell Death. Differ. 2011, 18, 1889–1903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindstrom, M.S. NPM1/B23: A Multifunctional Chaperone in Ribosome Biogenesis and Chromatin Remodeling. Biochem. Res. Int. 2011, 2011, 195209. [Google Scholar] [CrossRef] [Green Version]
- Lin, J.; Kato, M.; Nagata, K.; Okuwaki, M. Efficient DNA binding of NF-κB requires the chaperone-like function of NPM1. Nucleic Acids Res. 2017, 45, 3707–3723. [Google Scholar] [CrossRef] [Green Version]
- Rao, C.; Liu, B.; Huang, D.; Chen, R.; Huang, K.; Li, F.; Dong, N. Nucleophosmin contributes to vascular inflammation and endothelial dysfunction in atherosclerosis progression. J. Thorac. Cardiovasc. Surg. 2021, 161, e377–e393. [Google Scholar] [CrossRef]
- Wang, Z.; Gall, J.M.; Bonegio, R.; Havasi, A.; Illanes, K.; Schwartz, J.H.; Borkan, S.C. Nucleophosmin, a critical Bax cofactor in ischemia-induced cell death. Mol. Cell Biol. 2013, 33, 1916–1924. [Google Scholar] [CrossRef] [Green Version]
- Thompson, J.; Finlayson, K.; Salvo-Chirnside, E.; MacDonald, D.; McCulloch, J.; Kerr, L.; Sharkey, J. Characterisation of the Bax-nucleophosmin interaction: The importance of the Bax C-terminus. Apoptosis 2008, 13, 394–403. [Google Scholar] [CrossRef] [PubMed]
- Kerr, L.E.; Birse-Archbold, J.L.; Short, D.M.; McGregor, A.L.; Heron, I.; Macdonald, D.C.; Thompson, J.; Carlson, G.J.; Kelly, J.S.; McCulloch, J.; et al. Nucleophosmin is a novel Bax chaperone that regulates apoptotic cell death. Oncogene 2007, 26, 2554–2562. [Google Scholar] [CrossRef] [Green Version]
- Hochrainer, K.; Racchumi, G.; Zhang, S.; Iadecola, C.; Anrather, J. Monoubiquitination of nuclear RelA negatively regulates NF-κB activity independent of proteasomal degradation. Cell. Mol. Life Sci. 2012, 69, 2057–2073. [Google Scholar] [CrossRef] [Green Version]
- Hernandez-Verdun, D. Nucleolus: From structure to dynamics. Histochem. Cell Biol. 2006, 125, 127–137. [Google Scholar] [CrossRef] [PubMed]
- Yuan, X.; Zhao, J.; Zentgraf, H.; Hoffmann-Rohrer, U.; Grummt, I. Multiple interactions between RNA polymerase I, TIF-IA and TAF(I) subunits regulate preinitiation complex assembly at the ribosomal gene promoter. EMBO Rep. 2002, 3, 1082–1087. [Google Scholar] [CrossRef] [Green Version]
- Grummt, I. Life on a planet of its own: Regulation of RNA polymerase I transcription in the nucleolus. Genes Dev. 2003, 17, 1691–1702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pfister, A.S. Emerging Role of the Nucleolar Stress Response in Autophagy. Front. Cell NeuroSci. 2019, 13, 156. [Google Scholar] [CrossRef] [Green Version]
- Lindstrom, M.S.; Jurada, D.; Bursac, S.; Orsolic, I.; Bartek, J.; Volarevic, S. Nucleolus as an emerging hub in maintenance of genome stability and cancer pathogenesis. Oncogene 2018, 37, 2351–2366. [Google Scholar] [CrossRef]
- Hiscox, J.A.; Whitehouse, A.; Matthews, D.A. Nucleolar proteomics and viral infection. Proteomics 2010, 10, 4077–4086. [Google Scholar] [CrossRef]
- Rubbi, C.P.; Milner, J. Disruption of the nucleolus mediates stabilization of p53 in response to DNA damage and other stresses. EMBO J. 2003, 22, 6068–6077. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- James, A.; Wang, Y.; Raje, H.; Rosby, R.; DiMario, P. Nucleolar stress with and without p53. Nucleus 2014, 5, 402–426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woods, S.J.; Hannan, K.M.; Pearson, R.B.; Hannan, R.D. The nucleolus as a fundamental regulator of the p53 response and a new target for cancer therapy. Biochim. Biophys. Acta 2015, 1849, 821–829. [Google Scholar] [CrossRef] [PubMed]
- Russo, A.; Russo, G. Ribosomal Proteins Control or Bypass p53 during Nucleolar Stress. Int. J. Mol. Sci. 2017, 18, 140. [Google Scholar] [CrossRef] [Green Version]
- Pecoraro, A.; Virgilio, A.; Esposito, V.; Galeone, A.; Russo, G.; Russo, A. uL3 Mediated Nucleolar Stress Pathway as a New Mechanism of Action of Antiproliferative G-quadruplex TBA Derivatives in Colon Cancer Cells. Biomolecules 2020, 10, 583. [Google Scholar] [CrossRef] [Green Version]
- Russo, A.; Esposito, D.; Catillo, M.; Pietropaolo, C.; Crescenzi, E.; Russo, G. Human rpL3 induces G(1)/S arrest or apoptosis by modulating p21 (waf1/cip1) levels in a p53-independent manner. Cell Cycle 2013, 12, 76–87. [Google Scholar] [CrossRef] [Green Version]
- Bodem, J.; Dobreva, G.; Hoffmann-Rohrer, U.; Iben, S.; Zentgraf, H.; Delius, H.; Vingron, M.; Grummt, I. TIF-IA, the factor mediating growth-dependent control of ribosomal RNA synthesis, is the mammalian homolog of yeast Rrn3p. EMBO Rep. 2000, 1, 171–175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, R.; Zhou, W. TIF-IA: An oncogenic target of pre-ribosomal RNA synthesis. Biochim. Biophys. Acta 2016, 1866, 189–196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen le, X.T.; Mitchell, B.S. Akt activation enhances ribosomal RNA synthesis through casein kinase II and TIF-IA. Proc. Natl. Acad. Sci. USA 2013, 110, 20681–20686. [Google Scholar] [CrossRef] [Green Version]
- Bierhoff, H.; Dundr, M.; Michels, A.A.; Grummt, I. Phosphorylation by casein kinase 2 facilitates rRNA gene transcription by promoting dissociation of TIF-IA from elongating RNA polymerase I. Mol. Cell Biol. 2008, 28, 4988–4998. [Google Scholar] [CrossRef] [Green Version]
- Szymanski, J.; Mayer, C.; Hoffmann-Rohrer, U.; Kalla, C.; Grummt, I.; Weiss, M. Dynamic subcellular partitioning of the nucleolar transcription factor TIF-IA under ribotoxic stress. Biochim. Biophys. Acta 2009, 1793, 1191–1198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, X.; Zhou, Y.; Casanova, E.; Chai, M.; Kiss, E.; Grone, H.J.; Schutz, G.; Grummt, I. Genetic inactivation of the transcription factor TIF-IA leads to nucleolar disruption, cell cycle arrest, and p53-mediated apoptosis. Mol. Cell 2005, 19, 77–87. [Google Scholar] [CrossRef] [PubMed]
- Kreiner, G.; Bierhoff, H.; Armentano, M.; Rodriguez-Parkitna, J.; Sowodniok, K.; Naranjo, J.R.; Bonfanti, L.; Liss, B.; Schutz, G.; Grummt, I.; et al. A neuroprotective phase precedes striatal degeneration upon nucleolar stress. Cell Death Differ. 2013, 20, 1455–1464. [Google Scholar] [CrossRef] [PubMed]
- Parlato, R.; Kreiner, G.; Erdmann, G.; Rieker, C.; Stotz, S.; Savenkova, E.; Berger, S.; Grummt, I.; Schutz, G. Activation of an endogenous suicide response after perturbation of rRNA synthesis leads to neurodegeneration in mice. J. NeuroSci. 2008, 28, 12759–12764. [Google Scholar] [CrossRef] [Green Version]
- Thoms, H.C.; Dunlop, M.G.; Stark, L.A. p38-mediated inactivation of cyclin D1/cyclin-dependent kinase 4 stimulates nucleolar translocation of RelA and apoptosis in colorectal cancer cells. Cancer Res. 2007, 67, 1660–1669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fatyol, K.; Grummt, I. Proteasomal ATPases are associated with rDNA: The ubiquitin proteasome system plays a direct role in RNA polymerase I transcription. Biochim. Biophys. Acta 2008, 1779, 850–859. [Google Scholar] [CrossRef] [PubMed]
- Bailly, A.; Perrin, A.; Bou Malhab, L.J.; Pion, E.; Larance, M.; Nagala, M.; Smith, P.; O’Donohue, M.F.; Gleizes, P.E.; Zomerdijk, J.; et al. The NEDD8 inhibitor MLN4924 increases the size of the nucleolus and activates p53 through the ribosomal-Mdm2 pathway. Oncogene 2016, 35, 415–426. [Google Scholar] [CrossRef] [Green Version]
- Kato, T., Jr.; Delhase, M.; Hoffmann, A.; Karin, M. CK2 Is a C-Terminal IκB Kinase Responsible for NF-κB Activation during the UV Response. Mol. Cell 2003, 12, 829–839. [Google Scholar] [CrossRef]
- Jiang, H.Y.; Wek, R.C. GCN2 phosphorylation of eIF2alpha activates NF-κB in response to UV irradiation. Biochem. J. 2005, 385, 371–380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, H.Y.; Wek, S.A.; McGrath, B.C.; Scheuner, D.; Kaufman, R.J.; Cavener, D.R.; Wek, R.C. Phosphorylation of the alpha subunit of eukaryotic initiation factor 2 is required for activation of NF-κB in response to diverse cellular stresses. Mol. Cell Biol. 2003, 23, 5651–5663. [Google Scholar] [CrossRef] [Green Version]
- DuRose, J.B.; Scheuner, D.; Kaufman, R.J.; Rothblum, L.I.; Niwa, M. Phosphorylation of eukaryotic translation initiation factor 2alpha coordinates rRNA transcription and translation inhibition during endoplasmic reticulum stress. Mol. Cell Biol. 2009, 29, 4295–4307. [Google Scholar] [CrossRef] [Green Version]
- Russo, A.; Maiolino, S.; Pagliara, V.; Ungaro, F.; Tatangelo, F.; Leone, A.; Scalia, G.; Budillon, A.; Quaglia, F.; Russo, G. Enhancement of 5-FU sensitivity by the proapoptotic rpL3 gene in p53 null colon cancer cells through combined polymer nanoparticles. Oncotarget 2016, 7, 79670–79687. [Google Scholar] [CrossRef] [Green Version]
- Russo, A.; Saide, A.; Cagliani, R.; Cantile, M.; Botti, G.; Russo, G. rpL3 promotes the apoptosis of p53 mutated lung cancer cells by down-regulating CBS and NF-κB upon 5-FU treatment. Sci. Rep. 2016, 6, 38369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wan, F.; Anderson, D.E.; Barnitz, R.A.; Snow, A.; Bidere, N.; Zheng, L.; Hegde, V.; Lam, L.T.; Staudt, L.M.; Levens, D.; et al. Ribosomal protein S3: A KH domain subunit in NF-κB complexes that mediates selective gene regulation. Cell 2007, 131, 927–939. [Google Scholar] [CrossRef] [Green Version]
- Beji, S.; D’Agostino, M.; Gambini, E.; Sileno, S.; Scopece, A.; Vinci, M.C.; Milano, G.; Melillo, G.; Napolitano, M.; Pompilio, G.; et al. Doxorubicin induces an alarmin-like TLR4-dependent autocrine/paracrine action of Nucleophosmin in human cardiac mesenchymal progenitor cells. BMC Biol. 2021, 19, 124. [Google Scholar] [CrossRef]
- Chandra, T.; Kirschner, K. Chromosome organisation during ageing and senescence. Curr Opin Cell Biol. 2016, 40, 161–167. [Google Scholar] [CrossRef] [Green Version]
- Acosta, J.C.; Banito, A.; Wuestefeld, T.; Georgilis, A.; Janich, P.; Morton, J.P.; Athineos, D.; Kang, T.W.; Lasitschka, F.; Andrulis, M.; et al. A complex secretory program orchestrated by the inflammasome controls paracrine senescence. Nat. Cell Biol. 2013, 15, 978–990. [Google Scholar] [CrossRef]
- Tiku, V.; Antebi, A. Nucleolar Function in Lifespan Regulation. Trends Cell Biol. 2018, 28, 662–672. [Google Scholar] [CrossRef] [PubMed]
- Buchwalter, A.; Hetzer, M.W. Nucleolar expansion and elevated protein translation in premature aging. Nat. Commun 2017, 8, 328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosete, M.; Padros, M.R.; Vindrola, O. The nucleolus as a regulator of cellular senescence. Medicina (B Aires) 2007, 67, 183–194. [Google Scholar] [PubMed]
- Nishimura, K.; Kumazawa, T.; Kuroda, T.; Katagiri, N.; Tsuchiya, M.; Goto, N.; Furumai, R.; Murayama, A.; Yanagisawa, J.; Kimura, K. Perturbation of ribosome biogenesis drives cells into senescence through 5S RNP-mediated p53 activation. Cell Rep. 2015, 10, 1310–1323. [Google Scholar] [CrossRef] [Green Version]
- Osorio, F.G.; Soria-Valles, C.; Santiago-Fernandez, O.; Freije, J.M.; Lopez-Otin, C. NF-κB signaling as a driver of ageing. Int. Rev. Cell Mol. Biol. 2016, 326, 133–174. [Google Scholar] [CrossRef]
- Din, F.V.; Theodoratou, E.; Farrington, S.M.; Tenesa, A.; Barnetson, R.A.; Cetnarskyj, R.; Stark, L.; Porteous, M.E.; Campbell, H.; Dunlop, M.G. Effect of aspirin and NSAIDs on risk and survival from colorectal cancer. Gut 2010, 59, 1670–1679. [Google Scholar] [CrossRef]
- Burn, J.; Sheth, H.; Elliott, F.; Reed, L.; Macrae, F.; Mecklin, J.P.; Moslein, G.; McRonald, F.E.; Bertario, L.; Evans, D.G.; et al. Cancer prevention with aspirin in hereditary colorectal cancer (Lynch syndrome), 10-year follow-up and registry-based 20-year data in the CAPP2 study: A double-blind, randomised, placebo-controlled trial. Lancet 2020, 395, 1855–1863. [Google Scholar] [CrossRef]
- Walker, J.; Hutchison, P.; Ge, J.; Zhao, D.; Wang, Y.; Rothwell, P.M.; Gaziano, J.M.; Chan, A.; Burn, J.; Chia, J.; et al. Aspirin: 120 years of innovation. A report from the 2017 Scientific Conference of the International Aspirin Foundation, 14 September 2017, Charite, Berlin. Ecancermedicalscience 2018, 12, 813. [Google Scholar] [CrossRef]
- Chen, J.; Stark, L.A. Aspirin Prevention of Colorectal Cancer: Focus on NF-κB Signalling and the Nucleolus. Biomedicines 2017, 5, 43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van De, S.B.; Mao, X.; Zhai, Y.; Groot, A.J.; Vermeulen, J.F.; van der, W.E.; van Diest, P.J.; Hofker, M.H.; Wijmenga, C.; Klomp, L.W.; et al. COMMD1 disrupts HIF-1alpha/beta dimerization and inhibits human tumor cell invasion. J. Clin. Investig. 2010, 120, 2119–2130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peltonen, K.; Colis, L.; Liu, H.; Trivedi, R.; Moubarek, M.S.; Moore, H.M.; Bai, B.; Rudek, M.A.; Bieberich, C.J.; Laiho, M. A targeting modality for destruction of RNA polymerase I that possesses anticancer activity. Cancer Cell 2014, 25, 77–90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nunez Villacis, L.; Wong, M.S.; Ferguson, L.L.; Hein, N.; George, A.J.; Hannan, K.M. New Roles for the Nucleolus in Health and Disease. Bioessays 2018, 40, e1700233. [Google Scholar] [CrossRef]
- Myant, K.B.; Cammareri, P.; McGhee, E.J.; Ridgway, R.A.; Huels, D.J.; Cordero, J.B.; Schwitalla, S.; Kalna, G.; Ogg, E.L.; Athineos, D.; et al. ROS production and NF-κB activation triggered by RAC1 facilitate WNT-driven intestinal stem cell proliferation and colorectal cancer initiation. Cell Stem Cell 2013, 12, 761–773. [Google Scholar] [CrossRef] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Thoms, H.C.; Stark, L.A. The NF-κB Nucleolar Stress Response Pathway. Biomedicines 2021, 9, 1082. https://doi.org/10.3390/biomedicines9091082
Thoms HC, Stark LA. The NF-κB Nucleolar Stress Response Pathway. Biomedicines. 2021; 9(9):1082. https://doi.org/10.3390/biomedicines9091082
Chicago/Turabian StyleThoms, Hazel C., and Lesley A. Stark. 2021. "The NF-κB Nucleolar Stress Response Pathway" Biomedicines 9, no. 9: 1082. https://doi.org/10.3390/biomedicines9091082
APA StyleThoms, H. C., & Stark, L. A. (2021). The NF-κB Nucleolar Stress Response Pathway. Biomedicines, 9(9), 1082. https://doi.org/10.3390/biomedicines9091082