
Advances in Intracellular Calcium Signaling Reveal Untapped Targets for Cancer Therapy



Abstract
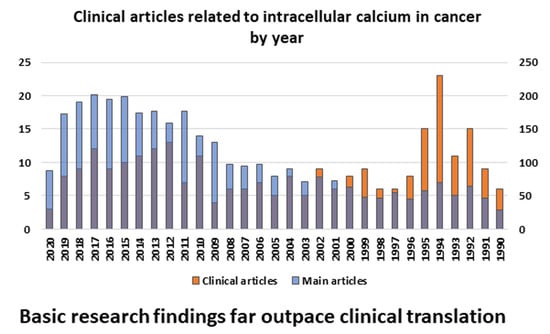
1. Introduction
2. Intracellular Ca2+ Buffers in Normal Cells
2.1. Soluble and Unbound Intracellular Proteins: Calmodulin, Calbindin, and Calretinin
2.2. Intramembranous Molecular Buffers: SERCA, PMCA, NCX, and TRP


2.3. Cellular Organelles
2.3.1. Endoplasmic Reticulum: STIM, ORAI, IP3Rs, and TRPC1 in SOCE and SOCIC Ca2+ Entry Models

2.3.2. Mitochondria and Acidic Vesicles (Mainly Lysosomes)


3. Redistribution of Intracellular Ca2+ and Hijack of Its Regulatory Machinery in Cancer Cells
3.1. Intracellular Ca2+ Pool in the Endoplasmic Reticulum and at the ER-PM Junction
3.2. Intracellular Ca2+ Pool at ER-Mitochondrial Junction
3.3. Intracellular Ca2+ Pool at ER-Lysosome Junction
4. Conclusions and Discussion
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Campbell, A.K. Intracellular Calcium; John Wiley & Sons: Hoboken, NJ, USA, 2014. [Google Scholar] [CrossRef]
- Carafoli, E.; Krebs, J. Why Calcium? How Calcium Became the Best Communicator. J. Biol. Chem. 2016, 291, 20849–20857. [Google Scholar] [CrossRef]
- Bagur, R.; Hajnóczky, G. Intracellular Ca2+ Sensing: Its Role in Calcium Homeostasis and Signaling. Mol. Cell 2017, 66, 780–788. [Google Scholar] [CrossRef]
- Schwaller, B. Cytosolic Ca2+ Buffers. Cold Spring Harb. Perspect. Biol. 2010, 2, a004051. [Google Scholar] [CrossRef]
- Prins, D.; Michalak, M. Organellar Calcium Buffers. Cold Spring Harb. Perspect. Biol. 2011, 3, a004069. [Google Scholar] [CrossRef]
- Chin, D.; Means, A.R. Calmodulin: A prototypical calcium sensor. Trends Cell Biol. 2000, 10, 322–328. [Google Scholar] [CrossRef]
- Nelson, M.R.; Chazin, W.J. An interaction-based analysis of calcium-induced conformational changes in Ca2+ sensor proteins. Protein Sci. 1998, 7, 270–282. [Google Scholar] [CrossRef]
- O’Day, D.H.; Eshak, K.; Myre, M.A. Calmodulin Binding Proteins and Alzheimer’s Disease. J. Alzheimer’s Dis. 2015, 46, 553–569. [Google Scholar] [CrossRef] [PubMed]
- Faas, C.; Raghavchri, S.; Lisman, J.E.; Mody, I. Calmodulin as a Direct Detector of Ca2+ Signals. Nat. Neurosci. 2011, 14, 301–304. [Google Scholar] [CrossRef] [PubMed]
- Villalobo, A.; Hiriaki, I.; Vogel, H.J.; Berchtold, M.W. Calmodulin as a protein linker and a regulator of adaptor/scaffold proteins. Biochim. Biophys. Acta (BBA)—Mol. Cell Res. 2018, 1865, 507–521. [Google Scholar] [CrossRef]
- Schmidt, H. Three functional facets of calbindin D-28k. Front. Mol. Neurosci. 2012, 5, 25. [Google Scholar] [CrossRef] [PubMed]
- Kawasaki, H.; Kretsinger, R.H. Structural and functional diversity of EF-hand proteins: Evolutionary perspectives. Protein Sci. 2017, 26, 1898–1920. [Google Scholar] [CrossRef] [PubMed]
- Rogers, J.H. Calretinin: A gene for a novel calcium-binding protein expressed principally in neurons. J. Cell Biol. 1987, 105, 1343–1353, Erratum in 1990, 110, 1845. [Google Scholar] [CrossRef] [PubMed]
- Lugli, A.; Forster, Y.; Haas, P.; Nocito, A.; Bucher, C.; Bissig, H.; Mirlacher, M.; Storz, M.; Mihatsch, M.J.; Sauter, G. Calretinin expression in human normal and neoplastic tissues: A tissue microarray analysis on 5233 tissue samples. Hum. Pathol. 2003, 34, 994–1000. [Google Scholar] [CrossRef]
- Marenholz, I.; Heizmann, C.W.; Fritz, G. S100 proteins in mouse and man: From evolution to function and pathology (Including an update of the nomenclature). Biochem. Biophys. Res. Commun. 2004, 322, 1111–1122. [Google Scholar] [CrossRef] [PubMed]
- Dudek, E.; Michalak, M. Calnexin and calreticulin. In Encyclopedia of Metalloproteins; Kretsinger, R.H., Uversky, V.N., Permyakov, E.A., Eds.; Springer: Berlin/Heidelberg, Germany, 2013; pp. 555–562. [Google Scholar] [CrossRef]
- Ben Johny, M.; Yang, P.S.; Bazzazi, H.; Yue, D.T. Dynamic switching of calmodulin interactions underlies Ca2+ regulation of CaV1.3 channels. Nat. Commun. 2013, 4, 1717. [Google Scholar] [CrossRef] [PubMed]
- Lambers, T.T.; Weidema, A.F.; Nilius, B.; Hoenderop, J.G.J.; Bindels, R.J.M. Regulation of the mouse epithelial Ca2+ channel trpv6 by the Ca2+-sensor calmodulin. J. Biol. Chem. 2004, 279, 28855–28861. [Google Scholar] [CrossRef] [PubMed]
- Nitsche, J.; Josts, I.; Heidemann, J.; Mertens, H.D.; Maric, S.; Moulin, M.; Haertlein, M.; Busch, S.; Forsyth, V.T.; Svergun, D.I.; et al. Structural basis for activation of plasma-membrane Ca2+-ATPase by calmodulin. Commun. Biol. 2018, 1, 206. [Google Scholar] [CrossRef]
- Kahl, C.R.; Means, A.R. Regulation of cell cycle progression by calcium/calmodulin-dependent pathways. Endocr. Rev. 2003, 24, 719–736. [Google Scholar] [CrossRef]
- Lambers, T.T.; Mahieu, F.; Oancea, E.; Hoofd, L.; de Lange, F.; Mensenkamp, A.R.; Voets, T.; Nilius, B.; Clapham, D.E.; Hoenderop, J.G.; et al. Calbindin-D28K dynamically controls TRPV5-mediated Ca2+ transport. EMBO J. 2006, 25, 2978–2988. [Google Scholar] [CrossRef]
- Berlin, J.R.; Bassani, J.W.; Bers, D.M. Intrinsic cytosolic calcium buffering properties of single rat cardiac myocytes. Biophys. J. 1994, 67, 1775–1787. [Google Scholar] [CrossRef]
- Chandrashekera, P.C.; Kargacin, M.E.; Deans, J.P.; Lytton, J. Determination of apparent calcium affinity for endogenously expressed human sarco(endo)plasmic reticulum calcium-ATPase isoform SERCA3. Am. J. Physiol. Cell Physiol. 2009, 296, C1105–C1114. [Google Scholar] [CrossRef]
- Gorski, P.A.; Ceholski, D.K.; Young, H.S. Structure-function relationship of the serca pump and its regulation by phospholamban and sarcolipin. In Membrane Dynamics and Calcium Signaling; Krebs, J., Ed.; Springer: Berlin/Heidelberg, Germany, 2017; pp. 77–119. [Google Scholar] [CrossRef]
- Vandecaetsbeek, I.; Trekels, M.; De Maeyer, M.; Ceulemans, H.; Lescrinier, E.; Raeymaekers, L.; Wuytack, F.; Vangheluwe, P. Structural basis for the high affinity of the ubiquitous SERCA2b Ca2+ pump. Proc. Natl. Acad. Sci. USA 2009, 106, 18533–18538. [Google Scholar] [CrossRef] [PubMed]
- Toyoshima, C.; Nakasako, M.; Nomura, H.; Ogawa, H. Crystal structure of the calcium pump of sarcoplasmic reticulum at 2.6 A resolution. Nature 2000, 405, 647–655. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Lewis, D.; Strock, C.; Inesi, G.; Nakasako, M.; Nomura, H.; Toyoshima, C. Detailed Characterization of the Cooperative Mechanism of Ca2+ Binding and Catalytic Activation in the Ca2+ Transport (SERCA) ATPase. Biochemistry 2000, 39, 8758–8767. [Google Scholar] [CrossRef] [PubMed]
- Inoue, M.; Skuta, N.; Watanabe, S.; Zhang, Y.; Yoshikaie, K.; Tanaka, Y.; Ushioda, R.; Kato, Y.; Takagi, J.; Tsukazaki, T.; et al. Structural Basis of Sarco/Endoplasmic Reticulum Ca2+-ATPase 2b Regulation via Transmembrane Helix Interplay. Cell Rep. 2019, 27, 1221–1230. [Google Scholar] [CrossRef] [PubMed]
- Anderson, D.M.; Makarewich, C.A.; Anderson, K.M.; Shelton, J.M.; Bezprozvannaya, S.; Bassel-Duby, R.; Olson, E.N. Widespread control of calcium signaling by a family of SERCA-inhibiting micropeptides. Sci. Signal. 2016, 9, ra119. [Google Scholar] [CrossRef]
- Zhang, Y.; Inoue, M.; Tsutsumi, A.; Watanabe, S.; Nishizawa, T.; Nagata, K.; Kikkawa, M.; Inaba, K. Cryo-EM structures of SERCA2b reveal the mechanism of regulation by the luminal extension tail. Sci. Adv. 2020, 6, eabb0147. [Google Scholar] [CrossRef]
- Dencher, N.A.; Choli, T.; Dresselhaus, D.; Fimmel, F.; Grzesiek, S.; Papadopoulos, G.; Wittmann-Liebold, B.; Büldt, G. Structure-function relationship of the light-driven proton pump bacteriorhodopsin. J. Protein Chem. 1989, 8, 340–343. [Google Scholar] [CrossRef] [PubMed]
- A buffering serca pump in models of calcium dynamics. Biophys. J. 2006, 91, 151–163. [CrossRef] [PubMed]
- Espinoza-Fonseca, L.M. Probing the effects of nonannular lipid binding on the stability of the calcium pump SERCA. Sci. Rep. 2019, 9, 3349. [Google Scholar] [CrossRef] [PubMed]
- Strehler, E.E.; Caride, A.J.; Filoteo, A.G.; Xiong, Y.; Penniston, J.T.; Enyedi, A. Plasma membrane Ca2+-ATPases as dynamic regulators of cellular calcium handling. Ann. N. Y. Acad. Sci. 2007, 1099. [Google Scholar] [CrossRef] [PubMed]
- Stafford, N.; Wilson, C.; Oceandy, D.; Neyses, L.; Cartwright, E.J. The plasma membrane calcium atp ases and their role as major new players in human disease. Physiol. Rev. 2017, 97, 1089–1125. [Google Scholar] [CrossRef]
- Padányi, R.; Pászty, K.; Hegedűs, L.; Varga, K.; Papp, B.; Penniston, J.T.; Enyedi, A. Multifaceted plasma membrane Ca2+ pumps: From structure to intracellular Ca2+ handling and cancer. Biochim. Biophys. Acta (BBA) Mol. Cell Res. 2016, 1863, 1351–1363. [Google Scholar] [CrossRef]
- Lopreiato, R.; Giacomello, M.; Carafoli, E. The plasma membrane calcium pump: New ways to look at an old enzyme. J. Biol. Chem. 2014, 289, 10261–10268. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Gordones, M.C.; Ramírez-Iglesias, J.R.; Cervino, V.; Uzcanga, G.L.; Benaim, G.; Mendoza, M. Evidence of the presence of a calmodulin-sensitive plasma membrane Ca2+-ATPase in Trypanosoma equiperdum. Mol. Biochem. Parasitol. 2017, 213, 1–11. [Google Scholar] [CrossRef]
- Bruce, J.I.E. Metabolic regulation of the PMCA: Role in cell death and survival. Cell Calcium 2018, 69, 28–36. [Google Scholar] [CrossRef]
- Strehler, E.E. Plasma membrane calcium atpases as novel candidates for therapeutic agent development. J. Pharm. Pharm. Sci. 2013, 16, 190. [Google Scholar] [CrossRef] [PubMed]
- Brini, M.; Carafoli, E. The Plasma Membrane Ca2+ ATPase and the Plasma Membrane Sodium Calcium Exchanger Cooperate in the Regulation of Cell Calcium. Cold Spring Harb Perspect Biol. 2011, 3, a004168. [Google Scholar] [CrossRef] [PubMed]
- Hilge, M. Ca2+ regulation of ion transport in the Na+/Ca2+ exchanger. J. Biol. Chem. 2012, 287, 31641–31649. [Google Scholar] [CrossRef]
- Giladi, M.; Lee, S.Y.; Ariely, Y.; Teldan, Y.; Granit, R.; Strulovich, R.; Haitin, Y.; Chung, K.Y.; Khananshvili, D. Structure-based dynamic arrays in regulatory domains of sodium-calcium exchanger (Ncx) isoforms. Sci. Rep. 2017, 7, 993. [Google Scholar] [CrossRef]
- Emery, L.; Whelan, S.; Hirschi, K.D.; Pittman, J.K. Protein phylogenetic analysis of Ca2+/cation antiporters and insights into their evolution in plants. Front. Plant. Sci. 2012, 3, 1. [Google Scholar] [CrossRef]
- Annunziato, L.; Pignataro, G.; Di Renzo, G.F. Pharmacology of brain Na+/Ca2+ exchanger: From molecular biology to therapeutic perspectives. Pharmacol. Rev. 2004, 56, 633–654. [Google Scholar] [CrossRef]
- Chemaly, E.R.; Troncone, L.; Lebeche, D. SERCA control of cell death and survival. Cell Calcium 2018, 69, 46–61. [Google Scholar] [CrossRef]
- Tsai, F.-C.; Seki, A.; Yang, H.W.; Hayer, A.; Carrasco, S.; Malmersjö, S.; Meyer, T. A polarized Ca2+, diacylglycerol and STIM1 signalling system regulates directed cell migration. Nat. Cell Biol. 2014, 16, 133–144. [Google Scholar] [CrossRef]
- Papin, J.; Zummo, F.P.; Pachera, N.; Guay, C.; Regazzi, R.; Cardozo, A.K.; Herchuelz, A. Na+/Ca2+ Exchanger a Druggable Target to Promote β-Cell Proliferation and Function. J. Endocr. Soc. 2018, 2, 631–645. [Google Scholar] [CrossRef] [PubMed]
- Molinaro, P.; Pannaccione, A.; Sisalli, M.J.; Secondo, A.; Cuomo, O.; Sirabella, R.; Cantile, M.; Ciccone, R.; Scorziello, A.; Di Renzo, G.; et al. A New Cell-penetrating Peptide That Blocks the Autoinhibitory XIP Domain of NCX1 and Enhances Antiporter Activity. Mol. Ther. 2015, 23, 465–476. [Google Scholar] [CrossRef] [PubMed]
- Khalil, M.; Alliger, K.; Weidinger, C.; Yerinde, C.; Wirtz, S.; Becker, C.; Engel, M.A. Functional Role of Transient Receptor Potential Channels in Immune Cells and Epithelia. Front. Immunol. 2018, 9, 174. [Google Scholar] [CrossRef] [PubMed]
- Schnitzler, M.M.; Wäring, J.; Gudermann, T.; Chubanov, V. Evolutionary determinants of divergent calcium selectivity of TRPM channels. FASEB J. 2008, 22, 1540–1551. [Google Scholar] [CrossRef]
- Voolstra, O.; Huber, A. Post-translational modifications of trp channels. Cells 2014, 3, 258–287. [Google Scholar] [CrossRef]
- van Goor, M.K.C.; Hoenderop, J.G.J.; van der Wijst, J. TRP channels in calcium homeostasis: From hormonal control to structure-function relationship of TRPV5 and TRPV6. Biochim. Biophys. Acta (BBA) Mol. Cell Res. 2017, 1864, 883–893. [Google Scholar] [CrossRef]
- Gees, M.; Colsoul, B.; Nilius, B. The Role of Transient Receptor Potential Cation Channels in Ca2+ Signaling. Cold Spring Harb. Perspect. Biol. 2010, 2, a003962. [Google Scholar] [CrossRef]
- Gaudet, R. Structural Insights into the Function of TRP Channels (Chapter 25). In TRP Ion Channel Function in Sensory Transduction and Cellular Signaling Cascades; CRC Press: Boca Raton, FL, USA; Taylor & Francis: Abingdon, UK, 2007. [Google Scholar]
- Cohen, M.; Moiseenkova-Bell, V.Y. Structure of Thermally Activated TRP Channels. Curr. Top. Membr. 2014, 74, 181–211. [Google Scholar]
- Gaudet, R. A primer on ankyrin repeat function in TRP channels and beyond. Mol. Biosyst. 2008, 4, 372–379. [Google Scholar] [CrossRef] [PubMed]
- Earley, S.; Brayden, J.E. Transient receptor potential channels in the vasculature. Physiol. Rev. 2015, 95, 645–690. [Google Scholar] [CrossRef] [PubMed]
- Clapham, D.E. TRP channels as cellular sensors. Nature 2003, 426, 517–524. [Google Scholar] [CrossRef]
- Muller, C.; Morales, P.; Reggio, P.H. Cannabinoid ligands targeting trp channels. Front. Mol. Neurosci. 2019, 11, 487. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Hackos, D.H. TRPA1 as a drug target—Promise and challenges. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2015, 388, 451–463. [Google Scholar] [CrossRef]
- Li, M.; Yu, Y.; Yang, J. Structural biology of trp channels. Adv. Exp. Med. Biol. 2011, 704, 1–23. [Google Scholar] [CrossRef]
- Zimova, L.; Barvikova, K.; Macikova, L.; Vyklicka, L.; Sinica, V.; Barvik, I.; Vlachova, V. Proximal c-terminus serves as a signaling hub for trpa1 channel regulation via its interacting molecules and supramolecular complexes. Front. Physiol. 2020, 11, 189. [Google Scholar] [CrossRef]
- Eder, P.; Schindl, R.; Romanin, C.; Groschner, K. Protein–protein interactions in trpc channel complexes. In TRP Ion Channel Function in Sensory Transduction and Cellular Signaling Cascades; Liedtke, W.B., Heller, S., Eds.; CRC Press: Boca Raton, FL, USA; Taylor & Francis: Abingdon, UK, 2007. [Google Scholar]
- Noyer, L.; Lemonnier, L.; Mariot, P.; Gkika, D. Partners in crime: Towards new ways of targeting calcium channels. Int. J. Mol. Sci. 2019, 20, 6344. [Google Scholar] [CrossRef]
- Shin, Y.-C.; Shin, S.-Y.; Chun, J.N.; Cho, H.S.; Lim, J.M.; Kim, H.-G.; So, I.; Kwon, D.; Jeon, J.-H. Trip database 2.0: A manually curated information hub for accessing trp channel interaction network. PLoS ONE 2012, 7. [Google Scholar] [CrossRef]
- Shin, Y.-C.; Shin, S.-Y.; So, I.; Kwon, D.; Jeon, J.-H. TRIP Database: A manually curated database of protein–protein interactions for mammalian TRP channels. Nucleic Acids Res. 2011, 39 (Suppl. 1), D356–D361. [Google Scholar] [CrossRef]
- Chun, J.N.; Lim, J.M.; Kang, Y.; Kim, E.H.; Shin, Y.-C.; Kim, H.-G.; Jang, D.; Kwon, D.; Shin, S.-Y.; So, I.; et al. A network perspective on unraveling the role of TRP channels in biology and disease. Pflügers Arch. Eur. J. Physiol. 2014, 466, 173–182. [Google Scholar] [CrossRef]
- Vangeel, L.; Voets, T. Transient receptor potential channels and calcium signaling. Cold Spring Harb. Perspect. Biol. 2019, 11. [Google Scholar] [CrossRef]
- Dong, X.; Wang, X.; Xu, H. Trp channels of intracellular membranes. J. Neurochem. 2010, 113, 313–328. [Google Scholar] [CrossRef]
- Pecze, L.; Blum, W.; Henzi, T.; Schwaller, B. Endogenous TRPV1 stimulation leads to the activation of the inositol phospholipid pathway necessary for sustained Ca2+ oscillations. Biochim. Biophys. Acta (BBA) Mol. Cell Res. 2016, 1863, 2905–2915. [Google Scholar] [CrossRef] [PubMed]
- Thillaiappan, N.B.; Chakraborty, P.; Hasan, G.; Taylor, C.W. IP3 receptors and Ca2+ entry. Biochim. Biophys. Acta (BBA) Mol. Cell Res. 2019, 1866, 1092–1100. [Google Scholar] [CrossRef] [PubMed]
- Cullen, P.J.; Steinberg, F. To degrade or not to degrade: Mechanisms and significance of endocytic recycling. Nat. Rev. Mol. Cell Biol. 2018, 19, 679–696. [Google Scholar] [CrossRef] [PubMed]
- Peinelt, C.; Beck, A.; Monteilh-Zoller, M.K.; Penner, R.; Fleig, A. IP3 receptor subtype-dependent activation of store-operated calcium entry through ICRAC. Cell Calcium 2009, 45, 326–330. [Google Scholar] [CrossRef] [PubMed]
- Rao, J.N.; Platoshyn, O.; Golovina, V.A.; Liu, L.; Zou, T.; Marasa, B.S.; Turner, D.J.; Yuan, J.X.-J.; Wang, J.-Y. TRPC1 functions as a store-operated Ca2+ channel in intestinal epithelial cells and regulates early mucosal restitution after wounding. Am. J. Physiol. Liver Physiol. 2006, 290, G782–G792. [Google Scholar] [CrossRef] [PubMed]
- Haustrate, A.; Prevarskaya, N.; Lehen’kyi, V. Role of the trpv channels in the endoplasmic reticulum calcium homeostasis. Cells 2020, 9, 317. [Google Scholar] [CrossRef]
- Firth, A.L.; Yuan, J.X.-J. Ion channels and transporters in the pulmonary vasculature: A focus on smooth muscle. In Textbook of Pulmonary Vascular Disease; Yuan, J.X.-J., Garcia, J.G.N., West, J.B., Hales, C.A., Rich, S., Archer, S.L., Eds.; Springer: Jersey, NJ, USA, 2011; pp. 223–244. [Google Scholar] [CrossRef]
- Sukumaran, P.; Schaar, A.; Sun, Y.; Singh, B.B. Functional role of TRP channels in modulating ER stress and Autophagy. Cell Calcium 2016, 60, 123–132. [Google Scholar] [CrossRef]
- Lam, A.K.; Galione, A. The endoplasmic reticulum and junctional membrane communication during calcium signaling. Biochim. Biophys. Acta (BBA)—Bioenerg. 2013, 1833, 2542–2559. [Google Scholar] [CrossRef]
- Michalak, M.; Robert Parker, J.M.; Opas, M. Ca2+ signaling and calcium binding chaperones of the endoplasmic reticulum. Cell Calcium 2002, 32, 269–278. [Google Scholar] [CrossRef] [PubMed]
- Berridge, M.J. The endoplasmic reticulum: A multifunctional signaling organelle. Cell Calcium 2002, 32, 235–249. [Google Scholar] [CrossRef] [PubMed]
- Burdakova, D.; Petersenb, O.H.; Verkhratskya, A. Intraluminal calcium as a primary regulator of endoplasmic reticulum function. Cell Calcium. 2005, 38, 303–310. [Google Scholar] [CrossRef]
- Raffaello, A.; Mammucari, C.; Gherardi, G.; Rizzuto, R. Calcium at the Center of Cell Signaling: Interplay between Endoplasmic Reticulum, Mitochondria, and Lysosomes. Trends Biochem. Sci. 2016, 41, 1035–1049. [Google Scholar] [CrossRef] [PubMed]
- Prakriya, M.; Lewis, R.S. Store-Operated Calcium Channels. Physiol. Rev. 2015, 85, 757–810. [Google Scholar] [CrossRef] [PubMed]
- Numaga-Tomita, T.; Putney, J.W. Role of STIM1- and Orai1-mediated Ca2+ entry in Ca2+-induced epidermal keratinocyte differentiation. J. Cell Sci. 2013, 126, 605–612. [Google Scholar] [CrossRef]
- Hodeify, R.; Yu, F.; Courjaret, R.; Nader, N.; Dib, M.; Sun, L.; Adap, E.; Hubrack, S.; Machaca, K. Regulation and role of store-operated Ca2+ entry in cellular proliferation. In Calcium Entry Channels in Non-Excitable Cells; Kozak, J.A., Putney, J.W., Eds.; CRC Press: Boca Raton, FL, USA; Taylor & Francis: Abingdon, UK, 2018. [Google Scholar] [CrossRef]
- Oritani, K.; Kincade, P.W. Identification of stromal cell products that interact with pre-B cells. J. Cell Biol. 1996, 134, 771–782. [Google Scholar] [CrossRef] [PubMed]
- Roos, J.; DiGregorio, P.J.; Yeromin, A.Y.; Ohlsen, K.; Lioudyno, M.; Zhang, S.; Safrina, O.; Kozak, J.A.; Wagner, S.L.; Cahalan, M.D.; et al. STIM1, an essential and conserved component of store-operated Ca2+ channel function. J. Cell Biol. 2005, 169, 435–445. [Google Scholar] [CrossRef]
- Liou, J.; Kim, M.L.; Heo, W.D.; Jones, J.T.; Myers, J.W.; Ferrell, J.E., Jr.; Meyer, T. STIM is a Ca2+ sensor essential for Ca2+-store-depletion-triggered Ca2+ influx. Curr. Biol. 2005, 15, 1235–2141. [Google Scholar] [CrossRef] [PubMed]
- Hewavitharana, T.; Deng, X.; Soboloff, J.; Gill, D.L. Role of Stim and Orai proteins in calcium signaling pathways. Cell Calcium 2007, 42, 173–182. [Google Scholar] [CrossRef]
- Zheng, L.; Stathopulos, P.B.; Schindl, R.; Li, G.-Y.; Romanin, C.; Ikura, M. Auto-inhibitory role of the EF-SAM domain of STIM proteins in store-operated calcium entry. Proc. Natl. Acad. Sci. USA 2011, 108, 1337–1342. [Google Scholar] [CrossRef]
- Stathopulos, P.B.; Zheng, L.; Ikura, M. Stromal Interaction Molecule (STIM) 1 and STIM2 Calcium Sensing Regions Exhibit Distinct Unfolding and Oligomerization Kinetics. J. Biol. Chem. 2009, 284, 728–732. [Google Scholar] [CrossRef] [PubMed]
- Brandman, O.; Liou, J.; Park, W.S.; Meyer, T. STIM2 is a feedback regulator that stabilizes basal cytosolic and endoplasmic reticulum Ca2+ levels. Cell 2007, 131, 1327–1339. [Google Scholar] [CrossRef]
- Soboloff, J.; Spassova, M.A.; Hewavitharana, T.; He, L.-P.; Xu, W.; Johnstone, L.S.; Dziadek, M.A.; Gill, D.L. STIM2 is an inhibitor of STIM1-mediated store-operated Ca2+ Entry. Curr. Biol. 2006, 16, 1465–1470. [Google Scholar] [CrossRef] [PubMed]
- Vig, M.; Peinelt, C.; Beck, A.; Koomoa, D.L.; Rabah, D.; Koblan-Huberson, M.; Kraft, S.; Turner, H.; Fleig, A.; Penner, R.; et al. Cracm1 is a plasma membrane protein essential for store-operated Ca2+ entry. Science 2006, 312, 1220–1223. [Google Scholar] [CrossRef]
- Lis, A.; Peinelt, C.; Beck, A.; Parvez, S.; Monteilh-Zoller, M.; Fleig, A.; Penner, R. CRACM1, CRACM2, and CRACM3 Are Store-Operated Ca2+ Channels with Distinct Functional Properties. Curr. Biol. 2007, 27, 794–800. [Google Scholar] [CrossRef]
- Prakriya, M. Store-Operated Orai Channels: Structure and Function. Curr. Top. Membr. 2013, 71, 1–32. [Google Scholar] [PubMed]
- Hogan, P.G.; Rao, A. Store-operated calcium entry: Mechanisms and modulation. Biochem. Biophys. Res. Commun. 2015, 460, 40–49. [Google Scholar] [CrossRef] [PubMed]
- Hoover, P.J.; Lewis, R.S. Stoichiometric requirements for trapping and gating of Ca2+ release-activated Ca2+ (Crac) channels by stromal interaction molecule 1 (Stim1). Proc. Natl. Acad. Sci. USA 2011, 108, 13299–13304. [Google Scholar] [CrossRef]
- Wu, M.M.; Covington, E.D.; Lewis, R.S. Single-molecule analysis of diffusion and trapping of STIM1 and Orai1 at endoplasmic reticulum—Plasma membrane junctions. Mol. Biol. Cell 2014, 25, 3672–3685. [Google Scholar] [CrossRef] [PubMed]
- Thompson, J.L.; Shuttleworth, T.J. Molecular basis of activation of the arachidonate-regulated Ca2+ (Arc) channel, a store-independent Orai channel, by plasma membrane STIM1. J. Physiol. 2013, 591, 3507–3523. [Google Scholar] [CrossRef]
- Facilitation of Orai3 targeting and store-operated function by Orai1. Biochim. Biophys. Acta (BBA) Mol. Cell Res. 2015, 1853, 1541–1550. [CrossRef]
- Putney, J.W.; McKay, R.R. Capacitative calcium entry channels. BioEssays 1999, 21, 38–46. [Google Scholar] [CrossRef]
- Mikoshiba, K.; Hattori, M. Ip3 receptor-operated calcium entry. Sci. Signal. 2000, 2000, pe1. [Google Scholar] [CrossRef]
- Sampieri, A.; Santoyo, K.; Asanov, A.; Vaca, L. Association of the IP3R to STIM1 provides a reduced intraluminal calcium microenvironment, resulting in enhanced store-operated calcium entry. Sci. Rep. 2018, 8, 13252. [Google Scholar] [CrossRef]
- Derler, I.; Jardin, I.; Romanin, C. Molecular mechanisms of STIM/Orai communication. Am. J. Physiol. Physiol. 2016, 310, C643–C662. [Google Scholar] [CrossRef] [PubMed]
- Tiffner, A.; Derler, I. Molecular Choreography and Structure of Ca2+ Release-Activated Ca2+ (CRAC) and KCa2+ Channels and Their Relevance in Disease with Special Focus on Cancer. Membranes 2020, 10, 425. [Google Scholar] [CrossRef] [PubMed]
- Derler, I.; Madl, J.; Schütz, G.; Romanin, C. Structure, regulation and biophysics of icrac, stim/orai1. In Calcium Signaling; Md. Islam, S., Ed.; Springer: Dordrecht, The Netherlands, 2012; pp. 383–410. [Google Scholar] [CrossRef]
- Chung, S.; Zhang, M.; Stathopulos, P. The 2β Splice Variation Alters the Structure and Function of the Stromal Interaction Molecule Coiled-Coil Domains. Int. J. Mol. Sci. 2018, 19, 3316. [Google Scholar] [CrossRef]
- Hamada, K.; Miyatake, H.; Terauchi, A.; Mikoshiba, K. IP3-mediated gating mechanism of the IP3 receptor revealed by mutagenesis and X-ray crystallography. Proc. Natl. Acad. Sci. USA 2017, 114, 4661–4666. [Google Scholar] [CrossRef] [PubMed]
- Narayanan, D.; Adebiyi, A.; Jaggar, J.H. Inositol trisphosphate receptors in smooth muscle cells. Am. J. Physiol. Circ. Physiol. 2012, 302, H2190–H2210. [Google Scholar] [CrossRef] [PubMed]
- Taylor, C.W.; da Fonseca, P.; Morris, E. IP3 receptors: The search for structure. Trends Biochem. Sci. 2004, 29, 210–219. [Google Scholar] [CrossRef] [PubMed]
- Serysheva, I.I. Toward a high-resolution structure of IP3R channel. Cell Calcium 2014, 56, 125–132. [Google Scholar] [CrossRef]
- Zhou, Y.; Nwokonko, R.M.; Baraniak, J.H., Jr.; Trebak, M.; Lee, K.P.K.; Gill, D.L. The remote allosteric control of Orai channel gating. PLoS Biol. 2019, 17, e3000413. [Google Scholar] [CrossRef] [PubMed]
- Cai, X.; Zhou, Y.; Nwokonko, R.; Loktionova, N.A.; Wang, X.; Xin, P.; Trebak, M.; Wang, Y.; Gill, D.L. The Orai1 Store-operated Calcium Channel Functions as a Hexamer. J. Biol. Chem. 2016, 291, 25764–25775. [Google Scholar] [CrossRef]
- Pan, Z.; Ma, J.; Zui, P.; Jianjie, M. Open Sesame: Treasure in store-operated calcium entry pathway for cancer therapy. Sci. China Life Sci. 2014, 58, 48–53. [Google Scholar] [CrossRef][Green Version]
- Zhai, X.; Sterea, A.M.; El Hiani, Y. Lessons from the Endoplasmic Reticulum Ca2+ Transporters—A Cancer Connection. Cells 2020, 9, 1536. [Google Scholar] [CrossRef] [PubMed]
- Emrich, S.M.; Yoast, R.E.; Xin, P.; Arige, V.; Wagner, L.E.; Hempel, N.; Gill, D.L.; Sneyd, J.; Yule, D.I.; Trebak, M. Omnitem-poral choreographies of IP3R and all five STIM/Orai underlie the complexity of mammalian Ca2+ signaling. BioRxiv 2020. [Google Scholar] [CrossRef]
- Korzeniowski, M.K.; Baird, B.; Holowka, D. STIM1 activation is regulated by a 14 amino acid sequence adjacent to the CRAC activation domain. AIMS Biophys. 2016, 3, 99–118. [Google Scholar] [CrossRef] [PubMed]
- Lopez, J.J.; Jardin, I.; Sanchez-Collado, J.; Salido, G.M.; Smani, T.; Rosado, J.A. TRPC Channels in the SOCE Scenario. Cells 2020, 9, 126. [Google Scholar] [CrossRef]
- Furuichi, T.; Yoshikawa, S.; Miyawaki, A.; Wada, K.; Maeda, N.; Mikoshiba, K. Primary structure and functional expression of the inositol 1,4,5-trisphosphate-binding protein P400. Nat. Cell Biol. 1989, 342, 32–38. [Google Scholar] [CrossRef] [PubMed]
- Taylor, C.W.; Tovey, S.C. IP3 Receptors: Toward Understanding Their Activation. Cold Spring Harb. Perspect. Biol. 2010, 2, a004010. [Google Scholar] [CrossRef]
- Hisatsune, C.; Mikoshiba, K. IP3receptor mutations and brain diseases in human and rodents. J. Neurochem. 2017, 141, 790–807. [Google Scholar] [CrossRef]
- Prole, D.L.; Taylor, C. Inositol 1,4,5-trisphosphate receptors and their protein partners as signalling hubs. J. Physiol. 2016, 594, 2849–2866. [Google Scholar] [CrossRef] [PubMed]
- Tu, J.C.; Xiao, B.; Yuan, J.P.; A Lanahan, A.; Leoffert, K.; Li, M.; Linden, D.J.; Worley, P.F. Homer Binds a Novel Proline-Rich Motif and Links Group 1 Metabotropic Glutamate Receptors with IP3 Receptors. Neuron 1998, 21, 717–726. [Google Scholar] [CrossRef]
- Li, C.; Enomoto, M.; Rossi, A.M.; Seo, M.-D.; Rahman, T.; Stathopulos, P.; Taylor, C.; Ikura, M.; Ames, J.B. CaBP1, a neuronal Ca2+ sensor protein, inhibits inositol trisphosphate receptors by clamping intersubunit interactions. Proc. Natl. Acad. Sci. 2013, 110, 8507–8512. [Google Scholar] [CrossRef]
- Hirota, J.; Michikawa, T.; Natsume, T.; Furuichi, T.; Mikoshiba, K. Calmodulin inhibits inositol 1,4,5-trisphosphate-induced calcium release through the purified and reconstituted inositol 1,4,5-trisphosphate receptor type 1. FEBS Lett. 1999, 456, 322–326. [Google Scholar] [CrossRef]
- Bourguignon, L.Y.; Jin, H.; Iida, N.; Brandt, N.R.; Zhang, S.H. The involvement of ankyrin in the regulation of inositol 1,4,5-trisphosphate receptor-mediated internal Ca2+ release from Ca2+ storage vesicles in mouse T-lymphoma cells. J. Biol. Chem. 1993, 268, 7290–7297. [Google Scholar] [CrossRef]
- Ando, H.; Mizutani, A.; Matsu-Ura, T.; Mikoshiba, K. IRBIT, a Novel Inositol 1,4,5-Trisphosphate (IP3) Receptor-binding Protein, Is Released from the IP3 Receptor upon IP3 Binding to the Receptor. J. Biol. Chem. 2003, 278, 10602–10612. [Google Scholar] [CrossRef]
- Uchida, K.; Miyauchi, H.; Furuichi, T.; Michikawa, T.; Mikoshiba, K. Critical Regions for Activation Gating of the Inositol 1,4,5-Trisphosphate Receptor. J. Biol. Chem. 2003, 278, 16551–16560. [Google Scholar] [CrossRef] [PubMed]
- Rahman, T. Dynamic clustering of IP3 receptors by IP3. Biochem. Soc. Trans. 2012, 40, 325–330. [Google Scholar] [CrossRef] [PubMed]
- Taylor, C.W.; Konieczny, V. IP3receptors: Take four IP3to open. Sci. Signal. 2016, 9, pe1. [Google Scholar] [CrossRef] [PubMed]
- Bezprozvanny, L.; Watras, J.; Ehrlich, B. Bell-shaped calcium-response curves of lns(l,4,5)P3- and calcium-gated channels from endoplasmic reticulum of cerebellum. Nat. Cell Biol. 1991, 351, 751–754. [Google Scholar] [CrossRef] [PubMed]
- Hisatsune, C.; Nakamura, K.; Kuroda, Y.; Nakamura, T.; Mikoshiba, K. Amplification of Ca2+ Signaling by Diacylglycerol-mediated Inositol 1,4,5-Trisphosphate Production. J. Biol. Chem. 2005, 280, 11723–11730. [Google Scholar] [CrossRef] [PubMed]
- Van Coppenolle, F.; Vanden Abeele, F.; Slomianny, C.; Flourakis, M.; Hesketh, J.; Dewailly, E.; Prevarskaya, N. Ribosome-translocon complex mediates calcium leakage from endoplasmic reticulum stores. J. Cell Sci. 2004, 117, 4135–4142. [Google Scholar] [CrossRef]
- Vaca, L. SOCIC: The store-operated calcium influx complex. Cell Calcium 2010, 47, 199–209. [Google Scholar] [CrossRef] [PubMed]
- Gudlur, A.; Hogan, P.G. The STIM-Orai Pathway: Orai, the Pore-Forming Subunit of the CRAC Channel. Adv. Exp. Med. Biol. 2017, 993, 39–57. [Google Scholar] [CrossRef]
- DeHaven, W.I.; Jones, B.F.; Petranka, J.G.; Smyth, J.T.; Tomita, T.; Bird, G.S.; Putney, J.W. TRPC channels function independently of STIM1 and Orai1. J. Physiol. 2009, 587, 2275–2298. [Google Scholar] [CrossRef]
- Cheng, K.T.; Ong, H.L.; Liu, X.; Ambudkar, I.S. Contribution and Regulation of TRPC Channels in Store-Operated Ca2+ Entry. Curr. Top. Membr. 2013, 71, 149–179. [Google Scholar] [CrossRef]
- De Souza, L.B.; Ong, H.L.; Liu, X.; Ambudkar, I.S. Fast endocytic recycling determines TRPC1–STIM1 clustering in ER–PM junctions and plasma membrane function of the channel. Biochim. Biophys. Acta (BBA)—Bioenerg. 2015, 1853, 2709–2721. [Google Scholar] [CrossRef]
- Ambudkar, I.S.; de Souza, L.B.; Ong, H.L. TRPC1, Orai1, and STIM1 in SOCE: Friends in tight spaces. Cell Calcium 2017, 63, 33–39. [Google Scholar] [CrossRef] [PubMed]
- Pacheco, J.; Vaca, L. Stim-trp pathways and microdomain organization: Auxiliary proteins of the stim/orai complex. In Store-Operated Ca2+ Entry (SOCE) Pathways: Emerging Signaling Concepts in Human (Patho)physiology; Groschner, K., Graier, W.F., Romanin, C., Eds.; Springer: Berlin/Heidelberg, Germany, 2017; pp. 189–210. [Google Scholar] [CrossRef]
- Redondo, P.C.; Harper, A.G.S.; Salido, G.M.; Pariente, J.A.; Sage, S.O.; Rosado, J.A. A role for SNAP-25 but not VAMPs in store-mediated Ca2+entry in human platelets. J. Physiol. 2004, 558, 99–109. [Google Scholar] [CrossRef]
- Sharma, A.; Ramena, G.; Yin, Y.; Premkumar, L.; Elble, R.C. CLCA2 is a positive regulator of store-operated calcium entry and TMEM16A. PLoS ONE 2018, 13, e0196512. [Google Scholar] [CrossRef]
- Lopez, J.J.; Albarran, L.; Gómez, L.J.; Smani, T.; Salido, G.M.; Rosado, J.A. Molecular modulators of store-operated calcium entry. Biochim. Biophys. Acta (BBA)—Bioenerg. 2016, 1863, 2037–2043. [Google Scholar] [CrossRef] [PubMed]
- Quintana, A.; Rajanikanth, V.; Farber-Katz, S.; Gudlur, A.; Zhang, C.; Jing, J.; Zhou, Y.; Rao, A.; Hogan, P.G. TMEM110 regulates the maintenance and remodeling of mammalian ER–plasma membrane junctions competent for STIM–ORAI signaling. Proc. Natl. Acad. Sci. USA 2015, 112, E7083–E7092. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Wang, L.; Li, S.; Xue, J.; Luo, D. Calsequestrin-1 Regulates Store-Operated Ca2+ Entry by Inhibiting STIM1 Aggregation. Cell. Physiol. Biochem. 2016, 38, 2183–2193. [Google Scholar] [CrossRef]
- Albarran, L.; Lopez, J.J.; Ben Amor, N.; Cano, F.E.M.; Erro, A.B.; Smani, T.; Salido, G.M.; Rosado, J.A. Dynamic interaction of SARAF with STIM1 and Orai1 to modulate store-operated calcium entry. Sci. Rep. 2016, 6, 24452. [Google Scholar] [CrossRef]
- Malli, R.; Frieden, M.; Trenker, M.; Graier, W.F. The Role of Mitochondria for Ca2+ Refilling of the Endoplasmic Reticulum. J. Biol. Chem. 2005, 280, 12114–12122. [Google Scholar] [CrossRef] [PubMed]
- DeLuca, H.F.; Engstrom, G.W. Calcium uptake by rat kidney mitochondria. Proc. Natl. Acad. Sci. USA 1961, 47, 1744–1750. [Google Scholar] [CrossRef]
- Finkel, T.; Menazza, S.; Holmström, K.; Parks, R.J.; Liu, J.; Sun, J.; Liu, J.; Pan, X.; Murphy, E. The Ins and Outs of Mitochondrial Calcium. Circ. Res. 2015, 116, 1810–1819. [Google Scholar] [CrossRef] [PubMed]
- Patergnani, S.; Suski, J.M.; Agnoletto, C.; Bononi, A.; Bonora, M.; De Marchi, E.; Giorgi, C.; Marchi, S.; Missiroli, S.; Poletti, F.; et al. Calcium signaling around Mitochondria Associated Membranes (MAMs). Cell Commun. Signal. 2011, 9, 19. [Google Scholar] [CrossRef]
- Alberts, B. (Ed.) Molecular Biology of the Cell, 4th ed.; Garland Science: New York, NY, USA, 2002. [Google Scholar]
- Hajnóczky, G.; Csordás, G.; Yi, M. Old players in a new role: Mitochondria-associated membranes, VDAC, and ryanodine receptors as contributors to calcium signal propagation from endoplasmic reticulum to the mitochondria. Cell Calcium 2002, 32, 363–377. [Google Scholar] [CrossRef] [PubMed]
- Bononi, A.; Missiroli, S.; Poletti, F.; Suski, J.M.; Agnoletto, C.; Bonora, M.; De Marchi, E.; Giorgi, C.; Marchi, S.; Patergnani, S.; et al. Mitochondria-associated membranes (Mams) as hotspot Ca2+ signaling units. In Calcium Signaling; Islam, M.S., Ed.; Springer: Dordrecht, The Netherlands, 2012; Volume 740, pp. 411–437. [Google Scholar] [CrossRef]
- Colombini, M. VDAC structure, selectivity, and dynamics. Biochim. Biophys. Acta (BBA) Biomembr. 2012, 1818, 1457–1465. [Google Scholar] [CrossRef]
- Mishra, J.; Jhun, B.S.; Hurst, S.; O-Uchi, J.; Csordás, G.; Sheu, S.-S. The mitochondrial Ca2+ uniporter: Structure, function, and pharmacology. In Pharmacology of Mitochondria; Singh, H., Sheu, S.-S., Eds.; Springer: Berlin/Heidelberg, Germany, 2017; Volume 240, pp. 129–156. [Google Scholar] [CrossRef]
- Paillard, M.; Csordás, G.; Szanda, G.; Golenár, T.; Debattisti, V.; Bartok, A.; Wang, N.; Moffat, C.; Seifert, E.L.; Spät, A.; et al. Tissue-specific mitochondrial decoding of cytoplasmic Ca2+ signals is controlled by the stoichiometry of micu1/2 and mcu. Cell Rep. 2017, 18, 2291–2300. [Google Scholar] [CrossRef] [PubMed]
- Sassano, M.L.; van Vliet, A.R.; Agostinis, P. Mitochondria-associated membranes as networking platforms and regulators of cancer cell fate. Front. Oncol. 2017, 7, 174. [Google Scholar] [CrossRef]
- Schrepfer, E.; Scorrano, L. Mitofusins, from Mitochondria to Metabolism. Mol. Cell. 2016, 61, 683–694. [Google Scholar] [CrossRef] [PubMed]
- Vallese, F.; Barazzuol, L.; Maso, L.; Brini, M.; Calì, T. Er-mitochondria calcium transfer, organelle contacts and neurodegen-erative diseases. In Calcium Signaling; Islam, M.S., Ed.; Springer: Berlin/Heidelberg, Germany, 2020; pp. 719–746. [Google Scholar] [CrossRef]
- Morgan, A.J.; Davis, L.C.; Wagner, S.K.T.Y.; Lewis, A.M.; Parrington, J.; Churchill, G.C.; Galione, A. Bidirectional Ca2+ signaling occurs between the endoplasmic reticulum and acidic organelles. J. Cell Biol. 2013, 200, 789–805. [Google Scholar] [CrossRef]
- Gao, P.; Yan, Z.; Zhu, Z. Mitochondria-associated endoplasmic reticulum membranes in cardiovascular diseases. Front. Cell Dev. Biol. 2020, 8, 1309. [Google Scholar] [CrossRef]
- van Vliet, A.R.; Agostinis, P. Mitochondria-associated membranes and er stress. In Coordinating Organismal Physiology through the Unfolded Protein Response; Wiseman, R.L., Haynes, C.M., Eds.; Springer: Berlin/Heidelberg, Germany, 2018; pp. 73–102. [Google Scholar] [CrossRef]
- Perrone, M.; Caroccia, N.; Genovese, I.; Missiroli, S.; Modesti, L.; Pedriali, G.; Vezzani, B.; Vitto, V.A.M.; Antenori, M.; Lebiedzinska-Arciszewska, M.; et al. The role of mitochondria-associated membranes in cellular homeostasis and diseases. Int. Rev. Cell Mol. Biol. 2020, 350, 119–196. [Google Scholar] [CrossRef]
- Betz, C.; Stracka, D.; Prescianotto-Baschong, C.; Frieden, M.; Demaurex, N.; Hall, M.N. Mtor complex 2-akt signaling at mitochondria-associated endoplasmic reticulum membranes (Mam) regulates mitochondrial physiology. Proc. Natl. Acad. Sci. USA 2013, 110, 12526–12534. [Google Scholar] [CrossRef]
- Ouyang, Y.-B.; Giffard, R.G. Er-mitochondria crosstalk during cerebral ischemia: Molecular chaperones and er-mitochondrial calcium transfer. Int. J. Cell Biol. 2012. [Google Scholar] [CrossRef]
- Matsuzaki, H.; Fujimoto, T.; Tanaka, M.; Shirasawa, S. Tespa1 is a novel component of mitochondria-associated endoplasmic reticulum membranes and affects mitochondrial calcium flux. Biochem. Biophys. Res. Commun. 2013, 433, 322–326. [Google Scholar] [CrossRef]
- Lee, S.; Min, K.-T. The interface between er and mitochondria: Molecular compositions and functions. Mol. Cells 2018, 41, 1000–1007. [Google Scholar] [CrossRef] [PubMed]
- Kilpatrick, B.S.; Eden, E.R.; Hockey, L.N.; Yates, E.; Futter, C.E.; Patel, S. An endosomal naadp-sensitive two-pore Ca2+ channel regulates er-endosome membrane contact sites to control growth factor signaling. Cell Rep. 2017, 18, 1636–1645. [Google Scholar] [CrossRef] [PubMed]
- Bernardi, P.; Di Lisa, F.; Fogolari, F.; Lippe, G. From ATP to PTP and back: A dual function for the mitochondrial ATP synthase. Circ. Res. 2015, 116, 1850–1862. [Google Scholar] [CrossRef] [PubMed]
- Vultur, A.; Gibhardt, C.S.; Stanisz, H.; Bogeski, I. The role of the mitochondrial calcium uniporter (Mcu) complex in cancer. Pflügers Arch. Eur. J. Physiol. 2018, 470, 1149–1163. [Google Scholar] [CrossRef]
- Halestrap, A.P. The c ring of the f1fo atp synthase forms the mitochondrial permeability transition pore: A critical appraisal. Front. Oncol. 2014, 4, 234. [Google Scholar] [CrossRef]
- Gomez-Suaga, P.; Paillusson, S.; Stoica, R.; Noble, W.; Hanger, D.P.; Miller, C.C.J. The ER-Mitochondria Tethering Complex VAPB-PTPIP51 Regulates Autophagy. Curr. Biol. 2017, 27, 371–385. [Google Scholar] [CrossRef]
- Lively Lysosomes. Available online: https://www.asbmb.org/asbmb-today/science/050116/lively-lysosomes (accessed on 20 September 2020).
- Yang, J.; Zhao, Z.; Gu, M.; Feng, X.; Xu, H. Release and uptake mechanisms of vesicular Ca2+ stores. Protein Cell 2019, 10, 8–19. [Google Scholar] [CrossRef]
- Karch, J.; Molkentin, J.D. Identifying the components of the elusive mitochondrial permeability transition pore. Proc. Natl. Acad. Sci. USA 2014, 111, 10396–10397. [Google Scholar] [CrossRef] [PubMed]
- Garrity, A.G.; Wang, W.; Collier, C.M.; Levey, S.A.; Gao, Q.; Xu, H. The endoplasmic reticulum, not the pH gradient, drives calcium refilling of lysosomes. eLife 2016, 5, e15887. [Google Scholar] [CrossRef]
- Natarajan, G.K.; Glait, L.; Mishra, J.; Stowe, D.F.; Camara, A.K.S.; Kwok, W.-M. Total Matrix Ca2+ Modulates Ca2+ Efflux via the Ca2+/H+ Exchanger in Cardiac Mitochondria. Front. Physiol. 2020, 11. [Google Scholar] [CrossRef]
- Pathak, T.; Trebak, M. Mitochondrial Ca2+ signaling. Pharmacol. Ther. 2018, 192, 112–123. [Google Scholar] [CrossRef]
- Palty, R.; Silverman, W.F.; Hershfinkel, M.; Caporale, T.; Sensi, S.L.; Parnis, J.; Nolte, C.; Fishman, D.; Shoshan-Barmatz, V.; Herrmann, S.; et al. NCLX is an essential component of mitochondrial Na+/Ca2+ exchange. Proc. Natl. Acad. Sci. USA 2010, 107, 436–441. [Google Scholar] [CrossRef]
- Bernardi, P.; Von Stockum, S. The permeability transition pore as a Ca2+ release channel: New answers to an old question. Cell Calcium 2012, 52, 22–27. [Google Scholar] [CrossRef]
- Massari, S.; Azzone, G.F. The equivalent pore radius of intact and damaged mitochondria and the mechanism of active shrinkage. Biochim. Biophys. Acta (BBA)—Bioenerg. 1972, 283, 23–29. [Google Scholar] [CrossRef]
- Di Lisa, F.; Carpi, A.; Giorgio, V.; Bernardi, P. The mitochondrial permeability transition pore and cyclophilin D in cardioprotection. Biochim. Biophys. Acta (BBA)—Bioenerg. 2011, 1813, 1316–1322. [Google Scholar] [CrossRef] [PubMed]
- Ichas, F.; Mazat, J.-P. From calcium signaling to cell death: Two conformations for the mitochondrial permeability transition pore. Switching from low- to high-conductance state. Biochim. Biophys. Acta (BBA)—Bioenerg. 1998, 1366, 33–50. [Google Scholar] [CrossRef]
- Tinel, H.; Cancela, J.M.; Mogami, H.; Gerasimenko, J.; Gerasimenko, O.; Tepikin, A.; Petersen, O. Active mitochondria surrounding the pancreatic acinar granule region prevent spreading of inositol trisphosphate-evoked local cytosolic Ca2+ signals. EMBO J. 1999, 18, 4999–5008. [Google Scholar] [CrossRef]
- Gerasimenko, J.; Sherwood, M.; Tepikin, A.; Petersen, O.; Gerasimenko, O. NAADP, cADPR and IP3 all release Ca2+ from the endoplasmic reticulum and an acidic store in the secretory granule area. J. Cell Sci. 2006, 119, 226–238. [Google Scholar] [CrossRef]
- Patel, S.; Ramakrishnan, L.; Rahman, T.; Hamdoun, A.; Marchant, J.; Taylor, C.; Brailoiu, E. The endo-lysosomal system as an NAADP-sensitive acidic Ca2+ store: Role for the two-pore channels. Cell Calcium 2011, 50, 157–167. [Google Scholar] [CrossRef]
- Faris, P.; Shekha, M.; Montagna, D.; Guerra, G.; Moccia, F. Endolysosomal Ca2+ signalling and cancer hallmarks: Two-pore channels on the move, trpml1 lags behind! Cancers 2019, 11, 27. [Google Scholar] [CrossRef]
- Phillips, M.J.; Voeltz, G.K. Structure and function of ER membrane contact sites with other organelles. Nat. Rev. Mol. Cell Biol. 2016, 17, 69–82. [Google Scholar] [CrossRef]
- López-Sanjurjo, C.I.; Tovey, S.C.; Prole, D.L.; Taylor, C.W. Lysosomes shape Ins(1,4,5) P 3 -evoked Ca2+ signals by selectively sequestering Ca2+ released from the endoplasmic reticulum. J. Cell Sci. 2013, 126, 289–300. [Google Scholar] [CrossRef] [PubMed]
- Sbano, L.; Bonora, M.; Marchi, S.; Baldassari, F.; Medina, D.L.; Ballabio, A.; Giorgi, C.; Pinton, P. TFEB-mediated increase in peripheral lysosomes regulates store-operated calcium entry. Sci. Rep. 2017, 7, 40797. [Google Scholar] [CrossRef]
- Carruthers, C.; Suntzeff, V. Calcium, Copper, and Zinc in the Epidermal Carcinogenesis of Mouse and Man. Cancer Res. 1946, 6, 296. [Google Scholar] [PubMed]
- Borowiec, A.S.; Bidaux, G.; Pigat, N.; Goffin, V.; Bernichtein, S.; Capiod, T. Calcium Channels, External Calcium Concentration and Cell Proliferation. Eur. J. Pharmacol. 2014, 739, 19–25. [Google Scholar] [CrossRef] [PubMed]
- Whitfield, J.F. Calcium signals and cancer. Crit. Rev. Oncog. 1992, 3, 55–90. [Google Scholar]
- Cook, S.J.; Lockyer, P.J. Recent advances in Ca2+-dependent Ras regulation and cell proliferation. Cell Calcium 2006, 39, 101–112. [Google Scholar] [CrossRef]
- Boynton, A.L.; Whitfield, J.F.; Isaacs, R.J. The different roles of serum and calcium in the control of proliferation of BALB/c 3T3 mouse cells. In Vitro-Plant 1976, 12, 120–123. [Google Scholar] [CrossRef]
- Hazelton, B.; Mitchell, B.; Tupper, J. Calcium, magnesium, and growth control in the WI-38 human fibroblast cell. J. Cell Biol. 1979, 83, 487–498. [Google Scholar] [CrossRef]
- Prevarskaya, N.; Ouadid-Ahidouch, H.; Skryma, R.; Shuba, Y. Remodelling of Ca2+ transport in cancer: How it contributes to cancer hallmarks? Philos. Trans. R. Soc. Lond. B Biol. Sci. 2014, 369, 20130097. [Google Scholar] [CrossRef]
- Humeau, J.; Bravo-San Pedro, J.M.; Vitale, I.; Nuñez, L.; Villalobos, C.; Kroemer, G.; Senovill, L. Calcium signaling and cell cycle: Progression or death. Cell Calcium. 2018, 70, 3–15. [Google Scholar] [CrossRef]
- Capiod, T.; Shuba, Y.; Skryma, R.; Prevarskaya, N. Calcium signalling and cancer cell growth. Subcell Biochem. 2007, 45, 405–427. [Google Scholar] [CrossRef]
- Clowes, G.; Frisbie, W. No. 32. On the relationship between the rate of growth, age, and potassium and calcium content of mouse tumors (adeno-carcinoma, jensen). Am. J. Physiol. Leg. Content 1905, 14, 173–192. [Google Scholar] [CrossRef]
- Carruthers, C.; Suntzeff, V. The role of calcium in carcinogenesis summary. Science 1944, 99, 245–247. [Google Scholar] [CrossRef] [PubMed]
- Miller, K. Calcium and cancer. Med. Hypotheses 1977, 3, 263–264. [Google Scholar] [CrossRef]
- Kadio, B.; Yaya, S.; Basak, A.; Djè, K.; Gomes, J.; Mesenge, C. Calcium role in human carcinogenesis: A comprehensive analysis and critical review of literature. Cancer Metastasis Rev. 2016, 35, 391–411. [Google Scholar] [CrossRef] [PubMed]
- Peterlik, M.; Grant, W.B.; Cross, H.S. Calcium, Vitamin D and Cancer. Anticancer Res. 2009, 29, 3687–3698. [Google Scholar]
- Pottle, J.; Sun, C.; Gray, L.; Li, M. Exploiting MCF-7 Cells’ calcium dependence with interlaced therapy. J. Cancer Ther. 2013, 4, 32–40. [Google Scholar] [CrossRef]
- Taylor, J.M.; Simpson, R.U. Inhibition of Cancer Cell Growth by Calcium Channel Antagonists in the Athymic Mouse. Cancer Res. 1992, 52, 2413–2418. [Google Scholar]
- Xu, M.M.; Seas, A.; Kiyani, M.; Ji, K.S.Y.; Bell, H.N. A temporal examination of calcium signaling in cancer- from tumorigenesis, to immune evasion, and metastasis. Cell Biosci. 2018, 8, 25. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Prevarskaya, N.; Skryma, R.; Shuba, Y. Ion Channels in cancer: Are cancer hallmarks oncochannelopathies? Physiol. Rev. 2018, 98, 559–621. [Google Scholar] [CrossRef] [PubMed]
- Monteith, G.R.; Prevarskaya, N.; Roberts-Thomson, S.J. The calcium–cancer signalling nexus. Nat. Rev. Cancer 2017, 17, 367–380. [Google Scholar] [CrossRef] [PubMed]
- Roberts-Thomson, S.J.; Chalmers, S.B.; Monteith, G.R. The Calcium-Signaling Toolkit in Cancer: Remodeling and Targeting. Cold Spring Harb. Perspect. Biol. 2019, 11, a035204. [Google Scholar] [CrossRef] [PubMed]
- Roderick, H.W.; Cook, S.J. Ca2+ signalling checkpoints in cancer: Remodelling Ca2+ for cancer cell proliferation and survival. Nat. Rev. Cancer 2008, 8, 361–375. [Google Scholar] [CrossRef]
- Phan, N.N.; Wang, C.Y.; Chen, C.F.; Sun, Z.; Lai, M.D.; Lin, Y.L. Voltage-gated calcium channels: Novel targets for cancer therapy. Oncol. Lett. 2017, 14, 2059–2074. [Google Scholar] [CrossRef]
- Wang, C.Y.; Lai, M.D.; Phan, N.N.; Sun, Z.; Lin, Y.C. Meta-analysis of public microarray datasets reveals voltage-gated calcium gene signatures in clinical cancer patients. PLoS ONE 2015, 10, e0125766. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.T.; Nagaba, Y.; Cross, H.S.; Wrba, F.; Zhang, L.; Guggino, S.E. The mRNA of L-type calcium channel elevated in colon cancer: Protein distribution in normal and cancerous colon. Am. J. Pathol. 2000, 157, 1549–1562. [Google Scholar] [CrossRef]
- Buchanan, P.J.; McCloskey, K.D. CaV channels and cancer: Canonical functions indicate benefits of repurposed drugs as cancer therapeutics. Eur. Biophys. J. 2016, 45, 621–633. [Google Scholar] [CrossRef]
- Taylor, J.T.; Huang, L.; Pottle, J.E.; Liu, K.; Yang, Y.; Zeng, X.; Keyser, B.M.; Agrawal, K.C.; Hansen, J.B.; Li, M. Selective blockade of T-type Ca2+ channels suppresses human breast cancer cell proliferation. Cancer Lett. 2008, 267, 116–124. [Google Scholar] [CrossRef]
- Azimi, I.; Roberts-Thomson, S.J.; Monteith, G.R. Calcium influx pathways in breast cancer: Opportunities for pharmacological intervention. Br. J. Pharmacol. 2014, 171, 945–960. [Google Scholar] [CrossRef]
- Antal, L.; Martin-Caraballo, M. T-Type Calcium Channels in Cancer. Cancers 2019, 11, 134. [Google Scholar] [CrossRef]
- Barceló, C.; Sisó, P.; Maiques, O.; de la Rosa, I.; Martí, R.M.; Macià, A. T-Type Calcium Channels in Cnacer: A Potential Target in Melanoma. Cancers 2020, 12, 391. [Google Scholar] [CrossRef] [PubMed]
- Choi, D.L.; Jang, S.J.; Cho, S.; Choi, H.E.; Rim, H.K.; Lee, K.T.; Lee, J.Y. Inhibition of Cellular Proliferation and Induction of Apoptosis in Human Lung Adenocarcinoma A549 Cells by T-type Calcium Channel Antagonist. Bioorg. Med. Chem. Lett. 2014, 24, 1565–1570. [Google Scholar] [CrossRef] [PubMed]
- Rim, H.K.; Cho, S.; Shin, D.H.; Chung, K.S.; Cho, Y.W.; Choi, J.H.; Lee, J.Y.; Lee, K.T. T-type Ca2+ channel blocker, KYS05090 induces autophagy and apoptosis in A549 cells through inhibiting glucose uptake. Molecules 2014, 19, 9864–9875. [Google Scholar] [CrossRef]
- Rim, H.K.; Lee, H.W.; Choi, I.S.; Park, J.Y.; Choi, H.W.; Choi, J.H.; Cho, Y.W.; Lee, J.Y.; Lee, K.T. T-type Ca2+ channel blocker, KYS05047 induces G1 phase cell cycle arrest by decreasing intracellular Ca2+ levels in human lung adenocarcinoma A549 cells. Bioorg. Med. Chem. Lett. 2012, 22, 7123–7126. [Google Scholar] [CrossRef]
- Arif, T.; Amsalem, Z.; Shoshan-Barmatz, V. Metabolic Reprograming Via Silencing of Mitochondrial VDAC1 Expression Encourages Differentiation of Cancer Cells. Mol. Ther.—Nucleic Acids 2019, 17, 24–37. [Google Scholar] [CrossRef]
- Thinnes, F.P. Neuroendocrine differentiation of LNCaP cells suggests: VDAC in the cell membrane is involved in the extrinsic apoptotic pathway. Mol. Genet. Metab. 2009, 97, 241–243. [Google Scholar] [CrossRef]
- Fourbon, Y.; Guéguinou, M.; Félix, R.; Constantin, B.; Uguen, A.; Fromont, G.; Lajoie, L.; Magaud, C.; LeComte, T.; Chamorey, E.; et al. Ca2+ protein alpha 1D of CaV1.3 regulates intracellular calcium concentration and migration of colon cancer cells through a non-canonical activity. Sci. Rep. 2017, 7, 14199. [Google Scholar] [CrossRef]
- Scarpellino, G.; Genova, T.; Avanzato, D.; Bernardini, M.; Bianco, S.; Petrillo, S.; Tolosano, E.; de Almeida Vieira, J.R.; Bussolati, B.; Fiorio Pla, A.; et al. Purinergic Calcium Signals in Tumor-Derived Endothelium. Cancers 2019, 11, 766. [Google Scholar] [CrossRef]
- Chong, J.-H.; Zheng, G.-G.; Zhu, X.-F.; Guo, Y.; Wang, L.; Ma, C.-H.; Liu, S.-Y.; Xu, L.-L.; Lin, Y.-M.; Wu, K.-F. Abnormal expression of P2X family receptors in Chinese pediatric acute leukemias. Biochem. Biophys. Res. Commun. 2010, 391, 498–504. [Google Scholar] [CrossRef] [PubMed]
- Vijayan, D.; Smyth, M.J.; Teng, M.W.L. Purinergic Receptors: Novel Targets for Cancer Immunotherapy. In Oncoimmunology; Zitvogel, L., Kroemer, G., Eds.; Springer: Cham, Switzerland, 2018; ISBN 978-3-319-62431-0. [Google Scholar] [CrossRef]
- Maehara, Y.; Kusumoto, H.; Anai, H.; Kusumoto, T.; Sugimachi, K. Human tumor tissues have higher ATP contents than normal tissues. Clin. Chim. Acta 1987, 169, 341–343. [Google Scholar] [CrossRef]
- Azimi, I.; Beilby, H.; Davis, F.M.; Marcial, D.L.; Kenny, P.A.; Thompson, E.W.; Roberts-Thomson, S.; Monteith, G.R. Altered purinergic receptor-Ca2+ signaling associated with hypoxia-induced epithelial-mesenchymal transition in breast cancer cells. Mol. Oncol. 2016, 10, 166–178. [Google Scholar] [CrossRef]
- Davis, F.M.; Kenny, P.A.; Soo, E.T.L.; van Denderen, B.J.W.; Thompson, E.W.; Cabot, P.J.; Parat, M.O.; Roberts-Thomson, S.J.; Monteith, G.R. Remodeling of Purinergic Receptor-Mediated Ca2+ Signaling as a Consequence of EGF-induced Epithelial-Mesenchymal Transition in Breast Cancer Cells. PLoS ONE 2011, 6, e23464. [Google Scholar] [CrossRef] [PubMed]
- Avanzato, D.; Genova, T.; Fiorio, A.; Bernardini, M.; Bianco, S.; Bussolati, B.; Mancardi, D.; Giraudo, E.; Maione, F.; Cassoni, P.; et al. Activation of P2X7 and P2Y11 purinergic receptors inhibits migration and normalizes tumor-derived endothelial cells via cAMP signaling. Sci. Rep. 2016, 6, 32602. [Google Scholar] [CrossRef]
- Feng, W.; Yang, X.; Wang, L.; Wang, R.; Yang, F.; Wang, H.; Liu, X.; Ren, Q.; Zhang, Y.; Zhu, X.; et al. P2X7 promotes the progression of MLL-AF9 induced acute myeloid leukemia by upregulation of Pbx3. Haematol. 2020, 106, 1278–1289. [Google Scholar] [CrossRef]
- Reisner, P.D.; Brandt, P.C.; Vanaman, T.C. Analysis of plasma membrane Ca2+-ATPase expression in control and SV40-transformed human fibroblasts. Cell Calcium 1997, 21, 53–62. [Google Scholar] [CrossRef]
- Usachev, Y.M.; Toutenhoofd, S.L.; Goellner, G.M.; Strehler, E.E.; Thayer, S.A. Differentiation induces up-regulation of plasma membrane Ca2+-ATPase and concomitant increase in Ca2+ efflux in human neuroblastoma cell line IMR-32. J. Neurochem. 2001, 76, 1756–1765. [Google Scholar] [CrossRef]
- Roberts-Thomson, S.J.; Curry, M.C.; Monteith, G.R. Plasma membrane calcium pumps and their emerging roles in cancer. World J. Biol. Chem. 2010, 1, 248–253. [Google Scholar] [CrossRef]
- Aung, C.S.; Ye, W.; Plowman, G.; Peters, A.A.; Monteith, G.; Roberts-Thomson, S.J. Plasma membrane calcium ATPase 4 and the remodeling of calcium homeostasis in human colon cancer cells. Carcinogenesis 2009, 30, 1962–1969. [Google Scholar] [CrossRef] [PubMed]
- Peterson, J.A.; Oblad, R.V.; Mecham, J.C.; Kenealey, J.D. Resveratrol inhibits plasma membrane Ca2+-ATPase inducing an increase in cytoplasmic calcium. Biochem. Biophys. Rep. 2016, 7, 253–258. [Google Scholar] [CrossRef]
- Rüschoff, J.H.; Brandenburger, T.; Strehler, E.E.; Filoteo, A.G.; Heinmöller, E.; Aumüller, G.; Wilhelm, B. Plasma Membrane Calcium ATPase Expression in Human Colon Multistep Carcinogenesis. Cancer Investig. 2012, 30, 251–257. [Google Scholar] [CrossRef]
- Ribiczey, P.; Papp, B.; Homolya, L.; Enyedi, Á.; Kovács, T. Selective upregulation of the expression of plasma membrane calcium ATPase isoforms upon differentiation and 1,25(OH)2D3-vitamin treatment of colon cancer cells. Biochem. Biophys. Res. Commun. 2015, 464, 189–194. [Google Scholar] [CrossRef]
- Varga, K.; Pászty, K.; Padányi, R.; Hegedűs, L.; Brouland, J.P.; Papp, B.; Enyedi, A. Histone deacetylase inhibitor- and PMA-induced upregulation of PMCA4b enhances Ca2+ clearance from MCF-7 breast cancer cells. Cell Calcium 2014, 55, 78–92. [Google Scholar] [CrossRef]
- James, A.D.; Patel, W.; Butt, Z.; Adiamah, M.; Dakhel, R.; Latif, A.; Uggenti, C.; Swanton, E.; Imamura, H.; Siriwardena, A.K.; et al. The Plasma Membrane Calcium Pump in Pancreatic Cancer Cells Exhibiting the Warburg Effect Relies on Glycolytic ATP. J. Biol. Chem. 2015, 290, 24760–24771. [Google Scholar] [CrossRef]
- Varga, K.; Hollósi, A.; Pászty, K.; Hegedűs, L.; Szakács, G.; Tímár, J.; Papp, B.; Enyedi, A.; Padányi, A. Expression of Calcium Pumps Is Differentially Regulated by Histone Deacetylase Inhibitors and Estrogen Receptor Alpha in Breast Cancer Cells. BMC Cancer 2018, 18, 1029. [Google Scholar] [CrossRef]
- Peters, A.A.; Milevskiy, M.J.G.; Lee, W.C.; Curry, M.C.; Smart, C.E.; Saunus, J.M.; Reid, L.; da Silva, L.; Marcial, D.L.; Dray, E.; et al. The calcium pump plasma membrane Ca2+-ATPase 2 (PMCA2) regulates breast cancer cell proliferation and sensitivity to doxorubicin. Sci. Rep. 2016, 6, 25505. [Google Scholar] [CrossRef] [PubMed]
- Balasubramaniam, S.L.; Gopalakrishnapillai, A.; Petrelli, N.J.; Barwe, S.P. Knockdown of Sodium-Calcium Exchanger 1 Induces Epithelial to Mesenchymal Transition in Kidney Epithelial Cells. J. Biol. Chem. 2017, 292, 11388–11399. [Google Scholar] [CrossRef] [PubMed]
- Mahdi, S.H.; Cheng, H.; Li, J.; Feng, R. The effect of TGF-beta induced epithelial-mesenchymal transition on the expression of intracellular calcium-handling proteins in T47D and MCF-7 human breast cancer cells. Arch. Biochem. Biophys. 2015, 42, 1240–1251. [Google Scholar] [CrossRef] [PubMed]
- Munoz, J.J.; Drigo, S.A.; Barros-Filho, M.C.; Marchi, F.A.; Scapulatempo-Neto, C.; Pessoa, G.S.; Guimaraes, G.C.; Trindade Filho, J.C.; Lopes, A.; Arruda, M.A.; et al. Down-Regulation of SLC8A1 as a Putative Apoptosis Evasion Mechanism by Modulation of Calcium Levels in Penile Carcinoma. J. Urol. 2015, 194, 245–251. [Google Scholar] [CrossRef]
- Pelzl, L.; Hosseinzadeh, Z.; al-Maghout, T.; Singh, Y.; Sahu, I.; Bissinger, R.; Schmidt, S.; Alkahtani, S.; Stournaras, C.; Toulany, M.; et al. Role of Na+/Ca2+ Exchangers in Therapy Resistance of Medulloblastoma Cells. Cell Physiol. Biochem. 2017, 42, 1240–1251. [Google Scholar] [CrossRef]
- Zheng, X.; Lu, S.; He, Z.; Huang, H.; Yao, Z.; Miao, Y.; Cai, C.; Zou, F. MCU-dependent negative sorting of miR-4488 to extracellular vesicles enhances angiogenesis and promotes breast cancer metastatic colonization. Oncogene 2020, 39, 6975–6989. [Google Scholar] [CrossRef]
- Kucukkaya, B.; Basoglu, H.; Erdag, D.; Akbas, F.; Susgun, S.; Yalcintepe, L. Calcium homeostasis in cisplatin resistant epithelial ovarian cancer. Gen. Physiol. Biophys. 2019, 38, 353–363. [Google Scholar] [CrossRef]
- Liskova, V.; Hudecova, S.; Lencesova, L.; Iuliano, F.; Sirova, M.; Ondrias, K.; Pastorekova, S.; Krizanova, O. Type 1 Sodium Calcium Exchanger Forms a Complex with Carbonic Anhydrase IX and Via Reverse Mode Activity Contributes to pH Control in Hypoxic Tumors. Cancers 2019, 11, 1139. [Google Scholar] [CrossRef] [PubMed]
- Svastova, E.; Witarski, W.; Csaderova, L.; Kosik, I.; Skvarkova, L.; Hulikova, A.; Zatovicova, M.; Barathova, M.; Kopacek, J.; Pastorek, J.; et al. Carbonic Anhydrase IX Interacts with Bicarbonate Transporters in Lamellipodia and Increases Cell Migration via Its Catalytic Domain. J. Biol. Chem. 2012, 287, 3392–3402. [Google Scholar] [CrossRef]
- Sennoune, S.R.; Santos, J.M.; Hussain, F.; Martínez-Zaguilán, R. Sodium calcium ex-changer operates in the reverse mode in metastatic human melanoma cells. Cell. Mol. Biol. 2015, 61, 40–49. [Google Scholar]
- Tojyo, Y.; Morita, T.; Nezu, A.; Tanimura, A. Key Components of Store-Operated Ca2+ Entry in Non-Excitable Cells. J. Pharmacol. Sci. 2014, 125, 340–346. [Google Scholar] [CrossRef]
- Chen, Y.F.; Lin, P.C.; Yeh, Y.M.; Chen, L.H.; Shen, M.R. Store-Operated Ca2+ Entry in Tumor Progression: From Molecular Mechanisms to Clinical Implications. Cancers 2019, 11, 899. [Google Scholar] [CrossRef]
- Stewart, T.A.; Yapa, K.T.D.S.; Monteith, G.R. Altered calcium signaling in cancer cells. Biochim. Biophys. Acta 2015, 1848, 2502–2511. [Google Scholar] [CrossRef]
- Pierro, C.; Sneyers, F.; Bultynck, G.; Roderick, H.W. ER Ca2+ Release and Store-Operated Ca2+ Entry—Partners in Crime or Independent Actors in Oncogenic Transformation? Cell Calcium 2019, 82, 102061. [Google Scholar] [CrossRef]
- Hoth, M.; Niemeyer, B.A. The neglected CRAC proteins: Orai2, Orai3, and STIM2. Curr. Top. Membr. 2013, 71, 237–271. [Google Scholar] [CrossRef] [PubMed]
- Azimi, I.; Bong, A.H.; Poo, G.X.H.; Armitage, K.; Lok, D.; Roberts-Thomson, S.J.; Monteith, G.R. Pharmacological Inhibition of Store-Operated Calcium Entry in MDA-MB-468 Basal A Breast Cancer Cells: Consequences on Calcium Signalling, Cell Migration and Proliferation. Cell Mol. Life Sci. 2018, 75, 4525–4537. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.W.; Chen, Y.F.; Chen, Y.T.; Chiu, W.T.; Shen, M.R. The STIM1-Orai1 pathway of store-operated Ca2+ entry controls the checkpoint in cell cycle G1/S transition. Sci. Rep. 2016, 6, 22142. [Google Scholar] [CrossRef]
- Jardin, I.; Lopez, J.J.; Salido, G.M.; Rosado, J.M. Store-Operated Ca2+ Entry in Breast Cancer Cells: Remodeling and Functional Role. Int. J. Mol. Sci. 2018, 19, 4053. [Google Scholar] [CrossRef]
- Yang, S.; Zhang, J.J.; Huang, X.Y. Orai1 and STIM1 are critical for breast tumor cell migration and metastasis. Cancer Cell 2009, 15, 124–134. [Google Scholar] [CrossRef]
- El Boustany, C.; Bidaux, G.; Enfissi, A.; Delcourt, P.; Prevarskaya, N.; Capiod, T. Capacitative Calcium Entry and Transient Receptor Potential Canonical 6 Expression Control Human Hepatoma Cell Proliferation. Hepatology 2008, 47, 2068–2077. [Google Scholar] [CrossRef]
- Feng, M.; Grice, D.M.; Faddy, H.M.; Nguyen, N.; Leitch, S.; Wang, Y.; Muend, S.; Kenny, P.A.; Sukumar, S.; Roberts-Thomson, S.J.; et al. Store-independent activation of Orai1 by SPCA2 in mammary tumors. Cell 2010, 143, 84–98. [Google Scholar] [CrossRef]
- Li, G.; Zhang, Z.; Wang, R.; Ma, W.; Yang, Y.; Wei, J.; Wei, Y. Suppression of STIM1 inhibits human glioblastoma cell proliferation and induces G0/G1 phase arrest. J. Exp. Clin. Cancer Res. 2013, 32, 20. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.F.; Chiu, W.T.; Chen, Y.T.; Lin, P.Y.; Huang, H.J.; Chou, C.Y.; Chang, H.C.; Tang, M.J.; Shen, M.R. Calcium store sensor stromal-interaction molecule 1-dependent signaling plays an important role in cervical cancer growth, migration, and angiogenesis. Proc. Natl. Acad. Sci. USA 2011, 31, 203–211. [Google Scholar] [CrossRef]
- Zang, J.; Zuo, D.; Shogren, K.L.; Gustafson, C.T.; Zhou, Z.; Thompson, M.A.; Guo, R.; Prakash, Y.S.; Lu, L.; Guo, W.; et al. STIM1 expression is associated with osteosarcoma cell survival. Chin J Cancer Res. 2019, 31, 203–211. [Google Scholar] [CrossRef]
- Cheng, H.; Wang, S.-Q.; Feng, R. STIM1 plays an important role in TGF-β-induced suppression of breast cancer cell proliferation. Oncotarget 2016, 7, 16866–16878. [Google Scholar] [CrossRef]
- Ge, C.; Zeng, B.; Li, R.; Li, Z.; Fu, Q.; Wang, W.; Wang, Z.; Dong, S.; Lai, Z.; Wang, Y.; et al. Knockdown of STIM1 expression inhibits non-small-cell lung cancer cell proliferation in vitro and in nude mouse xenografts. Bioengineered 2019, 10, 425–436. [Google Scholar] [CrossRef]
- Motiani, R.K.; Zhang, X.X.; Harmon, K.E.; Keller, R.S.; Matrougui, K.; Bennett, J.A.; Trebak, M. Orai3 is an estrogen receptor α-regulated Ca 2+ channel that promotes tumorigenesis. FASEB J. 2013, 27, 63–75. [Google Scholar] [CrossRef]
- Benzerdjeb, N.; Sevestre, H.; Ahidouch, A.; Ouadid-Ahidouch, H. Orai3 is a predictive marker of metastasis and survival in resectable lung adenocarcinoma. Oncotarget 2016, 7, 81588–81597. [Google Scholar] [CrossRef]
- Dubois, C.; Abeele, F.V.; Lehen’Kyi, V.; Gkika, D.; Guarmit, B.; Lepage, G.; Slomianny, C.; Borowiec, A.S.; Bidaux, G.; Benahmed, M.; et al. Remodeling of Channel-Forming ORAI Proteins Determines an Oncogenic Switch in Prostate Cancer. Cancer Cell 2014, 26, 19–32. [Google Scholar] [CrossRef]
- Stanisz, H.; Saul, S.; Müller, C.S.L.; Kappl, R.; Niemeyer, B.A.; Vogt, T.; Hoth, M.; Roesch, A.; Bogeski, I. Inverse regulation of melanoma growth and migration by Orai1/STIM2-dependent calcium entry. Pigment. Cell Melanoma Res. 2014, 27, 442–453. [Google Scholar] [CrossRef]
- Fiorio Pla, A.; Kondratska, K.; Prevarskaya, N. STIM and ORAI proteins: Crucial roles in hallmarks of cancer. Am. J. Physiol. Cell Physiol. 2016, 310, C509–C519. [Google Scholar] [CrossRef]
- Kondratska, K.; Kondratskyi, A.; Yassine, M.; Lemonnier, L.; Lepage, G.; Morabito, A.; Skryma, R.; Prevarskaya, N. Orai1 and STIM1 mediate SOCE and contribute to apoptotic resistance of pancreatic adenocarcinoma. Biochim. Biophys. Acta (BBA)—Bioenerg. 2014, 1843, 2263–2269. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, S.; Liu, G.; Liu, G.; Yang, W.; Honisch, S.; Pantelakos, S.; Stournaras, C.; Hönig, A.; Lang, F. Enhanced Orai1 and STIM1 expression as well as store operated Ca2+ entry in therapy resistant ovary carcinoma cells. Oncotarget 2014, 5, 4799–4810. [Google Scholar] [CrossRef]
- Gualdani, R.; De Clippele, M.; Ratbi, I.; Gailly, P.; Tajeddine, N. Store-Operated Calcium Entry Contributes to Cisplatin-Induced Cell Death in Non-Small Cell Lung Carcinoma. Cancers 2019, 11, 430. [Google Scholar] [CrossRef]
- Flourakis, M.; Lehen’Kyi, V.; Beck, B.; Raphael, M.; Vandenberghe, M.; Van Denabeele, F.; Roudbaraki, M.; Lepage, G.; Mauroy, B.; Romanin, C.; et al. Orai1 contributes to the establishment of an apoptosis-resistant phenotype in prostate cancer cells. Cell Death Dis. 2010, 1, e75. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Wang, J.; He, Y.-Q.; Feng, C.; Zhang, X.-J.; Sheng, J.-Q.; Li, P.-F. MicroRNA-185 regulates chemotherapeutic sensitivity in gastric cancer by targeting apoptosis repressor with caspase recruitment domain. Cell Death Dis. 2014, 5, e1197. [Google Scholar] [CrossRef] [PubMed]
- Fedida-Metula, S.; Feldman, B.; Koshelev, V.; Levin-Gromiko, U.; Voronov, E.; Fishman, D. Lipid rafts couple store-operated Ca2+ entry to constitutive activation of PKB/Akt in a Ca2+/calmodulin-, Src- and PP2A-mediated pathway and promote melanoma tumor growth. Carcinogenesis 2012, 33, 740–750. [Google Scholar] [CrossRef]
- Mimura, N.; Hideshima, T.; Shimomura, T.; Suzuki, R.; Ohguchi, H.; Rizq, O.; Kikuchi, S.; Yoshida, Y.; Cottini, F.; Jakubikova, J.; et al. Selective and potent Akt inhibition triggers anti-myeloma activities and enhances fatal endoplasmic reticulum stress induced by proteasome inhibition. Oncotarget 2014, 74, 4458–4469. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Ren, Y.; Wang, L.; Zhao, W.; Dong, X.; Pan, J.; Gao, X.; Tian, Y. Orai1 and Stim1 Mediate the Majority of Store-Operated Calcium Entry in Multiple Myeloma and Have Strong Implications for Adverse Prognosis. Cell. Physiol. Biochem. 2018, 48, 2273–2285. [Google Scholar] [CrossRef]
- Zhan, Z.Y.; Zhong, L.X.; Feng, M.; Wang, J.F.; Liu, D.B.; Xiong, J.P. Over-expression of Orai1 mediates cell proliferation and associates with poor prognosis in human non-small cell lung carcinoma. Int. J. Clin. Exp. Pathol. 2015, 8, 5080–5088. [Google Scholar] [PubMed]
- Khadra, N.; Bresson-Bepoldin, L.; Penna, A.; Chaigne-Delalande, B.; Ségui, B.; Levade, T.; Vacher, A.M.; Reiffers, J.; Ducret, T.; Moreau, J.F.; et al. CD95 triggers Orai1-mediated localized Ca2+ entry, regulates recruitment of protein kinase C (PKC) β2, and prevents death-inducing signaling complex formation. Proc. Natl. Acad. Sci. USA 2011, 108, 19072–19077. [Google Scholar] [CrossRef] [PubMed]
- Sobradillo, D.; Hernández-Morales, M.; Ubierna, D.; Moyer, M.P.; Núñez, L.; Villalobos, C. A Reciprocal Shift in Transient Receptor Potential Channel 1 (TRPC1) and Stromal Interaction Molecule 2 (STIM2) Contributes to Ca2+ Remodeling and Cancer Hallmarks in Colorectal Carcinoma Cells. J. Biol. Chem. 2014, 289, 28765–28782. [Google Scholar] [CrossRef]
- Hasna, J.; Hague, F.; Rodat-Despoix, L.; Geerts, D.; Leroy, C.; Tulasne, D.; Ouadid-Ahidouch, H.; Kischel, P. Orai3 calcium channel and resistance to chemotherapy in breast cancer cells: The p53 connection. Cell Death Differ. 2018, 25, 691–705. [Google Scholar] [CrossRef]
- Zhu, H.; Zhang, H.; Jin, F.; Fang, M.; Huang, M.; Yang, C.S.; Chen, T.; Fu, L.; Pan, Z. Elevated Orai1 Expression Mediates Tumor-Promoting Intracellular Ca2+ Oscillations in Human Esophageal Squamous Cell Carcinoma. Oncotarget 2014, 5, 3455–3471. [Google Scholar] [CrossRef] [PubMed]
- Didiasova, M.; Zakrzewicz, D.; Magdolen, V.; Nagaraj, C.; Bálint, Z.; Rohde, M.; Preissner, K.T.; Wygrecka, M. STIM1/ORAI1-mediated Ca2+ Influx Regulates Enolase-1 Exteriorization. J. Biol. Chem. 2015, 290, 11983–11999. [Google Scholar] [CrossRef]
- Chen, Y.F.; Hsu, K.F.; Shen, M.R. The Store-Operated Ca2+ Entry-Mediated Signaling Is Important for Cancer Spread. Biochim. Biophys. Acta 2016, 1863, 1427–1435. [Google Scholar] [CrossRef] [PubMed]
- Mo, P.; Yang, S. The Store-Operated Calcium Channels in Cancer Metastasis: From Cell Migration, Invasion to Metastatic Colonization. Front. Biosci. 2018, 23, 1241–1256. [Google Scholar] [CrossRef]
- McAndrew, D.; Grice, D.M.; Peters, A.A.; Davis, F.M.; Stewart, T.; Rice, M.; Smart, C.E.; Brown, M.A.; Kenny, P.A.; Roberts-Thomson, S.J.; et al. ORAI1-mediated Calcium Influx in Lactation and in Breast Cancer. Mol. Cancer Ther. 2011, 10, 448–460. [Google Scholar] [CrossRef]
- Moccia, F. Endothelial Ca2+ Signaling and the Resistance to Anticancer Treatments: Partners in Crime. Int. J. Mol. Sci. 2018, 19, 217. [Google Scholar] [CrossRef]
- Li, J.; Cubbon, R.M.; Wilson, L.A.; Amer, M.S.; McKeown, L.; Hou, B.; Majeed, Y.; Tumova, S.; Seymour, V.A.L.; Taylor, H.; et al. Orai1 and CRAC channel dependence of VEGF-activated Ca2entry and endothelial tube formation. Circ. Res. 2011, 108, 1190–1198. [Google Scholar] [CrossRef]
- Li, J.; Bruns, A.F.; Hou, B.; Rode, B.; Webster, P.J.; Bailey, M.A.; Appleby, H.L.; Moss, N.K.; Ritchie, J.E.; Yuldasheva, N.Y.; et al. Orai3 Surface Accumulation and Calcium Entry Evoked by Vascular Endothelial Growth Factor. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 1987–1994. [Google Scholar] [CrossRef]
- Hoth, M.; Penner, R. Depletion of intracellular calcium stores activates a calcium current in mast cells. Nature 1992, 355, 353–356. [Google Scholar] [CrossRef] [PubMed]
- Zweifach, A.; Lewis, R.S. Mitogen-regulated Ca2+ current of T lymphocytes is activated by depletion of intracellular Ca2+ stores. Proc. Natl. Acad. Sci. USA 1993, 90, 6295–6299. [Google Scholar] [CrossRef] [PubMed]
- Weidinger, C.; Shaw, P.J.; Feske, S. STIM1 and STIM2-mediated Ca2+ influx regulates antitumour immunity by CD8+ T cells. EMBO Mol. Med. 2013, 5, 1311–1321. [Google Scholar] [CrossRef]
- Zhou, X.; Friedmann, K.S.; Lyrmann, H.; Zhou, Y.; Schoppmeyer, R.; Knörck, A.; Mang, S.; Hoxha, C.; Angenendt, A.; Backes, C.S.; et al. A calcium optimum for cytotoxic T lymphocyte and natural killer cell cytotoxicity. J. Physiol. 2018, 596, 2681–2698. [Google Scholar] [CrossRef]
- Endo, M. Calcium Release from the Sarcoplasmic Reticulum. Physiol. Rev. 1977, 57, 71–108. [Google Scholar] [CrossRef]
- Courjaret, R.; Dib, M.; Machaca, K. Spatially restricted subcellular Ca2+ signaling downstream of store-operated calcium entry encoded by a cortical tunneling mechanism. Sci. Rep. 2018, 8, 11214. [Google Scholar] [CrossRef]
- Thillaiappan, N.B.; Chavda, A.P.; Tovey, S.C.; Prole, D.L.; Taylor, C.W. Ca2+ signals initiate at immobile IP3 receptors adjacent to ER-plasma membrane junctions. Nat. Commun. 2017, 8, 1505. [Google Scholar] [CrossRef]
- Rizzuto, R.; Pinton, P.; Carrington, W.; Fay, F.S.; Fogarty, K.E.; Lifshitz, L.M.; Tuft, R.A.; Pozzan, T. Close Contacts with the Endoplasmic Reticulum as Determinants of Mitochondrial Ca2+ Responses. Science 1998, 280, 1763–1766. [Google Scholar] [CrossRef]
- Mandeville, J.T.H.; Ghosh, R.N.; Maxfield, F.R. Intracellular calcium levels correlate with speed and persistent forward motion in migrating neutrophils. Biophys. J. 1995, 68, 1207–1217. [Google Scholar] [CrossRef]
- Ritaine, A.; Shapovalov, G.; Prevarskaya, N. Metabolic Disorders and Cancer: Store-Operated Ca2+ Entry in Cancer: Focus on IP 3 R-Mediated Ca2+ Release from Intracellular Stores and Its Role in Migration and Invasion. Adv. Exp. Med. Biol. 2017, 993, 623–637. [Google Scholar] [CrossRef] [PubMed]
- Wei, C.; Wang, X.; Chen, M.; Ouyang, K.; Song, L.S.; Cheng, H. Calcium flickers steer cell migration. Nature 2009, 457, 901–905. [Google Scholar] [CrossRef]
- Brundage, R.A.; Fogarty, K.E.; Tuft, R.A.; Fay, F.A. Calcium gradients underlying polarization and chemotaxis of eosinophils. Science 1991, 254, 703–706. [Google Scholar] [CrossRef]
- Okeke, E.; Parker, T.; Dingsdale, H.; Concannon, M.; Awais, M.; Voronina, S.; Molgó, J.; Begg, M.; Metcalf, D.; Knight, A.E.; et al. Epithelial–mesenchymal transition, IP3 receptors and ER–PM junctions: Translocation of Ca2+ signalling complexes and regulation of migration. Biochem. J. 2016, 473, 757–767. [Google Scholar] [CrossRef] [PubMed]
- Baljinnyam, E.; De Lorenzo, M.S.; Xie, L.H.; Iwatsubo, M.; Chen, S.; Goydos, J.S.; Nowycky, M.C.; Iwatsubo, K. Exchange Protein Directly Activated by Cyclic AMP Increases Melanoma Cell Migration by a Ca2+-dependent Mechanism. Cancer Res. 2010, 70, 5607–5617. [Google Scholar] [CrossRef]
- Jin, X.; Shah, S.; Liu, Y.; Zhang, H.; Lees, M.; Fu, Z.; Lippiat, J.D.; Beech, D.J.; Sivaprasadarao, A.; Baldwin, S.A.; et al. Activation of the Cl- Channel ANO1 by Localized Calcium Signals in Nociceptive Sensory Neurons Requires Coupling With the IP3 Receptor. Sci. Signal. 2013, 6, ra73. [Google Scholar] [CrossRef]
- Ruiz, C.; Martins, J.R.; Rudin, F.; Schneider, S.; Dietsche, T.; Fischer, C.A.; Tornillo, L.; Terracciano, L.M.; Schreiber, R.; Bubendorf, L.; et al. Enhanced Expression of ANO1 in Head and Neck Squamous Cell Carcinoma Causes Cell Migration and Correlates with Poor Prognosis. PLoS ONE. 2012, 7, e43265. [Google Scholar] [CrossRef]
- Zeng, X.; Pan, D.; Wu, H.; Chen, H.; Yuan, W.; Zhou, J.; Shen, Z.; Chen, S. Transcriptional Activation of ANO1 Promotes Gastric Cancer Progression. Biochem. Biophys. Res. Commun. 2019, 512, 131–136. [Google Scholar] [CrossRef]
- Lee, Y.S.; Lee, J.K.; Bae, Y.; Lee, B.S.; Kim, E.; Cho, C.H.; Ryoo, K.; Yoo, J.; Kim, C.H.; Yi, G.S.; et al. Suppression of 14-3-3γ-mediated surface expression of ANO1 inhibits cancer progression of glioblastoma cells. Sci. Rep. 2016, 6, 26413. [Google Scholar] [CrossRef]
- Crottès, D.; Lin, Y.H.T.; Peters, C.J.; Gilchrist, J.M.; Wiita, A.P.; Jan, Y.N.; Jan, L.Y. TMEM16A Controls EGF-induced Calcium Signaling Implicated in Pancreatic Cancer Prognosis. Proc. Natl. Acad. Sci. USA 2019, 116, 13026–13035. [Google Scholar] [CrossRef]
- Britschgi, A.; Bill, A.; Brinkhaus, H.; Rothwell, C.; Clay, I.; Duss, S.; Rebhan, M.; Raman, P.; Guy, C.T.; Wetzel, K.; et al. Calcium-activated chloride channel ANO1 promotes breast cancer progression by activating EGFR and CAMK signaling. Proc. Natl. Acad. Sci. USA 2013, 110, E1026–E1034. [Google Scholar] [CrossRef] [PubMed]
- Sakakura, C.; Hagiwara, A.; Fukuda, K.; Shimomura, K.; Takagi, T.; Kin, S.; Nakase, Y.; Fujiyama, J.; Mikoshiba, K.; Okazaki, Y.; et al. Possible involvement of inositol 1,4,5-trisphosphate receptor type 3 (IP3R3) in the peritoneal dissemination of gastric cancers. Anticancer Res. 2003, 23, 3691–3697. [Google Scholar]
- Courjaret, R.; Machaca, K. Mid-range Ca2+ Signalling Mediated by Functional Coupling Between Store-Operated Ca2+ Entry and IP3-dependent Ca2+ Release. Nat. Commun. 2014, 5, 3916. [Google Scholar] [CrossRef]
- Sui, Y.; Sun, M.; Wu, F.; Yang, L.; Di, W.; Zhang, G.; Zhong, L.; Ma, Z.; Zheng, J.; Fang, X.; et al. Inhibition of TMEM16A Expression Suppresses Growth and Invasion in Human Colorectal Cancer Cells. PLoS ONE 2014, 9, e115443. [Google Scholar] [CrossRef]
- Prevarskaya Na Zhang, L.; Barritt, G. TRP channels in cancer. Biochim. Biophys. Acta 2007, 1772, 937–946. [Google Scholar] [CrossRef]
- Pla, A.F.; Gkika, D. Emerging role of TRP channels in cell migration: From tumor vascularization to metastasis. Front. Physiol. 2013, 4, 311. [Google Scholar] [CrossRef]
- Shapovalov, G.; Ritaine, A.; Skryma, R.; Prevarskaya, N. Role of TRP Ion Channels in Cancer and Tumorigenesis. Semin. Immunopathol. 2016, 38, 357–369. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Cai, Y.; He, D.; Zou, C.; Zhang, P.; Lo, C.Y.; Xu, Z.; Chan, F.L.; Yu, S.; Chen, Y.; et al. Transient receptor potential channel TRPC5 is essential for P-glycoprotein induction in drug-resistant cancer cells. Proc. Natl. Acad. Sci. USA 2012, 109, 16282–16287. [Google Scholar] [CrossRef] [PubMed]
- Yang, D. Emerging role of transient receptor potential (TRP) channels in cancer progression. BMB Rep. 2020, 53, 125–132. [Google Scholar] [CrossRef]
- Zhou, K.; Zhang, S.-S.; Yan, Y.; Zhao, S. Overexpression of transient receptor potential vanilloid 2 is associated with poor prognosis in patients with esophageal squamous cell carcinoma. Med. Oncol. 2014, 31, 17. [Google Scholar] [CrossRef]
- Semenova, S.B.; Vassilieva, I.; Fomina, A.F.; Runov, A.L.; Negulyaev, Y. Endogenous expression of TRPV5 and TRPV6 calcium channels in human leukemia K562 cells. Am. J. Physiol. Physiol. 2009, 296, C1098–C1104. [Google Scholar] [CrossRef]
- Bödding, M.; Wissenbach, U.; Flockerzi, V. The Recombinant Human TRPV6 Channel Functions as Ca2+Sensor in Human Embryonic Kidney and Rat Basophilic Leukemia Cells. J. Biol. Chem. 2002, 277, 36656–36664. [Google Scholar] [CrossRef] [PubMed]
- Weber, L.V.; Al-Refae, K.; Wölk, G.; Bonatz, G.; Altmüller, J.; Becker, C.; Gisselmann, G.; Hatt, H. Expression and functionality of TRPV1 in breast cancer cells. Breast Cancer 2016, 8, 243–252. [Google Scholar] [CrossRef] [PubMed]
- Elzamzamy, O.M.; Penner, R.; Hazlehurst, L.A. The Role of TRPC1 in Modulating Cancer Progression. Cells 2020, 9, 388. [Google Scholar] [CrossRef] [PubMed]
- Grant, C.V.; Carver, C.M.; Hastings, S.D.; Ramachandran, K.; Muniswamy, M.; Risinger, A.L.; Beutler, J.A.; Mooberry, S.L. Triple-negative breast cancer cell line sensitivity to englerin A identifies a new, targetable subtype. Breast Cancer Res. Treat. 2019, 177, 345–355. [Google Scholar] [CrossRef]
- Santoni, G.; Maggi, F.; Morelli, M.B.; Santoni, M.; Marinelli, O. Transient Receptor Potential Cation Channels in Cancer Therapy. Med. Sci. 2019, 7, 108. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Hu, G.; Gong, Y.; Yu, Q.; He, B.; Li, W.; He, Z.; Hao, W.; He, Z.; Liu, Y. Silencing of TRPM8 inhibits aggressive tumor phenotypes and enhances gemcitabine sensitivity in pancreatic cancer. Pancreatology 2018, 18, 935–944. [Google Scholar] [CrossRef]
- Nazıroğlu, M.; Çiğ, B.; Blum, W.; Vizler, C.; Buhala, A.; Marton, A.; Katona, R.; Jósvay, K.; Schwaller, B.; Oláh, Z.; et al. Targeting breast cancer cells by MRS1477, a positive allosteric modulator of TRPV1 channels. PLoS ONE 2017, 12, e0179950. [Google Scholar] [CrossRef]
- Amantini, C.; Mosca, M.; Nabissi, M.; Lucciarini, R.; Caprodossi, S.; Arcella, A.; Giangaspero, F.; Santoni, G. Capsaicin-induced apoptosis of glioma cells is mediated by TRPV1 vanilloid receptor and requires p38 MAPK activation. J. Neurochem. 2007, 102, 977–990. [Google Scholar] [CrossRef]
- Canales, J.; Morales, D.; Blanco, C.; Rivas, J.; Díaz, N.; Angelopoulos, I.; Cerda, O. A TR(i)P to Cell Migration: New Roles of TRP Channels in Mechanotransduction and Cancer. Front. Physiol. 2019, 10, 757. [Google Scholar] [CrossRef]
- Chen, J.-P.; Wang, J.; Luan, Y.; Wang, C.-X.; Li, W.-H.; Zhang, J.-B.; Sha, D.; Shen, R.; Cui, Y.-G.; Zhang, Z.; et al. TRPM7 promotes the metastatic process in human nasopharyngeal carcinoma. Cancer Lett. 2015, 356, 483–490. [Google Scholar] [CrossRef] [PubMed]
- Rybarczyk, P.; Gautier, M.; Hague, F.; Dhennin-Duthille, I.; Chatelain, D.; Kerr-Conte, J.; Pattou, F.; Regimbeau, J.-M.; Sevestre, H.; Ouadid-Ahidouch, H. Transient receptor potential melastatin-related 7 channel is overexpressed in human pancreatic ductal adenocarcinomas and regulates human pancreatic cancer cell migration. Int. J. Cancer 2012, 131, E851–E861. [Google Scholar] [CrossRef]
- Middelbeek, J.; Kuipers, A.J.; Henneman, L.; Visser, D.; Eidhof, I.; Van Horssen, R.; Wieringa, B.; Canisius, S.V.; Zwart, W.; Wessels, L.F.; et al. TRPM7 Is Required for Breast Tumor Cell Metastasis. Cancer Res. 2012, 72, 4250–4261. [Google Scholar] [CrossRef]
- Lee, W.H.; Choong, L.Y.; Jin, T.H.; Mon, N.N.; Chong, S.; Liew, C.S.; Putti, T.; Lu, S.Y.; Harteneck, C.; Lim, Y.P. TRPV4 plays a role in breast cancer cell migration via Ca2+-dependent activation of AKT and downregulation of E-cadherin cell cortex protein. Oncog. 2017, 6, e338. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.H.; Choong, L.Y.; Mon, N.N.; Lu, S.; Lin, Q.; Pang, B.; Yan, B.; Krishna, V.S.R.; Singh, H.; Tan, T.Z.; et al. TRPV4 Regulates Breast Cancer Cell Extravasation, Stiffness and Actin Cortex. Sci. Rep. 2016, 6, 27903. [Google Scholar] [CrossRef] [PubMed]
- Lodish, H.; Kong, N.; Wikström, L. Calcium is required for folding of newly made subunits of the asialoglycoprotein receptor within the endoplasmic reticulum. J. Biol. Chem. 1992, 267, 12753–12760. [Google Scholar] [CrossRef]
- Dang, D.; Rao, R. Calcium-ATPases: Gene disorders and dysregulation in cancer. Biochim. Biophys. Acta (BBA)—Bioenerg. 2016, 1863, 1344–1350. [Google Scholar] [CrossRef] [PubMed]
- Chung, F.-Y.; Lin, S.-R.; Lu, C.-Y.; Yeh, C.-S.; Chen, F.-M.; Hsieh, J.-S.; Huang, T.-J.; Wang, J.-Y. Sarco/Endoplasmic Reticulum Calcium-ATPase 2 Expression as a Tumor Marker in Colorectal Cancer. Am. J. Surg. Pathol. 2006, 30, 969–974. [Google Scholar] [CrossRef]
- Gou, W.-F.; Niu, Z.-F.; Zhao, S.; Takano, Y.; Zheng, H.-C. Aberrant SERCA3 expression during the colorectal adenoma-adenocarcinoma sequence. Oncol. Rep. 2014, 31, 232–240. [Google Scholar] [CrossRef]
- Bergner, A.; Kellner, J.; Tufman, A.; Huber, R.M. Endoplasmic reticulum Ca2+-homeostasis is altered in small and non-small cell lung cancer cell lines. J. Exp. Clin. Cancer Res. 2009, 28, 25. [Google Scholar] [CrossRef]
- Arbabian, A.; Brouland, J.P.; Apáti, Á.; Pászty, K.; Hegedűs, L.; Enyedi, Á.; Chomienne, C.; Papp, B. Modulation of endoplasmic reticulum calcium pump expression during lung cancer cell differentiation. FEBS J. 2013, 280, 5408–5418. [Google Scholar] [CrossRef]
- O’Neill, J.P.; Velalar, C.N.; Lee, D.I.; Zhang, B.; Nakanishi, T.; Tang, Y.; Selaru, F.; Ross, D.; Meltzer, S.J.; Hussain, A. Thapsigargin resistance in human prostate cancer cells. Cancer 2006, 107, 649–659. [Google Scholar] [CrossRef]
- Brouland, J.P.; Gélébart, P.; Kovàcs, T.; Enouf, J.; Grossmann, J.; Papp, B. The Loss of Sarco/Endoplasmic Reticulum Calcium Transport ATPase 3 Expression Is an Early Event during the Multistep Process of Colon Carcinogenesis. Am. J. Pathol. 2005, 167, 233–242. [Google Scholar] [CrossRef]
- Launay, S.; Giannì, M.; Kovàcs, T.; Bredoux, R.; Bruel, A.; Gélébart, P.; Zassadowski, F.; Chomienne, C.; Enouf, J.; Papp, B. Lineage-specific Modulation of Calcium Pump Expression During Myeloid Differentiation. Blood 1999, 93, 4395–4405. [Google Scholar] [CrossRef]
- Gélébart, P.; Kovács, T.; Brouland, J.P.; van Gorp, R.; Grossmann, J.; Rivard, N.; Panis, Y.; Martin, V.; Bredoux, R.; Enouf, J.; et al. Expression of Endomembrane Calcium Pumps in Colon and Gastric Cancer Cells. J. Biol. Chem. 2002, 277, 26310–26320. [Google Scholar] [CrossRef]
- Roti, G.; Carlton, A.; Ross, K.N.; Markstein, M.; Pajcini, K.; Su, A.H.; Perrimon, N.; Pear, W.S.; Kung, A.; Blacklow, S.C.; et al. Complementary Genomic Screens Identify SERCA as a Therapeutic Target in NOTCH1 Mutated Cancer. Cancer Cell 2013, 23, 390–405. [Google Scholar] [CrossRef]
- Park, K.C.; Kim, S.W.; Jeon, J.Y.; Jo, A.R.; Choi, H.J.; Kim, J.; Lee, H.G.; Kim, Y.; Mills, G.; Noh, S.H.; et al. Survival of Cancer Stem-Like Cells Under Metabolic Stress via CaMK2α-mediated Upregulation of Sarco/Endoplasmic Reticulum Calcium ATPase Expression. Clin. Cancer Res. 2017, 24, 1677–1690. [Google Scholar] [CrossRef]
- Mahalingam, D.; Peguero, J.; Cen, P.; Arora, S.P.; Sarantopoulos, J.; Rowe, J.; Allgood, V.; Tubb, B.; Campos, L. A Phase II, Multicenter, Single-Arm Study of Mipsagargin (G-202) as a Second-Line Therapy Following Sorafenib for Adult Patients with Progressive Advanced Hepatocellular Carcinoma. Cancers 2019, 11, 833. [Google Scholar] [CrossRef]
- Berchtold, M.W.; Villalobo, A. The many faces of calmodulin in cell proliferation, programmed cell death, autophagy, and cancer. Biochim. Biophys. Acta (BBA)—Bioenerg. 2014, 1843, 398–435. [Google Scholar] [CrossRef]
- Colomer, J.; Agell, N.; Engel, P.; Bachs, O. Expression of calmodulin and calmodulin binding proteins in lymphoblastoid cells. J. Cell. Physiol. 1994, 159, 542–550. [Google Scholar] [CrossRef]
- Ye, Q.; Wei, Y.; Fischer, R.; Borner, C.; Berchtold, M.W. Expression of calmodulin and calmodulin binding proteins in rat fibroblasts stably transfected with protein kinase C and oncogenes. Biochim. Biophys. Acta (BBA)—Bioenerg. 1997, 1359, 89–96. [Google Scholar] [CrossRef]
- Krishnaraju, K.; Murugesan, K.; Vij, U.; Kapur, B.; Farooq, A. Calmodulin levels in oestrogen receptor positive and negative human breast tumours. Br. J. Cancer 1991, 63, 346–347. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Wei, J.W.; Morris, H.P.; A Hickie, R. Positive correlation between calmodulin content and hepatoma growth rates. Cancer Res. 1982, 42, 2571–2574. [Google Scholar] [PubMed]
- Veigl, M.L.; Vanaman, T.C.; E Branch, M.; Sedwick, W.D. Differences in calmodulin levels of normal and transformed cells as determined by culture conditions. Cancer Res. 1984, 44, 3184–3189. [Google Scholar] [PubMed]
- Wang, R.; Zhang, H.; Li, S.; Xue, S. Intracellular levels of calmodulin are increased in transformed cells. Cell Res. 1992, 2, 119–127. [Google Scholar] [CrossRef]
- Yokokura, S.; Yurimoto, S.; Matsuoka, A.; Imataki, O.; Dobashi, H.; Bandoh, S.; Matsunaga, T. Calmodulin antagonists induce cell cycle arrest and apoptosis in vitro and inhibit tumor growth in vivo in human multiple myeloma. BMC Cancer 2014, 14, 1–11. [Google Scholar] [CrossRef]
- Skelding, K.A.; Rostas, J.A.P.; Verrills, N.M. Controlling the cell cycle: The role of calcium/calmodulin-stimulated protein kinases I and II. Cell Cycle 2011, 10, 631–639. [Google Scholar] [CrossRef]
- Kahl, C.R.; Means, A.R. Regulation of Cyclin D1/Cdk4 Complexes by Calcium/Calmodulin-dependent Protein Kinase I. J. Biol. Chem. 2004, 279, 15411–15419. [Google Scholar] [CrossRef]
- Patel, R.; Holt, M.; Philipova, R.; Moss, S.; Schulman, H.; Hidaka, H.; Whitaker, M. Calcium/Calmodulin-dependent Phosphorylation and Activation of Human Cdc25-C at the G2/M Phase Transition in HeLa Cells. J. Biol. Chem. 1999, 274, 7958–7968. [Google Scholar] [CrossRef]
- Wang, Y.; Zhao, R.; Zhe, H. The emerging role of CaMKII in cancer. Oncotarget 2015, 6, 11725–11734. [Google Scholar] [CrossRef]
- Brzozowski, J.S.; Skelding, K.A. The Multi-Functional Calcium/Calmodulin Stimulated Protein Kinase (CaMK) Family: Emerging Targets for Anti-Cancer Therapeutic Intervention. Pharmaceuticals 2019, 12, 8. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Joyal, J.L.; Sacks, D. Calmodulin Enhances the Stability of the Estrogen Receptor. J. Biol. Chem. 2001, 276, 17354–17360. [Google Scholar] [CrossRef]
- Li, Z.; Zhang, Y.; Hedman, A.C.; Ames, J.B.; Sacks, D.B. Calmodulin Lobes Facilitate Dimerization and Activation of Estrogen Receptor-α. J. Biol. Chem. 2017, 292, 4614–4622. [Google Scholar] [CrossRef] [PubMed]
- Cifuentes, E.; Mataraza, J.M.; Yoshida, B.A.; Menon, M.; Sacks, D.; Barrack, E.R.; Reddy, G.P.-V. Physical and functional interaction of androgen receptor with calmodulin in prostate cancer cells. Proc. Natl. Acad. Sci. USA 2004, 101, 464–469. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wu, G.; Yang, Y.; Fu, S.; Liu, X.; Kang, H.; Yang, X.; Su, X.-C.; Shen, Y. Calmodulin dissociates the STIM1-Orai1 complex and STIM1 oligomers. Nat. Commun. 2017, 8, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Rossi, A.M.; Rahman, T.; Taylor, C. Activation of IP3 receptors requires an endogenous 1-8-14 calmodulin-binding motif. Biochem. J. 2012, 449, 39–49. [Google Scholar] [CrossRef][Green Version]
- Li, S.; Xue, J.; Sun, Z.; Liu, T.; Zhang, L.; Wang, L.; You, H.; Fan, Z.; Zheng, Y.; Luo, D. CaMKII Potentiates Store-Operated Ca2+ Entry Through Enhancing STIM1 Aggregation and Interaction with Orai1. Cell. Physiol. Biochem. 2018, 46, 1042–1054. [Google Scholar] [CrossRef] [PubMed]
- Nussinov, R.; Wang, G.; Tsai, C.-J.; Jang, H.; Lu, S.; Banerjee, A.; Zhang, J.; Gaponenko, V. Calmodulin and PI3K Signaling in KRAS Cancers. Trends Cancer 2017, 3, 214–224. [Google Scholar] [CrossRef] [PubMed]
- Umemura, M.; Baljinnyam, E.; Feske, S.; De Lorenzo, M.S.; Xie, L.-H.; Feng, X.; Oda, K.; Makino, A.; Fujita, T.; Yokoyama, U.; et al. Store-Operated Ca2+ Entry (SOCE) Regulates Melanoma Proliferation and Cell Migration. PLoS ONE 2014, 9, e89292. [Google Scholar] [CrossRef] [PubMed]
- Villalobo, A.; Berchtold, M.W. The Role of Calmodulin in Tumor Cell Migration, Invasiveness, and Metastasis. Int. J. Mol. Sci. 2020, 21, 765. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Yi, L.; Hai, L.; Ma, H.; Tao, Z.; Zhang, C.; Abeysekera, I.; Zhao, K.; Yang, Y.; Wang, W.; et al. The interactome and spatial redistribution feature of Ca2+ receptor protein calmodulin reveals a novel role in invadopodia-mediated invasion. Cell Death Dis. 2018, 9, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.S.; Kim, D.-A.; Chung, H.; Park, I.H.; Kim, B.H.; Oh, E.-S.; Kang, D.-H. Screening of breast cancer stem cell inhibitors using a protein kinase inhibitor library. Cancer Cell Int. 2017, 17, 1–12. [Google Scholar] [CrossRef]
- Kaur, A.; Raghavan, M. A Calreticulin Tail: C-terminal Mutants of Calreticulin Allow Cancer Cells to Evade Phagocytosis. Mol. Cell 2020, 77, 683–685. [Google Scholar] [CrossRef] [PubMed]
- Venkateswaran, K.; Verma, A.; Bhatt, A.N.; Shrivastava, A.; Manda, K.; Raj, H.G.; Prasad, A.; Len, C.; Parmar, V.S.; Dwarakanath, B.S. Emerging Roles of Calreticulin in Cancer: Implications for Therapy. Curr. Protein Pept. Sci. 2018, 19, 344–357. [Google Scholar] [CrossRef] [PubMed]
- Han, A.; Li, C.; Zahed, T.; Wong, M.; Smith, I.; Hoedel, K.; Green, D.; Boiko, A.D. Calreticulin is a Critical Cell Survival Factor in Malignant Neoplasms. PLoS Biol. 2019, 17, e3000402. [Google Scholar] [CrossRef]
- Jin, C.; Lin, T.; Shan, L. Downregulation of Calbindin 1 by miR-454-3p Suppresses Cell Proliferation in Nonsmall Cell Lung Cancer In Vitro. Cancer Biother. Radiopharm. 2019, 34, 119–127. [Google Scholar] [CrossRef]
- Pfyffer, G.E.; Humbel, B.; Sträuli, P.; Mohrmann, I.; Murer, H.; Heizmann, C.W. Calcium-binding proteins in carcinoma, neuroblastoma and glioma cell lines. Virchows Archiv 1987, 412, 135–144. [Google Scholar] [CrossRef]
- Lu, Y.-C.; Weng, W.-C.; Lee, H. Functional Roles of Calreticulin in Cancer Biology. BioMed Res. Int. 2015, 2015, 1–9. [Google Scholar] [CrossRef]
- Sun, J.; Mu, H.; Dai, K.; Yi, L. Calreticulin: A potential anti-cancer therapeutic target. Die Pharm. 2017, 72, 503–510. [Google Scholar]
- Villagomez, M.; Szabo, E.; Podcheko, A.; Feng, T.; Papp, S.; Opas, M. Calreticulin and focal-contact-dependent adhesion. Biochem. Cell Biol. 2009, 87, 545–556. [Google Scholar] [CrossRef]
- Huang, G.; Sun, Z.; Wu, J.; Shui, S.; Han, X.; Guo, D.; Li, T. Calreticulin Promotes Proliferation and Migration but Inhibits Apoptosis in Schwann Cells. Med. Sci. Monit. 2016, 22, 4516–4522. [Google Scholar] [CrossRef]
- Clark, R.A.; Li, S.-L.; Pearson, D.W.; Leidal, K.G.; Clark, J.R.; Denning, G.M.; Reddick, R.; Krause, K.-H.; Valente, A.J. Regulation of Calreticulin Expression during Induction of Differentiation in Human Myeloid Cells. J. Biol. Chem. 2002, 277, 32369–32378. [Google Scholar] [CrossRef]
- Martins, I.; Kepp, O.; Galluzzi, L.; Senovilla, L.; Schlemmer, F.; Adjemian, S.; Menger, L.; Michaud, M.; Zitvogel, L.; Kroemer, G. Surface-exposed calreticulin in the interaction between dying cells and phagocytes. Ann. N. Y. Acad. Sci. 2010, 1209, 77–82. [Google Scholar] [CrossRef]
- Raghavan, M.; Wijeyesakere, S.J.; Peters, L.R.; Del Cid, N. Calreticulin in the immune system: Ins and outs. Trends Immunol. 2013, 34, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Zamanian, M.; Hamadneh, L.A.Q.; Veerakumarasivam, A.; Rahman, S.A.; Shohaimi, S.; Rosli, R. Calreticulin mediates an invasive breast cancer phenotype through the transcriptional dysregulation of p53 and MAPK pathways. Cancer Cell Int. 2016, 16, 56. [Google Scholar] [CrossRef] [PubMed]
- Banerjea, A.; Ahmed, S.; E Hands, R.; Huang, F.; Han, X.; Shaw, P.M.; Feakins, R.; A Bustin, S.; Dorudi, S. Colorectal cancers with microsatellite instability display mRNA expression signatures characteristic of increased immunogenicity. Mol. Cancer 2004, 3, 21. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Harada, K.; Takenawa, T.; Ferdous, T.; Kuramitsu, Y.; Ueyama, Y. Calreticulin is a novel independent prognostic factor for oral squamous cell carcinoma. Oncol. Lett. 2017, 13, 4857–4862. [Google Scholar] [CrossRef]
- Chiang, W.-F.; Hwang, T.-Z.; Hour, T.-C.; Wang, L.-H.; Chiu, C.-C.; Chen, H.-R.; Wu, Y.-J.; Wang, C.-C.; Wang, L.-F.; Chien, C.-Y.; et al. Calreticulin, an endoplasmic reticulum-resident protein, is highly expressed and essential for cell proliferation and migration in oral squamous cell carcinoma. Oral Oncol. 2013, 49, 534–541. [Google Scholar] [CrossRef] [PubMed]
- Hacken, E.; Gounari, M.; Back, J.W.; Shimanovskaya, E.; Scarfò, L.; Kim, E.; Burks, J.; Ponzoni, M.; Ramirez, G.A.; Wierda, W.G.; et al. Calreticulin as a novel B-cell receptor antigen in chronic lymphocytic leukemia. Haematologica 2017, 102, e394–e396. [Google Scholar] [CrossRef]
- Lwin, Z.-M.; Guo, C.; Salim, A.; Yip, G.W.-C.; Chew, F.-T.; Nan, J.; Thike, A.A.; Tan, P.-H.; Bay, B.-H. Clinicopathological significance of calreticulin in breast invasive ductal carcinoma. Mod. Pathol. 2010, 23, 1559–1566. [Google Scholar] [CrossRef]
- Liu, R.; Gong, J.; Chen, J.; Li, Q.; Song, C.; Zhang, J.; Li, Y.; Liu, Z.; Dong, Y.; Chen, L.; et al. Calreticulin as a potential diagnostic biomarker for lung cancer. Cancer Immunol. Immunother. 2011, 61, 855–864. [Google Scholar] [CrossRef]
- Alur, M.; Nguyen, M.M.; Eggener, S.E.; Jiang, F.; Dadras, S.S.; Stern, J.; Kimm, S.; Roehl, K.; Kozlowski, J.; Pins, M.; et al. Suppressive Roles of Calreticulin in Prostate Cancer Growth and Metastasis. Am. J. Pathol. 2009, 175, 882–890. [Google Scholar] [CrossRef] [PubMed]
- Hsu, W.-M.; Hsieh, F.J.; Jeng, Y.-M.; Kuo, M.L.; Chen, C.-N.; Lai, D.M.; Wang, B.T.; Tsao, P.-N.; Lee, H.; Lin, M.T.; et al. Calreticulin expression in neuroblastoma—A novel independent prognostic factor. Ann. Oncol. 2005, 16, 314–321. [Google Scholar] [CrossRef] [PubMed]
- Sheng, W.; Chen, C.; Dong, M.; Zhou, J.; Liu, Q.; Dong, Q.; Li, F. Overexpression of Calreticulin Contributes to the Development and Progression of Pancreatic Cancer. J. Cell. Physiol. 2014, 229, 887–897. [Google Scholar] [CrossRef]
- Lee, P.-C.; Chiang, J.-C.; Chen, C.-Y.; Chien, Y.-C.; Chen, W.-M.; Huang, C.-W.; Weng, W.-C.; Chen, C.-I.; Lee, P.-H.; Chen, C.-N.; et al. Calreticulin regulates vascular endothelial growth factor-A mRNA stability in gastric cancer cells. PLoS ONE 2019, 14, e0225107. [Google Scholar] [CrossRef] [PubMed]
- Fadel, M.P.; Szewczenko-Pawlikowski, M.; Leclerc, P.; Dziak, E.; Symonds, J.M.; Blaschuk, O.; Michalak, M.; Opas, M. Calreticulin Affects β-Catenin-associated Pathways. J. Biol. Chem. 2001, 276, 27083–27089. [Google Scholar] [CrossRef]
- Coppolino, M.G.; Woodside, M.J.; Demaurex, N.; Grinstein, S.; St-Arnaud, R.; Dedhar, S. Calreticulin is essential for integrin-mediated calcium signalling and cell adhesion. Nat. Cell Biol. 1997, 386, 843–847. [Google Scholar] [CrossRef]
- Goicoechea, S.; Pallero, M.A.; Eggleton, P.; Michalak, M.; Murphy-Ullrich, J.E. The Anti-adhesive Activity of Thrombospondin Is Mediated by the N-terminal Domain of Cell Surface Calreticulin. J. Biol. Chem. 2002, 277, 37219–37228. [Google Scholar] [CrossRef]
- Hayashida, Y.; Urata, Y.; Muroi, E.; Kono, T.; Miyata, Y.; Nomata, K.; Kanetake, H.; Kondo, T.; Ihara, Y. Calreticulin Represses E-cadherin Gene Expression in Madin-Darby Canine Kidney Cells via Slug. J. Biol. Chem. 2006, 281, 32469–32484. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Su, T.; Lu, Y.; Lee, H. Calreticulin regulates cell proliferation and migration in gastric cancer cell line AGS. FASEB J. 2007, 21, A1318. [Google Scholar] [CrossRef]
- Yi, L.; Shan, J.; Chen, X.; Li, G.; Li, L.; Tan, H.; Su, Q. Involvement of calreticulin in cell proliferation, invasion and differentiation in diallyl disulfide-treated HL-60 cells. Oncol. Lett. 2016, 12, 1861–1867. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.-C.; Chen, C.-N.; Wang, B.; Hsu, W.-M.; Chen, S.-T.; Chang, K.-J.; Chang, C.-C.; Lee, H. Changes in Tumor Growth and Metastatic Capacities of J82 Human Bladder Cancer Cells Suppressed by Down-Regulation of Calreticulin Expression. Am. J. Pathol. 2011, 179, 1425–1433. [Google Scholar] [CrossRef]
- de Bruyn, M.; Wiersma, V.R.; Helfrich, W.; Eggleton, P.; Bremer, E. The Ever-Expanding Immunomodulatory Role of Calreticulin in Cancer Immunity. Front. Oncol. 2015, 5, 35. [Google Scholar] [CrossRef] [PubMed]
- Okunaga, T.; Urata, Y.; Goto, S.; Matsuo, T.; Mizota, S.; Tsutsumi, K.; Nagata, I.; Kondo, T.; Ihara, Y.; Kendall, H.E.; et al. Calreticulin, a Molecular Chaperone in the Endoplasmic Reticulum, Modulates Radiosensitivity of Human Glioblastoma U251MG Cells. Cancer Res. 2006, 66, 8662–8671. [Google Scholar] [CrossRef] [PubMed]
- Matsukuma, S.; Yoshimura, K.; Ueno, T.; Oga, A.; Inoue, M.; Watanabe, Y.; Kuramasu, A.; Fuse, M.; Tsunedomi, R.; Nagaoka, S.; et al. Calreticulin is highly expressed in pancreatic cancer stem-like cells. Cancer Sci. 2016, 107, 1599–1609. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Fan, G.; Wang, D. Downregulation of calbindin 1, a calcium-binding protein, reduces the proliferation of osteosarcoma cells. Oncol. Lett. 2017, 13, 3727–3733. [Google Scholar] [CrossRef]
- Favier, J.; Brière, J.-J.; Burnichon, N.; Rivière, J.; Vescovo, L.; Benit, P.; Giscos-Douriez, I.; De Reyniès, A.; Bertherat, J.; Badoual, C.; et al. The Warburg Effect Is Genetically Determined in Inherited Pheochromocytomas. PLoS ONE 2009, 4, e7094. [Google Scholar] [CrossRef]
- Seyfried, T.N.; Shelton, L.M. Cancer as a metabolic disease. Nutr. Metab. 2010, 7, 1–9. [Google Scholar] [CrossRef]
- Hirschey, M.D.; DeBerardinis, R.J.; Diehl, A.M.E.; Drew, J.E.; Frezza, C.; Green, M.; Jones, L.W.; Ko, Y.H.; Le, A.; Lea, M.A.; et al. Dysregulated metabolism contributes to oncogenesis. Semin. Cancer Biol. 2015, 35, S129–S150. [Google Scholar] [CrossRef]
- Amuthan, G.; Biswas, G.; Ananadatheerthavarada, H.K.; Vijayasarathy, C.; Shephard, H.M.; Avadhani, N.G. Mitochondrial stress-induced calcium signaling, phenotypic changes and invasive behavior in human lung carcinoma A549 cells. Oncogene 2002, 21, 7839–7849. [Google Scholar] [CrossRef]
- Rizzuto, R.; Brini, M.; Murgia, M.; Pozzan, T. Microdomains with high Ca2+ close to IP3-sensitive channels that are sensed by neighboring mitochondria. Science 1993, 262, 744–747. [Google Scholar] [CrossRef]
- Kania, E.; Roest, G.; Vervliet, T.; Parys, J.B.; Bultynck, G. IP3 Receptor-Mediated Calcium Signaling and Its Role in Autophagy in Cancer. Front. Oncol. 2017, 7, 140. [Google Scholar] [CrossRef]
- Parys, J.B.; Decuypere, J.-P.; Bultynck, G. Role of the inositol 1,4,5-trisphosphate receptor/Ca2+-release channel in autophagy. Cell Commun. Signal. 2012, 10, 17. [Google Scholar] [CrossRef]
- Vicencio, J.M.; Ortiz, C.; Criollo, A.; E Jones, A.W.; Kepp, O.; Galluzzi, L.; Joza, N.; Vitale, I.; Morselli, E.; Tailler, M.; et al. The inositol 1,4,5-trisphosphate receptor regulates autophagy through its interaction with Beclin 1. Cell Death Differ. 2009, 16, 1006–1017. [Google Scholar] [CrossRef]
- Choi, K.S. Autophagy and cancer. Exp. Mol. Med. 2012, 44, 109–120. [Google Scholar] [CrossRef] [PubMed]
- Degenhardt, K.; Mathew, R.; Beaudoin, B.; Bray, K.; Anderson, D.; Chen, G.; Mukherjee, C.; Shi, Y.; Gélinas, C.; Fan, Y.; et al. Autophagy promotes tumor cell survival and restricts necrosis, inflammation, and tumorigenesis. Cancer Cell 2006, 10, 51–64. [Google Scholar] [CrossRef] [PubMed]
- Zheng, K.; He, Z.; Kitazato, K.; Wang, Y. Selective Autophagy Regulates Cell Cycle in Cancer Therapy. Theranostics 2019, 9, 104–125. [Google Scholar] [CrossRef]
- Jones, R.G.; Plas, D.R.; Kubek, S.; Buzzai, M.; Mu, J.; Xu, Y.; Birnbaum, M.; Thompson, C.B. AMP-Activated Protein Kinase Induces a p53-Dependent Metabolic Checkpoint. Mol. Cell 2005, 18, 283–293. [Google Scholar] [CrossRef] [PubMed]
- Mathiassen, S.G.; De Zio, D.; Cecconi, F. Autophagy and the Cell Cycle: A Complex Landscape. Front. Oncol. 2017, 7, 51. [Google Scholar] [CrossRef]
- Sorokina, I.V.; Denisenko, T.V.; Imreh, G.; Tyurin-Kuzmin, P.A.; Kaminskyy, V.; Gogvadze, V.; Zhivotovsky, B. Involvement of autophagy in the outcome of mitotic catastrophe. Sci. Rep. 2017, 7, 1–12. [Google Scholar] [CrossRef]
- Blandino, G.; Di Agostino, S. New therapeutic strategies to treat human cancers expressing mutant p53 proteins. J. Exp. Clin. Cancer Res. 2018, 37, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.-Y.; White, E. Role of Autophagy in Cancer Prevention. Cancer Prev. Res. 2011, 4, 973–983. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zhou, Y.; Li, Y.; Yang, L.; Ma, Y.; Peng, X.; Yang, S.; Liu, J.; Li, H. Autophagy: A novel mechanism of chemoresistance in cancers. Biomed. Pharmacother. 2019, 119, 109415. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.; MacLeod, K.F. Autophagy, cancer stem cells and drug resistance. J. Pathol. 2019, 247, 708–718. [Google Scholar] [CrossRef]
- Sui, X.; Chen, R.; Wang, Z.; Huang, Z.; Kong, N.; Zhang, M.; Han, W.; Lou, F.; Yang, J.; Zhang, Q.; et al. Autophagy and chemotherapy resistance: A promising therapeutic target for cancer treatment. Cell Death Dis. 2013, 4, e838. [Google Scholar] [CrossRef]
- Liu, E.Y.; Ryan, K.M. Autophagy and cancer—Issues we need to digest. J. Cell Sci. 2012, 125, 2349–2358. [Google Scholar] [CrossRef]
- Patergnani, S.; Missiroli, S.; Marchi, S.; Giorgi, C. Mitochondria-associated endoplasmic reticulum membranes microenvironment: Targeting autophagic and apoptotic pathways in cancer therapy. Front. Oncol. 2015, 5, 173. [Google Scholar] [CrossRef]
- Pihán, P.; Carreras-Sureda, A.; Hetz, C. BCL-2 family: Integrating stress responses at the ER to control cell demise. Cell Death Differ. 2017, 24, 1478–1487. [Google Scholar] [CrossRef]
- Lallemand, V.; Zhu, J.; Puvion, F.; Koken, M.; Honoré, N.; Doubeikovsky, A.; Duprez, E.; Pandolfi, P.P.; Puvion, E.; Freemont, P.; et al. Role of Promyelocytic Leukemia (Pml) Sumolation in Nuclear Body Formation, 11s Proteasome Recruitment, and as2O3-Induced Pml or Pml/Retinoic Acid Receptor α Degradation. J. Exp. Med. 2001, 193, 1361–1372. [Google Scholar] [CrossRef] [PubMed]
- Missiroli, S.; Bonora, M.; Patergnani, S.; Poletti, F.; Perrone, M.; Gafà, R.; Magri, E.; Raimondi, A.; Lanza, G.; Tacchetti, C.; et al. PML at Mitochondria-Associated Membranes Is Critical for the Repression of Autophagy and Cancer Development. Cell Rep. 2016, 16, 2415–2427. [Google Scholar] [CrossRef]
- Endoplasmic Reticulum-Mitochondria Ca 2+ Crosstalk in the Control of the Tumor Cell Fate. Biochim. Biophys. Acta Mol. Cell Res. 2017, 1864, 858–864. [CrossRef]
- Bultynck, G.; Campanella, M. Tumor suppressive Ca2+ signaling is driven by IP3 receptor fitness. Cell Stress 2017, 1, 73–78. [Google Scholar] [CrossRef]
- Hedgepeth, S.C.; Garcia, M.I.; Wagner, L.E., II; Rodriguez, A.M.; Chintapalli, S.V.; Snyder, R.R.; Hankins, G.D.V.; Henderson, B.R.; Brodie, K.M.; Yule, D.I.; et al. The BRCA1 Tumor Suppressor Binds to Inositol 1,4,5-Trisphosphate Receptors to Stimulate Apoptotic Calcium Release. J. Biol. Chem. 2015, 290, 7304–7313. [Google Scholar] [CrossRef] [PubMed]
- Nussinov, R.; Muratcioglu, S.; Tsai, C.-J.; Jang, H.; Gursoy, A.; Keskin, O. K-Ras4B/calmodulin/PI3Kα: A promising new adenocarcinoma-specific drug target? Expert Opin. Ther. Targets 2016, 20, 831–842. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; An, P.; Quan, X.-J.; Zhang, J.; Zhou, Z.-Y.; Zou, L.-P.; Luo, H.-S. Ca2+/calmodulin-dependent protein kinase II regulates colon cancer proliferation and migration via ERK1/2 and p38 pathways. World J. Gastroenterol. 2017, 23, 6111–6118. [Google Scholar] [CrossRef] [PubMed]
- Szabadkai, G.; Bianchi, K.; Várnai, P.; De Stefani, D.; Wieckowski, M.; Cavagna, D.; Nagy, A.I.; Balla, T.; Rizzuto, R. Chaperone-mediated coupling of endoplasmic reticulum and mitochondrial Ca2+ channels. J. Cell Biol. 2006, 175, 901–911. [Google Scholar] [CrossRef]
- Rizzuto, R.; De Stefani, D.; Raffaello, A. Cristina Mammucari Mitochondria as sensors and regulators of calcium signaling. Nat. Rev. Mol. Cell Biol. 2012, 13, 566–578. [Google Scholar] [CrossRef] [PubMed]
- Shoshan-Barmatz, V.; Mizrachi, D.; Mizrachi, D. VDAC1: From structure to cancer therapy. Front. Oncol. 2012, 2, 164. [Google Scholar] [CrossRef]
- Shoshan-Barmatz, V.; Krelin, Y.; Shteinfer-Kuzmine, A.; Arif, T. Voltage-Dependent Anion Channel 1 As an Emerging Drug Target for Novel Anti-Cancer Therapeutics. Front. Oncol. 2017, 7, 154. [Google Scholar] [CrossRef]
- Mathupala, S.P.; Ko, Y.H.; Pedersen, P.L. Hexokinase II: Cancer’s double-edged sword acting as both facilitator and gatekeeper of malignancy when bound to mitochondria. Oncogene 2006, 25, 4777–4786. [Google Scholar] [CrossRef]
- Abu-Hamad, S.; Arbel, N.; Calo, D.; Arzoine, L.; Israelson, A.; Keinan, N.; Ben-Romano, R.; Friedman, O.; Shoshan-Barmatz, V. The VDAC1 N-terminus is essential both for apoptosis and the protective effect of anti-apoptotic proteins. J. Cell Sci. 2009, 122, 1906–1916. [Google Scholar] [CrossRef] [PubMed]
- Mallilankaraman, K.; Doonan, P.; Cárdenas, C.; Chandramoorthy, H.C.; Müller, M.; Miller, R.; Hoffman, N.E.; Gandhirajan, R.K.; Molgó, J.; Birnbaum, M.J.; et al. MICU1 is an essential gatekeeper for MCU-mediated mitochondrial Ca(2+) uptake that regulates cell survival. Cell 2012, 151, 630–644. [Google Scholar] [CrossRef] [PubMed]
- Plovanich, M.; Bogorad, R.L.; Sancak, Y.; Kamer, K.J.; Strittmatter, L.; Li, A.A.; Girgis, H.S.; Kuchimanchi, S.; De Groot, J.; Speciner, L.; et al. MICU2, a paralog of MICU1, resides within the mitochondrial uniporter complex to regulate calcium handling. PLoS ONE 2013, 8, e55785. [Google Scholar] [CrossRef]
- Graier, W.F.; Malli, R. Mitochondrial calcium: A crucial hub for cancer cell metabolism? Transl. Cancer Res. 2017, 6, S1124–S1127. [Google Scholar] [CrossRef]
- Curry, M.C.; Peters, A.A.; Kenny, P.A.; Roberts-Thomson, S.J.; Monteith, G.R. Mitochondrial calcium uniporter silencing potentiates caspase-independent cell death in MDA-MB-231 breast cancer cells. Biochem. Biophys. Res. Commun. 2013, 434, 695–700. [Google Scholar] [CrossRef] [PubMed]
- Tosatto, A.; Sommaggio, R.; Kummerow, C.; Bentham, R.B.; Blacker, T.; Berecz, T.; Duchen, M.; Rosato, A.; Bogeski, I.; Szabadkai, G.; et al. The mitochondrial calcium uniporter regulates breast cancer progression via HIF -1α. EMBO Mol. Med. 2016, 8, 569–585. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, R.E.; Zoncu, R. The lysosome as a cellular centre for signalling, metabolism and quality control. Nat. Cell Biol. 2019, 21, 133–142. [Google Scholar] [CrossRef] [PubMed]
- Galione, A. A primer of NAADP-mediated Ca2+ signalling: From sea urchin eggs to mammalian cells. Cell Calcium 2015, 58, 27–47. [Google Scholar] [CrossRef] [PubMed]
- Piao, S.; Amaravadi, R.K. Targeting the lysosome in cancer. Ann. N. Y. Acad. Sci. 2016, 1371, 45–54. [Google Scholar] [CrossRef]
- Yang, S.; Wang, X.; Contino, G.; Liesa, M.; Sahin, E.; Ying, H.; Bause, A.; Li, Y.; Stommel, J.M.; Dell’Antonio, G.; et al. Pancreatic cancers require autophagy for tumor growth. Genes Dev. 2011, 25, 717–729. [Google Scholar] [CrossRef]
- Hämälistö, S.; Jäättelä, M. Lysosomes in cancer—living on the edge (of the cell). Curr. Opin. Cell Biol. 2016, 39, 69–76. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, S.; Jha, A.; Li, Q.; Soyombo, A.; Dickinson, G.; Churamani, D.; Brailoiu, E.; Patel, S.; Muallem, S. Transient Receptor Potential Mucolipin 1 (TRPML1) and Two-pore Channels Are Functionally Independent Organellar Ion Channels. J. Biol. Chem. 2011, 286, 22934–22942. [Google Scholar] [CrossRef]
- Favia, A.; Pafumi, I.; Desideri, M.; Padula, F.; Montesano, C.; Passeri, D.; Nicoletti, C.; Orlandi, A.; Del Bufalo, D.; Sergi, M.; et al. NAADP-Dependent Ca2+ Signaling Controls Melanoma Progression, Metastatic Dissemination and Neoangiogenesis. Sci. Rep. 2016, 6, 18925. [Google Scholar] [CrossRef]
- Nguyen, O.N.P.; Grimm, C.; Schneider, L.S.; Chao, Y.-K.; Atzberger, C.; Bartel, K.; Watermann, A.; Ulrich, M.; Mayr, D.; Wahl-Schott, C.; et al. Two-Pore Channel Function Is Crucial for the Migration of Invasive Cancer Cells. Cancer Res. 2017, 77, 1427–1438. [Google Scholar] [CrossRef]
- Kern, U.; Wischnewski, V.; Biniossek, M.L.; Schilling, O.; Reinheckel, T. Lysosomal protein turnover contributes to the acquisition of TGFβ-1 induced invasive properties of mammary cancer cells. Mol. Cancer 2015, 14, 39. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Cheng, X.; Yu, L.; Yang, J.; Calvo, R.; Patnaik, S.; Hu, X.; Gao, Q.; Yang, M.; Lawas, M.; et al. MCOLN1 is a ROS sensor in lysosomes that regulates autophagy. Nat. Commun. 2016, 7, 12109. [Google Scholar] [CrossRef]
- Settembre, C.; Di Malta, C.; Polito, V.A.; Arencibia, M.G.; Vetrini, F.; Serkan, E.; Erdin, S.U.; Huynh, T.; Medina, D.; Colella, P.; et al. TFEB Links Autophagy to Lysosomal Biogenesis. Science 2011, 332, 1429–1433. [Google Scholar] [CrossRef] [PubMed]
- Durinck, S.; Stawiski, E.W.; Pavía-Jiménez, A.; Modrusan, Z.; Kapur, P.; Jaiswal, B.S.; Zhang, N.; Toffessi-Tcheuyap, V.; Nguyen, T.; Pahuja, K.B.; et al. Spectrum of diverse genomic alterations define non–clear cell renal carcinoma subtypes. Nat. Genet. 2015, 47, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Calcagni, A.; Kors, L.; Verschuren, E.; De Cegli, R.; Zampelli, N.; Nusco, E.; Confalonieri, S.; Bertalot, G.; Pece, S.; Settembre, C.; et al. Modelling TFE renal cell carcinoma in mice re-veals a critical role of WNT signaling. eLife 2016, 5, e17047. [Google Scholar] [CrossRef]
- Perera, R.M.; Stoykova, S.; Nicolay, B.N.; Ross, K.N.; Fitamant, J.; Boukhali, M.; Lengrand, J.; Deshpande, V.; Selig, M.K.; Ferrone, C.R.; et al. Transcriptional control of autophagy–lysosome function drives pancreatic cancer metabolism. Nat. Cell Biol. 2015, 524, 361–365. [Google Scholar] [CrossRef]
- Medina, D.L.; Di Paola, S.; Peluso, I.; Armani, A.; De Stefani, D.; Venditti, R.; Montefusco, S.; Rosato, A.S.; Prezioso, C.; Forrester, A.; et al. Lysosomal calcium signalling regulates autophagy through calcineurin and TFEB. Nat. Cell Biol. 2015, 17, 288–299. [Google Scholar] [CrossRef]
- Giatromanolaki, A.; Kalamida, D.; Sivridis, E.; Karagounis, I.V.; Gatter, K.C.; Harris, A.L.; Koukourakis, M.I. Increased expression of transcription factor EB (TFEB) is associated with autophagy, migratory phenotype and poor prognosis in non-small cell lung cancer. Lung Cancer 2015, 90, 98–105. [Google Scholar] [CrossRef]
- Liang, J.; Jia, X.; Wang, K.; Zhao, N. High expression of TFEB is associated with aggressive clinical features in colorectal cancer. OncoTargets Ther. 2018, 11, 8089–8098. [Google Scholar] [CrossRef]
- Slade, L.; Biswas, D.; Ihionu, F.; El Hiani, Y.; Kienesberger, P.C.; Pulinilkunnil, T. A Lysosome Independent Role for TFEB in Activating DNA Repair and Inhibiting Apoptosis in Breast Cancer Cells. Biochem. J. 2020, 477, 137–160. [Google Scholar] [CrossRef] [PubMed]
- Cui, C.; Chang, Y.; Zhang, X.; Choi, S.; Tran, H.; Penmetsa, K.V.; Viswanadha, S.; Fu, L.; Pan, Z. Targeting Orai1-mediated store-operated calcium entry by RP4010 for anti-tumor activity in esophagus squamous cell carcinoma. Cancer Lett. 2018, 432, 169–179. [Google Scholar] [CrossRef]
- Serafini, M.; Cordero-Sanchez, C.; Di Paola, R.; Bhela, I.P.; Aprile, S.; Purghè, B.; Fusco, R.; Cuzzocrea, S.; Genazzani, A.A.; Riva, B.; et al. Store-Operated Calcium Entry as a Therapeutic Target in Acute Pancreatitis: Discovery and Development of Drug-Like SOCE Inhibitors. J. Med. Chem. 2020, 63, 14761–14779. [Google Scholar] [CrossRef] [PubMed]
- Waldherr, L.; Tiffner, A.; Mishra, D.; Sallinger, M.; Schober, R.; Frischauf, I.; Schmidt, T.; Handl, V.; Sagmeister, P.; Köckinger, M.; et al. Blockage of Store-Operated Ca2+ Influx by Synta66 is Mediated by Direct Inhibition of the Ca2+ Selective Orai1 Pore. Cancers 2020, 12, 2876. [Google Scholar] [CrossRef] [PubMed]
- Marchesini, M.; Gherli, A.; Montanaro, A.; Patrizi, L.; Sorrentino, C.; Pagliaro, L.; Rompietti, C.; Kitara, S.; Heit, S.; Olesen, C.E.; et al. Blockade of Oncogenic NOTCH1 with the SERCA Inhibitor CAD204520 in T Cell Acute Lymphoblastic Leukemia. Cell Chem. Biol. 2020, 27, 678–697.e13. [Google Scholar] [CrossRef]
- Pagliaro, L.; Marchesini, M.; Roti, G. Targeting oncogenic Notch signaling with SERCA inhibitors. J. Hematol. Oncol. 2021, 14, 1–17. [Google Scholar] [CrossRef]
- Patergnani, S.; Danese, A.; Bouhamida, E.; Aguiari, G.; Previati, M.; Pinton, P.; Giorgi, C. Various Aspects of Calcium Signaling in the Regulation of Apoptosis, Autophagy, Cell Proliferation, and Cancer. Int. J. Mol. Sci. 2020, 21, 8323. [Google Scholar] [CrossRef]
- Prasad, V.; Mailankody, S. Research and development spending to bring a single cancer drug to market and revenues after approval. JAMA Internal Med. 2017, 177, 1569–1575. [Google Scholar] [CrossRef]
- Senapati, S.; Mahanta, A.K.; Kumar, S.; Maiti, P. Controlled drug delivery vehicles for cancer treatment and their performance. Signal Transduct. Target. Ther. 2018, 3, 7. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.; Hong, J.H. Physiological application of nanoparticles in calcium-related proteins and channels. Nanomedicine 2019, 14, 2479–2486. [Google Scholar] [CrossRef] [PubMed]
- Colone, M.; Calcabrini, A.; Stringaro, A. Drug Delivery Systems of Natural Products in Oncology. Molecules 2020, 25, 4560. [Google Scholar] [CrossRef]
- Bong, A.H.; Monteith, G.R. Calcium signaling and the therapeutic targeting of cancer cells. Biochim. Biophys. Acta (BBA)—Bioenerg. 2018, 1865, 1786–1794. [Google Scholar] [CrossRef]
- Shen, Y.; Li, X.; Dong, D.; Zhang, B.; Xue, Y.; Shang, P. Transferrin receptor 1 in cancer: A new sight for cancer therapy. Am. J. Cancer Res. 2018, 8, 916–931. [Google Scholar] [PubMed]
- Choudhary, D.; Goykar, H.; Karanwad, T.; Kannaujia, S.; Gadekar, V.; Misra, M. An understanding of mitochondria and its role in targeting nanocarriers for diagnosis and treatment of cancer. Asian J. Pharm. Sci. 2020. [Google Scholar] [CrossRef]
- Rahman, S.; Rahman, T. Unveiling some FDA-approved drugs as inhibitors of the store-operated Ca2+ entry pathway. Sci. Rep. 2017, 7, 12881. [Google Scholar] [CrossRef]
- Frandsen, S.K.; Vissing, M.; Gehl, J. A Comprehensive Review of Calcium Electroporation—A Novel Cancer Treatment Modality. Cancers 2020, 12, 290. [Google Scholar] [CrossRef]
- Munk, M.; Alcalde, J.; Lorentzen, L.; Villalobo, A.; Berchtold, M.W.; Panina, S. The impact of calmodulin on the cell cycle analyzed in a novel human cellular genetic system. Cell Calcium 2020, 88, 102207. [Google Scholar] [CrossRef]
- Chai, S.; Xu, X.; Wang, Y.; Zhou, Y.; Zhang, C.; Yang, Y.; Xu, H.; Xu, R.; Wang, K.; Yang, Y. Ca2+/calmodulin-dependent protein kinase IIγ enhances stem-like traits and tumorigenicity of lung cancer cells. Oncotarget 2015, 6, 16069–16083. [Google Scholar] [CrossRef] [PubMed]
- Chi, M.; Evans, H.; Gilchrist, J.; Mayhew, J.; Hoffman, A.; Pearsall, E.A.; Jankowski, H.; Brzozowski, J.; Skelding, K.A. Phosphorylation of calcium/calmodulin-stimulated protein kinase II at T286 enhances invasion and migration of human breast cancer cells. Sci. Rep. 2016, 6, 33132. [Google Scholar] [CrossRef]
- Daft, P.G.; Yang, Y.; Napierala, D.; Zayzafoon, M. The Growth and Aggressive Behavior of Human Osteosarcoma Is Regulated by a CaMKII-Controlled Autocrine VEGF Signaling Mechanism. PLoS ONE 2015, 10, e0121568. [Google Scholar] [CrossRef] [PubMed]
- Jung, H.J.; Kim, J.H.; Shim, J.S.; Kwon, H.J. A Novel Ca2+/Calmodulin Antagonist HBC Inhibits Angiogenesis and Down-regulates Hypoxia-inducible Factor. J. Biol. Chem. 2010, 285, 25867–25874. [Google Scholar] [CrossRef]
- Can, G.; Akpinar, B.; Baran, Y.; Zhivotovsky, B.; Olsson, M. 5-Fluorouracil signaling through a calcium–calmodulin-dependent pathway is required for p53 activation and apoptosis in colon carcinoma cells. Oncogene 2013, 32, 4529–4538. [Google Scholar] [CrossRef] [PubMed]
- Ren, T.; Tang, Y.; Wang, M.; Wang, H.; Liu, Y.; Qian, X.; Chang, C.; Chen, M. Triptolide induces apoptosis through the calcium/calmodulin-dependent protein kinase kinaseβ/AMP-activated protein kinase signaling pathway in non-small cell lung cancer cells. Oncol. Rep. 2020, 44, 2288–2296. [Google Scholar] [CrossRef] [PubMed]
- Karacosta, L.G.; Foster, B.A.; Azabdaftari, G.; Feliciano, D.; Edelman, A.M. A Regulatory Feedback Loop Between Ca2+/Calmodulin-dependent Protein Kinase Kinase 2 (CaMKK2) and the Androgen Receptor in Prostate Cancer Progression. J. Biol. Chem. 2012, 287, 24832–24843. [Google Scholar] [CrossRef]
- Chimote, A.A.; Gawali, V.; Newton, H.S.; Wise-Draper, T.M.; Conforti, L. A Compartmentalized Reduction in Membrane-Proximal Calmodulin Reduces the Immune Surveillance Capabilities of CD8+ T Cells in Head and Neck Cancer. Front. Pharmacol. 2020, 11, 143. [Google Scholar] [CrossRef]
- Racioppi, L.; Nelson, E.R.; Huang, W.; Mukherjee, D.; Lawrence, S.A.; Lento, W.; Masci, A.M.; Jiao, Y.; Park, S.; York, B.; et al. CaMKK2 in myeloid cells is a key regulator of the immune-suppressive microenvironment in breast cancer. Nat. Commun. 2019, 10, 1–16. [Google Scholar] [CrossRef]
- Blum, W.; Schwaller, B. Calretinin is essential for mesothelioma cell growth/survival in vitro: A potential new target for malignant mesothelioma therapy? Int. J. Cancer 2013, 133, 2077–2088. [Google Scholar] [CrossRef]
- Winn, B.; Tavares, R.; Fanion, J.; Noble, L.; Gao, J.; Sabo, E.; Resnick, M.B. Differentiating the undifferentiated: Immunohistochemical profile of medullary carcinoma of the colon with an emphasis on intestinal differentiation. Hum. Pathol. 2009, 40, 398–404. [Google Scholar] [CrossRef]
- Wörthmüller, J.; Oberson, A.; Salicio, V.; Blum, W.; Schwaller, B. Calretinin Functions in Malignant Mesothelioma Cells Cannot Be Replaced by the Closely Related Ca2+-Binding Proteins Calbindin-D28k and Parvalbumin. Int. J. Mol. Sci. 2018, 19, 4015. [Google Scholar] [CrossRef]
- Dodla, P.; Bhoopalan, V.; Khoo, S.K.; Miranti, C.; Sridhar, S. Gene expression analysis of human prostate cell lines with and without tumor metastasis suppressor CD82. BMC Cancer 2020, 20, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Bignotti, E.; Tassi, R.A.; Calza, S.; Ravaggi, A.; Bandiera, E.; Rossi, E.; Donzelli, C.; Pasinetti, B.; Pecorelli, S.; Santin, A.D. Gene expression profile of ovarian serous papillary carcinomas: Identification of metastasis-associated genes. Am. J. Obstet. Gynecol. 2007, 196, 245.e1–245.e11. [Google Scholar] [CrossRef] [PubMed]
- Wörthmüller, J.; Blum, W.; Pecze, L.; Salicio, V.; Schwaller, B. Calretinin promotes invasiveness and EMT in malignant mesothelioma cells involving the activation of the FAK signaling pathway. Oncotarget 2018, 9, 36256–36272. [Google Scholar] [CrossRef] [PubMed]
- Gordillo, C.H.; Sandoval, P.; Muñoz-Hernández, P.; Pascual-Antón, L.; López-Cabrera, M.; Jiménez-Heffernan, J.A. Mesothelial-to-Mesenchymal Transition Contributes to the Generation of Carcinoma-Associated Fibroblasts in Locally Advanced Primary Colorectal Carcinomas. Cancers 2020, 12, 499. [Google Scholar] [CrossRef] [PubMed]
- Jung, E.M.; Choi, K.C.; Jeung, E.B. Expression of calbindin-D28k is inversely correlated with proapototic gene expression in hydrogen peroxide-induced cell death in endometrial cancer cells. Int. J. Oncol. 2011, 38, 1059–1066. [Google Scholar] [CrossRef] [PubMed]
- Stevenson, L.; Allen, W.L.; Proutski, I.; Stewart, G.; Johnston, L.; McCloskey, K.; Wilson, P.M.; Longley, D.B.; Johnston, P.G. Calbindin 2 (CALB2) Regulates 5-Fluorouracil Sensitivity in Colorectal Cancer by Modulating the Intrinsic Apoptotic Pathway. PLoS ONE 2011, 6, e20276. [Google Scholar] [CrossRef]
- Sergeev, I.N. Vitamin D and cellular Ca2+ signaling in breast cancer. Anticancer. Res. 2012, 32, 299–302. [Google Scholar]
- Kim, J.H.; sil Hong, B.; Heo, W.; Han, J.M.; Han, W.; Noh, D.-Y.; Moon, H.-G. Abstract 33: Calsequestrin 2 regulates prolif-eration, migration, and invasion in triple-negative breast cancer cells. Cancer Res. 2018, 78 (Suppl. 13), 33. [Google Scholar] [CrossRef]
- Toquet, C.; Jarry, A.; Bou-Hanna, C.; Bach, K.; Denis, M.G.; Mosnier, J.F.; Laboisse, C.L. Altered Calreticulin expression in human colon cancer: Maintenance of Calreticulin expression is associated with mucinous differentiation. Oncol. Rep. 2007, 17, 1101–1107. [Google Scholar] [CrossRef]
- Vanoverberghe, K.; Abeele, F.V.; Mariot, P.; Lepage, G.; Roudbaraki, M.; Bonnal, J.L.; Mauroy, B.; Shuba, Y.; Skryma, R.; Prevarskaya, N. Ca2+ homeostasis and apoptotic resistance of neuroendocrine-differentiated prostate cancer cells. Cell Death Differ. 2003, 11, 321–330. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Liao, Q.; Wang, H.; Rao, S.; Yi, P.; Tang, L.; Tian, Y.; Oyang, L.; Wang, H.; Shi, Y.; et al. High expression of calreticulin indicates poor prognosis and modulates cell migration and invasion via activating Stat3 in nasopharyngeal carcinoma. J. Cancer 2019, 10, 5460–5468. [Google Scholar] [CrossRef] [PubMed]
- Pouliquen, D.L.; Boissard, A.; Coqueret, O.; Guette, C. Biomarkers of tumor invasiveness in proteomics (Review). Int. J. Oncol. 2020, 57, 409–432. [Google Scholar] [CrossRef]
- Amuthan, G.; Biswas, G.; Zhang, S.; Klein-Szanto, A.; Vijayasarathy, C.; Avadhani, N.G. Mitochondria-to-nucleus stress signaling induces phenotypic changes, tumor progression and cell invasion. EMBO J. 2001, 20, 1910–1920. [Google Scholar] [CrossRef]
- Pike, S.E.; Yao, L.; Setsuda, J.; Jones, K.D.; Cherney, B.; Appella, E.; Sakaguchi, K.; Nakhasi, H.; Atreya, C.D.; Teruya-Feldstein, J.; et al. Calreticulin and calreticulin fragments are endothelial cell inhibitors that suppress tumor growth. Blood 1999, 94, 2461–2468. [Google Scholar] [CrossRef] [PubMed]
- Delom, F.; Emadali, A.; Cocolakis, E.; Lebrun, J.-J.; Nantel, A.; Chevet, E. Calnexin-dependent regulation of tunicamycin-induced apoptosis in breast carcinoma MCF-7 cells. Cell Death Differ. 2006, 14, 586–596. [Google Scholar] [CrossRef] [PubMed]
- Li, D.-D.; Xie, B.; Wu, X.-J.; Li, J.-J.; Ding, Y.; Wen, X.-Z.; Zhang, X.; Zhu, S.-G.; Liu, W.; Zhang, X.-S.; et al. Late-stage inhibition of autophagy enhances calreticulin surface exposure. Oncotarget 2016, 7, 80842–80854. [Google Scholar] [CrossRef] [PubMed]
- Obeid, M.; Tesniere, A.; Ghiringhelli, F.; Fimia, G.M.; Apetoh, L.; Perfettini, J.-L.; Castedo, M.; Mignot, G.; Panaretakis, T.; Casares, N.; et al. Calreticulin exposure dictates the immunogenicity of cancer cell death. Nat. Med. 2007, 13, 54–61. [Google Scholar] [CrossRef]
- Chen, Y.; Ma, D.; Wang, X.; Fang, J.; Liu, X.; Song, J.; Li, X.; Ren, X.; Li, Q.; Li, Q.; et al. Calnexin Impairs the Antitumor Immunity of CD4+ and CD8+ T Cells. Cancer Immunol. Res. 2018, 7, 123–135. [Google Scholar] [CrossRef]
- Loulousis, M.; Krager, S.L.; Darcy, Y.L.; Tischkau, S.A.; Copello, J.A. Drugs that inhibit the sarcoplasmic reticulum Ca2+ atpase (Serca) and prevention of breast cancer cell proliferation. FASEB J. 2016, 30, 768.4. [Google Scholar] [CrossRef]
- Lee, W.J.; Robinson, J.A.; Holman, N.A.; McCall, M.N.; Roberts-Thomson, S.; Monteith, G. Antisense-mediated Inhibition of the Plasma Membrane Calcium-ATPase Suppresses Proliferation of MCF-7 Cells. J. Biol. Chem. 2005, 280, 27076–27084. [Google Scholar] [CrossRef]
- Chovancova, B.; Liskova, V.; Babula, P.; Krizanova, O. Role of Sodium/Calcium Exchangers in Tumors. Biomolecules 2020, 10, 1257. [Google Scholar] [CrossRef]
- Sritangos, P.; Alarcon, E.P.; James, A.; Sultan, A.; Richardson, D.A.; Bruce, J.I.E. Plasma Membrane Ca2+ ATPase Isoform 4 (PMCA4) Has an Important Role in Numerous Hallmarks of Pancreatic Cancer. Cancers 2020, 12, 218. [Google Scholar] [CrossRef]
- Fondello, C.; Agnetti, L.; Glikin, G.C.; Finocchiaro, L.M. Mechanisms Enhancing the Cytotoxic Effects of Bleomycin plus Suicide or Interferon-β Gene Lipofection in Metastatic Human Melanoma Cells. Anti-Cancer Agents Med. Chem. 2019, 18, 1338–1348. [Google Scholar] [CrossRef]
- Szadvari, I.; Hudecova, S.; Chovancova, B.; Matuskova, M.; Cholujova, D.; Lencesova, L.; Valerian, D.; Ondrias, K.; Babula, P.; Krizanova, O. Sodium/calcium exchanger is involved in apoptosis induced by H2S in tumor cells through decreased levels of intracellular pH. Nitric Oxide 2019, 87, 1–9. [Google Scholar] [CrossRef]
- Ho, P.-C.; Bihuniak, J.D.; Macintyre, A.; Staron, M.; Liu, X.; Amezquita, R.; Tsui, Y.-C.; Cui, G.; Micevic, G.; Perales, J.C.; et al. Phosphoenolpyruvate Is a Metabolic Checkpoint of Anti-tumor T Cell Responses. Cell 2015, 162, 1217–1228. [Google Scholar] [CrossRef] [PubMed]
- James, A.; Richardson, D.A.; Oh, I.-W.; Sritangos, P.; Attard, T.; Barrett, L.; Bruce, J.I.E. Cutting off the fuel supply to calcium pumps in pancreatic cancer cells: Role of pyruvate kinase-M2 (PKM2). Br. J. Cancer 2020, 122, 266–278. [Google Scholar] [CrossRef] [PubMed]
- Márián, T.; Szabó-Péli, J.; Németh, E.; Trón, L.; Friedländer, E.; Szabó, A.; Balkay, L.; Veress, G.; Krasznai, Z. Na+/Ca2+ exchanger inhibitors modify the accumulation of tumor-diagnostic PET tracers in cancer cells. Eur. J. Pharm. Sci. 2007, 30, 56–63. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Adhikary, A.; Chakraborty, S.; Nandi, P.; Mohanty, S.; Chakraborty, S.; Bhattacharjee, P.; Mukherjee, S.; Putatunda, S.; Chakraborty, S.; et al. Nifetepimine, a Dihydropyrimidone, Ensures CD4+ T Cell Survival in a Tumor Microenvironment by Maneuvering Sarco(endo)plasmic Reticulum Ca2+ ATPase (SERCA). J. Biol. Chem. 2012, 287, 32881–32896. [Google Scholar] [CrossRef]
- Bhargava, A.; Saha, S. T-Type voltage gated calcium channels: A target in breast cancer? Breast Cancer Res. Treat. 2019, 173, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Xie, R.; Xu, J.; Wen, G.; Jin, H.; Liu, X.; Yang, Y.; Ji, B.; Jiang, Y.; Song, P.; Dong, H.; et al. The P2Y2 Nucleotide Receptor Mediates the Proliferation and Migration of Human Hepatocellular Carcinoma Cells Induced by ATP. J. Biol. Chem. 2014, 289, 19137–19149. [Google Scholar] [CrossRef] [PubMed]
- Adinolfi, E.; Melchiorri, L.; Falzoni, S.; Chiozzi, P.; Morelli, A.; Tieghi, A.; Cuneo, A.; Castoldi, G.; Di Virgilio, F.; Baricordi, O.R. P2X7 receptor expression in evolutive and indolent forms of chronic B lymphocytic leukemia. Blood 2002, 99, 706–708. [Google Scholar] [CrossRef]
- Ledur, P.F.; Villodre, E.S.; Paulus, R.; Cruz, L.A.; Flores, D.G.; Lenz, G. Extracellular ATP reduces tumor sphere growth and cancer stem cell population in glioblastoma cells. Purinergic Signal. 2011, 8, 39–48. [Google Scholar] [CrossRef] [PubMed]
- Conigrave, A.D.; van der Weyden, L.; Holt, L.; Jiang, L.; Wilson, P.; I Christopherson, R.; Morris, M.B. Extracellular ATP-dependent suppression of proliferation and induction of differentiation of human HL-60 leukemia cells by distinct mechanisms. Biochem. Pharmacol. 2000, 60, 1585–1591. [Google Scholar] [CrossRef]
- Maiques, O.; Macià, A.; Moreno, S.; Barceló, C.; Santacana, M.; Vea, A.; Herreros, J.; Gatius, S.; Ortega, E.; Valls, J.; et al. Immunohistochemical analysis of T-type calcium channels in acquired melanocytic naevi and melanoma. Br. J. Dermatol. 2016, 176, 1247–1258. [Google Scholar] [CrossRef]
- Zhou, X.; Wang, W.; Zhang, S.; Wang, X.; Tang, Z.; Gu, J.; Li, J.; Huang, J. CACNA1B (Cav2.2) Overexpression and Its Association with Clinicopathologic Characteristics and Unfavorable Prognosis in Non-Small Cell Lung Cancer. Dis. Markers 2017, 2017, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, D.; Strilic, B.; Sivaraj, K.K.; Wettschureck, N.; Offermanns, S. Platelet-Derived Nucleotides Promote Tumor-Cell Transendothelial Migration and Metastasis via P2Y2 Receptor. Cancer Cell 2013, 24, 130–137. [Google Scholar] [CrossRef]
- Takai, E.; Tsukimoto, M.; Harada, H.; Kojima, S. Autocrine signaling via release of ATP and activation of P2X7 receptor influences motile activity of human lung cancer cells. Purinergic Signal. 2014, 10, 487–497. [Google Scholar] [CrossRef]
- Rumjahn, S.M.; A Javed, M.; Wong, N.; E Law, W.; O Buxton, I.L. Purinergic regulation of angiogenesis by human breast carcinoma-secreted nucleoside diphosphate kinase. Br. J. Cancer 2007, 97, 1372–1380. [Google Scholar] [CrossRef]
- Dziegielewska, B.; Gray, L.S.; Dziegielewski, J. T-type calcium channels blockers as new tools in cancer therapies. Pflügers Archiv—Eur. J. Physiol. 2014, 466, 801–810. [Google Scholar] [CrossRef]
- Gilbert, S.M.; Oliphant, C.J.; Hassan, S.; Peille, A.L.; Bronsert, P.; Falzoni, S.; Di Virgilio, F.; McNulty, S.; Lara, R. ATP in the tumour microenvironment drives expression of nfP2X7, a key mediator of cancer cell survival. Oncogene 2019, 38, 194–208. [Google Scholar] [CrossRef] [PubMed]
- Höpfner, M.; Maaser, K.; Barthel, B.; Von Lampe, B.; Hanski, C.; Riecken, E.-O.; Zeitz, M.; Scherübl, H. Growth inhibition and apoptosis induced by P2Y2 receptors in human colorectal carcinoma cells: Involvement of intracellular calcium and cyclic adenosine monophosphate. Int. J. Color. Dis. 2001, 16, 154–166. [Google Scholar] [CrossRef]
- Sallán, M.C.; Visa, A.; Shaikh, S.; Nàger, M.; Herreros, J.; Cantí, C. T-type Ca2+ Channels: T for Targetable. Cancer Res. 2018, 78, 603–609. [Google Scholar] [CrossRef]
- Amoroso, F.S.; Capece, M.; Rotondo, A.; Cangelosi, D.; Ferracin, M.; Franceschini, A.; Raffaghello, L.; Pistoia, V.; Varesio, L.; Adinolfi, E. The P2X7 receptor is a key modulator of the PI3K/GSK3β/VEGF signaling network: Evidence in experimental neuroblastoma. Oncogene 2015, 34, 5240–5251. [Google Scholar] [CrossRef]
- Pfaffenzeller, M.S.; Franciosi, M.L.M.; Cardoso, A.M. Purinergic signaling and tumor microenvironment in cervical Cancer. Purinergic Signal. 2020, 16, 123–135. [Google Scholar] [CrossRef]
- Wu, L.; Lin, W.; Liao, Q.; Wang, H.; Lin, C.; Tang, L.; Lian, W.; Chen, Z.; Li, K.; Xu, L.; et al. Calcium Channel Blocker Nifedipine Suppresses Colorectal Cancer Progression and Immune Escape by Preventing NFAT2 Nuclear Translocation. Cell Rep. 2020, 33, 108327. [Google Scholar] [CrossRef] [PubMed]
- Karacicek, B.; Erac, Y.; Tosun, M. Functional consequences of enhanced expression of STIM1 and Orai1 in Huh-7 hepatocellular carcinoma tumor-initiating cells. BMC Cancer 2019, 19, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Faouzi, M.; Kischel, P.; Hague, F.; Ahidouch, A.; Benzerdjeb, N.; Sevestre, H.; Penner, R.; Ouadid-Ahidouch, H. ORAI3 silencing alters cell proliferation and cell cycle progression via c-myc pathway in breast cancer cells. Biochim. Biophys. Acta 2013, 1833, 752–760. [Google Scholar] [CrossRef] [PubMed]
- Hou, M.-F.; Kuo, H.-C.; Li, J.-H.; Wang, Y.-S.; Chang, C.-C.; Chen, K.-C.; Chen, W.-C.; Chiu, C.-C.; Yang, S.; Chang, W.-C. Orai1/CRACM1 overexpression suppresses cell proliferation via attenuation of the store-operated calcium influx-mediated signalling pathway in A549 lung cancer cells. Biochim. Biophys. Acta (BBA)—Gen. Subj. 2011, 1810, 1278–1284. [Google Scholar] [CrossRef]
- Diez-Bello, R.; Jardin, I.; Salido, G.M.; Rosado, J. Orai1 and Orai2 mediate store-operated calcium entry that regulates HL60 cell migration and FAK phosphorylation. Biochim. Biophys. Acta (BBA)—Bioenerg. 2017, 1864, 1064–1070. [Google Scholar] [CrossRef]
- Zhang, Z.; Liu, X.; Feng, B.; Liu, N.; Wu, Q.; Han, Y.; Nie, Y.; Wu, K.; Shi, Y.; Fan, D. STIM1, a direct target of microRNA-185, promotes tumor metastasis and is associated with poor prognosis in colorectal cancer. Oncogene 2015, 34, 4808–4820. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H.; Rigas, N.K.; Lee, C.-R.; Bang, A.; Srikanth, S.; Gwack, Y.; Kang, M.K.; Kim, R.H.; Park, N.-H.; Shin, K.-H. Orai1 promotes tumor progression by enhancing cancer stemness via NFAT signaling in oral/oropharyngeal squamous cell carcinoma. Oncotarget 2016, 7, 43239–43255. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Chen, M.; Huang, J.; Zhang, F.; Lv, Z.; Jia, Y.; Cui, Y.-Z.; Sun, L.-Z.; Wang, Y.; Tang, Y.; et al. ORAI2 Promotes Gastric Cancer Tumorigenicity and Metastasis through PI3K/Akt Signaling and MAPK-Dependent Focal Adhesion Disassembly. Cancer Res. 2021, 81, 986–1000. [Google Scholar] [CrossRef] [PubMed]
- Miao, Y.; Shen, Q.; Zhang, S.; Huang, H.; Meng, X.; Zheng, X.; Yao, Z.; He, Z.; Lu, S.; Cai, C.; et al. Calcium-sensing stromal interaction molecule 2 upregulates nuclear factor of activated T cells 1 and transforming growth factor-β signaling to promote breast cancer metastasis. Breast Cancer Res. 2019, 21, 1–12. [Google Scholar] [CrossRef]
- Sun, J.; Lu, F.; He, H.; Shen, J.; Messina, J.; Mathew, R.; Wang, D.; Sarnaik, A.A.; Chang, W.-C.; Kim, M.; et al. STIM1- and Orai1-mediated Ca2+ oscillation orchestrates invadopodium formation and melanoma invasion. J. Cell Biol. 2014, 207, 535–548. [Google Scholar] [CrossRef] [PubMed]
- Pan, S.; Zhao, X.; Shao, C.; Fu, B.; Huang, Y.; Zhang, N.; Dou, X.; Zhang, Z.; Qiu, Y.; Wang, R.; et al. STIM1 promotes angiogenesis by reducing exosomal miR-145 in breast cancer MDA-MB-231 cells. Cell Death Dis. 2021, 12, 1–15. [Google Scholar] [CrossRef]
- Liu, X.; Wan, X.; Kan, H.; Wang, Y.; Yu, F.; Feng, L.; Jin, J.; Zhang, P.; Ma, X. Hypoxia-induced upregulation of Orai1 drives colon cancer invasiveness and angiogenesis. Eur. J. Pharmacol. 2018, 832, 1–10. [Google Scholar] [CrossRef]
- Sun, X.; Wei, Q.; Cheng, J.; Bian, Y.; Tian, C.; Hu, Y.; Li, H. Enhanced Stim1 expression is associated with acquired chemo-resistance of cisplatin in osteosarcoma cells. Hum. Cell 2017, 30, 216–225. [Google Scholar] [CrossRef]
- Faouzi, M.; Hague, F.; Potier, M.; Ahidouch, A.; Sevestre, H.; Ouadid-Ahidouch, H. Down-regulation of Orai3 arrests cell-cycle progression and induces apoptosis in breast cancer cells but not in normal breast epithelial cells. J. Cell. Physiol. 2011, 226, 542–551. [Google Scholar] [CrossRef]
- Liu, H.; Hughes, J.D.; Rollins, S.; Chen, B.; Perkins, E. Calcium entry via ORAI1 regulates glioblastoma cell proliferation and apoptosis. Exp. Mol. Pathol. 2011, 91, 753–760. [Google Scholar] [CrossRef]
- Zhao, H.; Yan, G.; Zheng, L.; Zhou, Y.; Sheng, H.; Wu, L.; Zhang, Q.; Lei, J.; Zhang, J.; Xin, R.; et al. STIM1 is a metabolic checkpoint regulating the invasion and metastasis of hepatocellular carcinoma. Theranostics 2020, 10, 6483–6499. [Google Scholar] [CrossRef] [PubMed]
- Frisch, J.; Angenendt, A.; Hoth, M.; Prates Roma, L.; Lis, A. STIM-Orai Channels and Reactive Oxygen Species in the Tumor Microenvironment. Cancers 2019, 11, 457. [Google Scholar] [CrossRef]
- Tang, B.-D.; Xia, X.; Lv, X.-F.; Yu, B.-X.; Yuan, J.-N.; Mai, X.-Y.; Shang, J.-Y.; Zhou, J.-G.; Liang, S.-J.; Pang, R.-P. Inhibition of Orai1-mediated Ca2+entry enhances chemosensitivity of HepG2 hepatocarcinoma cells to 5-fluorouracil. J. Cell. Mol. Med. 2016, 21, 904–915. [Google Scholar] [CrossRef] [PubMed]
- Fleur-Lominy, S.S.; Maus, M.; Vaeth, M.; Lange, I.; Zee, I.; Suh, D.; Liu, C.; Wu, X.; Tikhonova, A.; Aifantis, I.; et al. STIM1 and STIM2 Mediate Cancer-Induced Inflammation in T Cell Acute Lymphoblastic Leukemia. Cell Rep. 2018, 24, 3045–3060.e5. [Google Scholar] [CrossRef]
- Xu, Y.; Zhang, S.; Niu, H.; Ye, Y.; Hu, F.; Chen, S.; Li, X.; Luo, X.; Jiang, S.; Liu, Y.; et al. STIM1 accelerates cell senescence in a remodeled microenvironment but enhances the epithelial-to-mesenchymal transition in prostate cancer. Sci. Rep. 2015, 5, 11754. [Google Scholar] [CrossRef]
- Szatkowski, C.; Parys, J.B.; Ouadid-Ahidouch, H.; Matifat, F. Inositol 1,4,5-trisphosphate-induced Ca2+ signalling is involved in estradiol-induced breast cancer epithelial cell growth. Mol. Cancer 2010, 9, 156. [Google Scholar] [CrossRef]
- Nougarède, A.; Popgeorgiev, N.; Kassem, L.; Omarjee, S.; Borel, S.; Mikaelian, I.; Lopez, J.; Gadet, R.; Marcillat, O.; Treilleux, I.; et al. Breast Cancer Targeting through Inhibition of the Endoplasmic Reticulum-Based Apoptosis Regulator Nrh/BCL2L10. Cancer Res. 2018, 78, 1404–1417. [Google Scholar] [CrossRef] [PubMed]
- Thoppil, R.J.; Adapala, R.K.; Cappelli, H.C.; Kondeti, V.; Dudley, A.C.; Gary Meszaros, J.; Paruchuri, S.; Thodeti, C.K. TRPV4 channel activation selectively inhibits tumor endothelial cell proliferation. Sci. Rep. 2015, 5, 14257. [Google Scholar] [CrossRef]
- Bomben, V.C.; Sontheimer, H. Disruption of transient receptor potential canonical channel 1 causes incomplete cytokinesis and slows the growth of human malignant gliomas. Glia 2010, 58, 1145–1156. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Li, M.-H.; Inoue, K.; Chu, X.-P.; Seeds, J.; Xiong, Z.-G. Transient Receptor Potential Melastatin 7–like Current in Human Head and Neck Carcinoma Cells: Role in Cell Proliferation. Cancer Res. 2007, 67, 10929–10938. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.; Shui, B.; Zhao, W.; Liu, H.; Li, W.; Lee, J.C.; Doran, R.; Lee, F.K.; Sun, T.; Shen, Q.S.; et al. Central role of IP3R2-mediated Ca2+ oscillation in self-renewal of liver cancer stem cells elucidated by high-signal ER sensor. Cell Death Dis. 2019, 10, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Bidaux, G.; Flourakis, M.; Thébault, S.; Zholos, A.; Beck, B.; Gkika, D.; Roudbaraki, M.; Bonnal, J.-L.; Mauroy, B.; Shuba, Y.; et al. Prostate cell differentiation status determines transient receptor potential melastatin member 8 channel subcellular localization and function. J. Clin. Investig. 2007, 117, 1647–1657. [Google Scholar] [CrossRef]
- Jiang, H.-N.; Zeng, B.; Zhang, Y.; Daskoulidou, N.; Fan, H.; Qu, J.-M.; Xu, S.-Z. Involvement of TRPC Channels in Lung Cancer Cell Differentiation and the Correlation Analysis in Human Non-Small Cell Lung Cancer. PLoS ONE 2013, 8, e67637. [Google Scholar] [CrossRef]
- Middelbeek, J.; Visser, D.; Henneman, L.; Kamermans, A.; Kuipers, A.J.; Hoogerbrugge, P.M.; Jalink, K.; Van Leeuwen, F.N. TRPM7 maintains progenitor-like features of neuroblastoma cells: Implications for metastasis formation. Oncotarget 2015, 6, 8760–8776. [Google Scholar] [CrossRef]
- Kang, S.S.; Han, K.-S.; Ku, B.M.; Lee, Y.K.; Hong, J.; Shin, H.Y.; Almonte, A.; Woo, D.H.; Brat, D.J.; Hwang, E.M.; et al. Caffeine-Mediated Inhibition of Calcium Release Channel Inositol 1,4,5-Trisphosphate Receptor Subtype 3 Blocks Glioblastoma Invasion and Extends Survival. Cancer Res. 2010, 70, 1173–1183. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Jin, M.; Chen, Y.-X.; Wang, J.; Chang, Y.; Yuan, Q.; Yao, K.-T.; Ji, G. ERP44 inhibits human lung cancer cell migration mainly via IP3R2. Aging 2016, 8, 1276–1286. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Xu, N.; Zhang, D.; Chen, J.; He, G.; Gao, L. Low expression of ryanodine receptor 2 is associated with poor prognosis in thyroid carcinoma. Oncol. Lett. 2019, 18, 3605–3612. [Google Scholar] [CrossRef] [PubMed]
- De Quirós, S.B.; Merlo, A.; Secades, P.; Zambrano, I.; de Santa María, I.S.; Ugidos, N.; Jantus-Lewintre, E.; Sirera, R.; Suarez, C.; Chiara, M.-D. Identification of TRPC6 as a possible candidate target gene within an amplicon at 11q21-q22.2 for migratory capacity in head and neck squamous cell carcinomas. BMC Cancer 2013, 13, 116. [Google Scholar] [CrossRef]
- Ramer, R.; Merkord, J.; Rohde, H.; Hinz, B. Cannabidiol inhibits cancer cell invasion via upregulation of tissue inhibitor of matrix metalloproteinases-1. Biochem. Pharmacol. 2010, 79, 955–966. [Google Scholar] [CrossRef]
- Gaunt, H.J.; Vasudev, N.S.; Beech, D.J. Transient receptor potential canonical 4 and 5 proteins as targets in cancer therapeutics. Eur. Biophys. J. 2016, 45, 611–620. [Google Scholar] [CrossRef] [PubMed]
- Kanugula, A.K.; Adapala, R.K.; Midha, P.; Cappelli, H.C.; Meszaros, J.G.; Paruchuri, S.; Chilian, W.M.; Thodeti, C.K. Novel noncanonical regulation of soluble VEGF/VEGFR2 signaling by mechanosensitive ion channel TRPV4. FASEB J. 2019, 33, 195–203. [Google Scholar] [CrossRef] [PubMed]
- Bernardini, M.; Brossa, A.; Chinigo, G.; Grolez, G.P.; Trimaglio, G.; Allart, L.; Hulot, A.; Marot, G.; Genova, T.; Joshi, A.; et al. Transient Receptor Potential Channel Expression Signatures in Tumor-Derived Endothelial Cells: Functional Roles in Prostate Cancer Angiogenesis. Cancers 2019, 11, 956. [Google Scholar] [CrossRef] [PubMed]
- Santoni, G.; Morelli, M.; Santoni, M.; Nabissi, M. New deals on the transcriptional and post-transcriptional regulation of TRP channel target genes during the angiogenesis of glioma. J. Exp. Integr. Med. 2011, 1, 221–234. [Google Scholar] [CrossRef]
- Mariot, P.; Prevarskaya, N.; Roudbaraki, M.M.; Le Bourhis, X.; Van Coppenolle, F.; Vanoverberghe, K.; Skryma, R. Evidence of functional ryanodine receptor involved in apoptosis of prostate cancer (Lncap) cells. Prostate 2000, 43, 205–214. [Google Scholar] [CrossRef]
- Shin, D.-H.; Leem, D.-G.; Shin, J.-S.; Kim, J.-I.; Kim, K.-T.; Choi, S.Y.; Lee, M.-H.; Choi, J.-H.; Lee, K.-T. Compound K induced apoptosis via endoplasmic reticulum Ca2+ release through ryanodine receptor in human lung cancer cells. J. Ginseng Res. 2018, 42, 165–174. [Google Scholar] [CrossRef]
- Rezuchova, I.; Hudecova, S.; Soltysova, A.; Matuskova, M.; Durinikova, E.; Chovancova, B.; Zuzcak, M.; Cihova, M.; Burikova, M.; Penesova, A.; et al. Type 3 inositol 1,4,5-trisphosphate receptor has antiapoptotic and proliferative role in cancer cells. Cell Death Dis. 2019, 10, 186. [Google Scholar] [CrossRef]
- Akl, H.; Monaco, G.; La Rovere, R.; Welkenhuyzen, K.; Kiviluoto, S.; Vervliet, T.; Molgó, J.; Distelhorst, C.W.; Missiaen, L.; Mikoshiba, K.; et al. IP3R2 levels dictate the apoptotic sensitivity of diffuse large B-cell lymphoma cells to an IP3R-derived peptide targeting the BH4 domain of Bcl-2. Cell Death Dis. 2013, 4, e632. [Google Scholar] [CrossRef] [PubMed]
- Boutin, B.; Tajeddine, N.; Monaco, G.; Molgo, J.; Vertommen, D.; Rider, M.; Parys, J.; Bultynck, G.; Gailly, P. Endoplasmic reticulum Ca2+ content decrease by PKA-dependent hyperphosphorylation of type 1 IP3 receptor contributes to prostate cancer cell resistance to androgen deprivation. Cell Calcium 2015, 57, 312–320. [Google Scholar] [CrossRef]
- Kiss, F.; Pohóczky, K.; Szállási, A.; Helyes, Z. Transient Receptor Potential (TRP) Channels in Head-and-Neck Squamous Cell Carcinomas: Diagnostic, Prognostic, and Therapeutic Potentials. Int. J. Mol. Sci. 2020, 21, 6374. [Google Scholar] [CrossRef]
- Monet, M.; Gkika, D.; Lehen’Kyi, V.; Pourtier, A.; Abeele, F.V.; Bidaux, G.; Juvin, V.; Rassendren, F.; Humez, S.; Prevarsakaya, N. Lysophospholipids stimulate prostate cancer cell migration via TRPV2 channel activation. Biochim. Biophys. Acta (BBA)—Bioenerg. 2009, 1793, 528–539. [Google Scholar] [CrossRef]
- Liu, X.; Zou, J.; Su, J.; Lu, Y.; Zhang, J.; Li, L.; Yin, F. Downregulation of transient receptor potential cation channel, subfamily C, member 1 contributes to drug resistance and high histological grade in ovarian cancer. Int. J. Oncol. 2015, 48, 243–252. [Google Scholar] [CrossRef]
- Cardenas, C.; Lovy, A.; Silva-Pavez, E.; Urra, F.; Mizzoni, C.; Ahumada-Castro, U.; Bustos, G.; Jaňa, F.; Cruz, P.; Farias, P.; et al. Cancer cells with defective oxidative phosphorylation require endoplasmic reticulum–to–mitochondria Ca2+transfer for survival. Sci. Signal. 2020, 13, eaay1212. [Google Scholar] [CrossRef]
- Law, B.Y.K.; Michelangeli, F.; Qu, Y.Q.; Xu, S.-W.; Han, Y.; Mok, S.W.F.; de Seabra Rodrigues Dias, I.R.; Javed, M.-H.; Chan, W.-K.; Xue, W.-W.; et al. Neferine induces autophagy-dependent cell death in apoptosis-resistant cancers via ryanodine receptor and Ca2+-dependent mechanism. Sci. Rep. 2019, 9, 20034. [Google Scholar] [CrossRef] [PubMed]
- Fels, B.; Bulk, E.; Pethő, Z.; Schwab, A. The Role of TRP Channels in the Metastatic Cascade. Pharmaceuticals 2018, 11, 48. [Google Scholar] [CrossRef]
- Liu, X.; Zhang, P.; Xie, C.; Sham, K.W.-Y.; Ng, S.S.M.; Chen, Y.; Cheng, C.H.K. Activation of PTEN by inhibition of TRPV4 suppresses colon cancer development. Cell Death Dis. 2019, 10, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Li, S.; Jia, Z.; Zhao, W.; Zhou, C.; Zhang, R.; Ali, D.W.; Michalak, M.; Chen, X.-Z.; Tang, J. Transient Receptor Potential Melastatin 8 (TRPM8) Channel Regulates Proliferation and Migration of Breast Cancer Cells by Activating the AMPK-ULK1 Pathway to Enhance Basal Autophagy. Front. Oncol. 2020, 10, 2645. [Google Scholar] [CrossRef]
- Lan, X.; Zhao, J.; Song, C.; Yuan, Q.; Liu, X. TRPM8 facilitates proliferation and immune evasion of esophageal cancer cells. Biosci. Rep. 2019, 39, BSR20191878. [Google Scholar] [CrossRef]
- Yu, S.; Huang, S.; Ding, Y.; Wang, W.; Wang, A.; Lu, Y. Transient receptor potential ion-channel subfamily V member 4: A potential target for cancer treatment. Cell Death Dis. 2019, 10, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, M.; Horie, I.; Isohama, Y.; Tsukimoto, M. Activation of TRPV4 Channel Regulates Differentiation to and Function of Myeloid-Derived Suppressor Cells. BPB Rep. 2020, 3, 70–75. [Google Scholar] [CrossRef]
- Eliaa, S.G.; Al-Karmalawy, A.A.; Saleh, R.M.; ElShal, M.F. Empagliflozin and Doxorubicin Synergistically Inhibit the Survival of Triple-Negative Breast Cancer Cells via Interfering with the mTOR Pathway and Inhibition of Calmodulin: In Vitro and Molecular Docking Studies. ACS Pharmacol. Transl. Sci. 2020, 3, 1330–1338. [Google Scholar] [CrossRef]
- Pawar, P.; Ma, L.; Byon, C.H.; Liu, H.; Ahn, E.-Y.; Jhala, N.; Arnoletti, J.P.; McDonald, J.M.; Chen, Y. Molecular Mechanisms of Tamoxifen Therapy for Cholangiocarcinoma: Role of Calmodulin. Clin. Cancer Res. 2009, 15, 1288–1296. [Google Scholar] [CrossRef]
- Mine, N.; Yamamoto, S.; Saito, N.; Yamazaki, S.; Suda, C.; Ishigaki, M.; Kufe, D.W.; Von Hoff, D.D.; Kawabe, T. CBP501-Calmodulin Binding Contributes to Sensitizing Tumor Cells to Cisplatin and Bleomycin. Mol. Cancer Ther. 2011, 10, 1929–1938. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.; Wu, Q.; Feng, J.; Yan, L.; Sun, Y.; Liu, S.; Xiang, Y.; Zhang, M.; Pan, T.; Chen, X.; et al. Erianin, a novel dibenzyl compound in Dendrobium extract, inhibits lung cancer cell growth and migration via calcium/calmodulin-dependent ferroptosis. Signal Transduct. Target. Ther. 2020, 5, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Merlot, A.M.; Shafie, N.H.; Yu, Y.; Richardson, V.; Jansson, P.J.; Sahni, S.; Lane, D.; Kovacevic, Z.; Kalinowski, D.S.; Richardson, D. Mechanism of the induction of endoplasmic reticulum stress by the anti-cancer agent, di-2-pyridylketone 4,4-dimethyl-3-thiosemicarbazone (Dp44mT): Activation of PERK/eIF2α, IRE1α, ATF6 and calmodulin kinase. Biochem. Pharmacol. 2016, 109, 27–47. [Google Scholar] [CrossRef] [PubMed]
- Rewcastle, G.W.; Baguley, B.C.; Atwell, G.J.; Denny, W.A. Potential antitumor agents. 52. Carbamate analogs of amsacrine with in vivo activity against multidrug-resistant P388 leukemia. J. Med. Chem. 1987, 30, 1576–1581. [Google Scholar] [CrossRef]
- Mayur, Y.C.; Jagadeesh, S.; Thimmaiah, K.N. Targeting calmodulin in reversing multi drug resistance in cancer cells. Mini-Reviews Med. Chem. 2006, 6, 1383–1389. [Google Scholar] [CrossRef]
- Kim, C.; Kim, B. Anti-Cancer Natural Products and Their Bioactive Compounds Inducing ER Stress-Mediated Apoptosis: A Review. Nutrients 2018, 10, 1021. [Google Scholar] [CrossRef]
- Koido, S.; Kan, S.; Yoshida, K.; Yoshizaki, S.; Takakura, K.; Namiki, Y.; Tsukinaga, S.; Odahara, S.; Kajihara, M.; Okamoto, M.; et al. Immunogenic modulation of cholangiocarcinoma cells by chemoimmunotherapy. Anticancer. Res. 2014, 34, 6353–6361. [Google Scholar] [PubMed]
- Humeau, J.; Sauvat, A.; Cerrato, G.; Xie, W.; Loos, F.; Iannantuoni, F.; Bezu, L.; Lévesque, S.; Paillet, J.; Pol, J.; et al. Inhibition of transcription by dactinomycin reveals a new characteristic of immunogenic cell stress. EMBO Mol. Med. 2020, 12, e11622. [Google Scholar] [CrossRef] [PubMed]
- Charlier, H.A., Jr.; Olson, R.D.; Thornock, C.M.; Mercer, W.K.; Olson, D.R.; Broyles, T.S.; Muhlestein, D.J.; Larson, C.L.; Cusack, B.J.; Shadle, S.E. Investigations of Calsequestrin as a Target for Anthracyclines: Comparison of Functional Effects of Daunorubicin, Daunorubicinol, and Trifluoperazine. Mol. Pharmacol. 2005, 67, 1505–1512. [Google Scholar] [CrossRef]
- Gorini, S.; De Angelis, A.; Berrino, L.; Malara, N.; Rosano, G.; Ferraro, E. Chemotherapeutic Drugs and Mitochondrial Dysfunction: Focus on Doxorubicin, Trastuzumab, and Sunitinib. Oxidative Med. Cell. Longev. 2018, 2018, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Tadini-Buoninsegni, F.; Smeazzetto, S.; Gualdani, R.; Moncelli, M.R. Drug Interactions With the Ca2+-ATPase From Sarco(Endo)Plasmic Reticulum (SERCA). Front. Mol. Biosci. 2018, 5. [Google Scholar] [CrossRef]
- Yiallouris, A.; Patrikios, I.; Johnson, E.O.; Sereti, E.; Dimas, K.; De Ford, C.; Fedosova, N.U.; Graier, W.F.; Sokratous, K.; Kyriakou, K.; et al. Annonacin promotes selective cancer cell death via NKA-dependent and SERCA-dependent pathways. Cell Death Dis. 2018, 9, 1–13. [Google Scholar] [CrossRef]
- Varghese, E.; Samuel, S.M.; Sadiq, Z.; Kubatka, P.; Liskova, A.; Benacka, J.; Pazinka, P.; Kruzliak, P.; Büsselberg, D. Anti-Cancer Agents in Proliferation and Cell Death: The Calcium Connection. Int. J. Mol. Sci. 2019, 20, 3017. [Google Scholar] [CrossRef]
- Stenius, U.; Miraglia, E.; Högberg, J. Statins exhibit anticancer effects through modifications of the pAkt signaling pathway. Int. J. Oncol. 2011, 40, 867–875. [Google Scholar] [CrossRef] [PubMed]
- Stokes, L.; Bidula, S.; Bibič, L.; Allum, E. To Inhibit or Enhance? Is There a Benefit to Positive Allosteric Modulation of P2X Receptors? Front. Pharmacol. 2020, 11, 627. [Google Scholar] [CrossRef] [PubMed]
- Mitrugno, A.; Sylman, J.L.; Rigg, R.A.; Yunga, S.T.; Shatzel, J.J.; Williams, C.D.; Mccarty, O.J. Carpe low-dose aspirin: The new anti-cancer face of an old anti-platelet drug. Platelets 2017, 29, 773–778. [Google Scholar] [CrossRef] [PubMed]
- Ballerini, P.; Dovizio, M.; Bruno, A.; Tacconelli, S.; Patrignani, P. P2Y12 Receptors in Tumorigenesis and Metastasis. Front. Pharmacol. 2018, 9, 66. [Google Scholar] [CrossRef] [PubMed]
- Bentham Science Publisher Bentham Science Publisher Pharmacological Inhibition of Voltage-gated Ca2+ Channels for Chronic Pain Relief. Curr. Neuropharmacol. 2013, 11, 606–620. [CrossRef]
- Vashisht, A.; Trebak, M.; Motiani, R.K. STIM and Orai proteins as novel targets for cancer therapy. A Review in the Theme: Cell and Molecular Processes in Cancer Metastasis. Am. J. Physiol. Physiol. 2015, 309, C457–C469. [Google Scholar] [CrossRef]
- Echakraborty, S.; Eghosh, S.; Ebanerjee, B.; Santra, A.; Eadhikary, A.; Misra, A.K.; Sen, P.C. Phemindole, a Synthetic Di-indole Derivative Maneuvers the Store Operated Calcium Entry (SOCE) to Induce Potent Anti-Carcinogenic Activity in Human Triple Negative Breast Cancer Cells. Front. Pharmacol. 2016, 7, 114. [Google Scholar] [CrossRef]
- Ali, S.; Doan, D.; Ojong, T.; Solomon, H.; Corpuz, E.; Huynh, T.; Haziq, M.; Shahid, M.; Siddiqui, M.R.; Newaz, M.; et al. Metformin Attenuates High Glucose-Induced Coronary Vascular Endothelial Hyper Permeability Via Inhibition of Orai-1 Mediated Store-Operated Calcium Entry. FASEB J. 2020, 34, 1. [Google Scholar] [CrossRef]
- Song, M.; Chen, D.; Yu, S.P. The TRPC channel blocker SKF 96365 inhibits glioblastoma cell growth by enhancing reverse mode of the Na+/Ca2+exchanger and increasing intracellular Ca2+. Br. J. Pharmacol. 2014, 171, 3432–3447. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, T.; Sieglitz, F.; Bernardes, G. Natural product modulators of transient receptor potential (TRP) channels as potential anti-cancer agents. Chem. Soc. Rev. 2016, 45, 6130–6137. [Google Scholar] [CrossRef] [PubMed]
- Distelhorst, C.W.; Bootman, M. Creating a New Cancer Therapeutic Agent by Targeting the Interaction between Bcl-2 and IP3Receptors. Cold Spring Harb. Perspect. Biol. 2019, 11, a035196. [Google Scholar] [CrossRef]
- Liu, Y.; Jin, M.; Wang, Y.; Zhu, J.; Tan, R.; Zhao, J.; Ji, X.; Jin, C.; Jia, Y.; Ren, T.; et al. MCU-induced mitochondrial calcium uptake promotes mitochondrial biogenesis and colorectal cancer growth. Signal Transduct. Target. Ther. 2020, 5, 1–13. [Google Scholar] [CrossRef]
- Marchi, S.; Corricelli, M.; Branchini, A.; Vitto, V.A.M.; Missiroli, S.; Morciano, G.; Perrone, M.; Ferrarese, M.; Giorgi, C.; Pinotti, M.; et al. Akt-mediated phosphorylation of MICU 1 regulates mitochondrial Ca 2+ levels and tumor growth. EMBO J. 2019, 38, e99435. [Google Scholar] [CrossRef]
- Marchi, S.; Vitto, V.A.M.; Danese, A.; Wieckowski, M.; Giorgi, C.; Pinton, P. Mitochondrial calcium uniporter complex modulation in cancerogenesis. Cell Cycle 2019, 18, 1068–1083. [Google Scholar] [CrossRef]
- Li, X.; Spelat, R.; Bartolini, A.; Cesselli, D.; Ius, T.; Skrap, M.; Caponnetto, F.; Manini, I.; Yang, Y.; Torre, V. Mechanisms of malignancy in glioblastoma cells are linked to MCU upregulation and higher intracellular calcium level. J. Cell Sci. 2020, 133, 237503. [Google Scholar] [CrossRef]
- Rao, G.; Dwivedi, S.K.D.; Zhang, Y.; Dey, A.; Shameer, K.; Karthik, R.; Srikantan, S.; Hossen, N.; Wren, J.D.; Madesh, M.; et al. Micro RNA -195 controls MICU 1 expression and tumor growth in ovarian cancer. EMBO Rep. 2020, 21, 48483. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Song, X.; Cheng, G.; Zhang, J.; Dong, L.; Bai, J.; Luo, D.; Xiong, Y.; Li, S.; Liu, F.; et al. The Regulatory Mechanism and Biological Significance of Mitochondrial Calcium Uniporter in the Migration, Invasion, Angiogenesis and Growth of Gastric Cancer. OncoTargets Ther. 2020, 13, 11781–11794. [Google Scholar] [CrossRef]
- Jin, M.; Wang, J.; Ji, X.; Cao, H.; Zhu, J.; Chen, Y.; Yang, J.; Zhao, Z.; Ren, T.; Xing, J. MCUR1 facilitates epithelial-mesenchymal transition and metastasis via the mitochondrial calcium dependent ROS/Nrf2/Notch pathway in hepatocellular carcinoma. J. Exp. Clin. Cancer Res. 2019, 38, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Marchi, S.; Lupini, L.; Patergnani, S.; Rimessi, A.; Missiroli, S.; Bonora, M.; Bononi, A.; Corrà, F.; Giorgi, C.; De Marchi, E.; et al. Downregulation of the Mitochondrial Calcium Uniporter by Cancer-Related miR-25. Curr. Biol. 2013, 23, 58–63. [Google Scholar] [CrossRef] [PubMed]
- Ren, T.; Wang, J.; Zhang, H.; Yuan, P.; Zhu, J.; Wu, Y.; Huang, Q.; Guo, X.; Zhang, J.; Jiaojiao, W.; et al. MCUR1-Mediated Mitochondrial Calcium Signaling Facilitates Cell Survival of Hepatocellular Carcinoma via Reactive Oxygen Species-Dependent P53 Degradation. Antioxidants Redox Signal. 2018, 28, 1120–1136. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.; Wang, Y.; Peng, J.; Shen, Q.; Chen, M.; Tang, W.; Li, X.; Cai, C.; Wang, B.; Cai, S.; et al. Mitochondrial calcium uniporter as a target of microRNA-340 and promoter of metastasis via enhancing the Warburg effect. Oncotarget 2017, 8, 83831–83844. [Google Scholar] [CrossRef]
- Chakraborty, P.K.; Mustafi, S.B.; Xiong, X.; Dwivedi, S.K.D.; Nesin, V.; Saha, S.; Zhang, M.; Dhanasekaran, D.; Jayaraman, M.; Mannel, R.; et al. MICU1 drives glycolysis and chemoresistance in ovarian cancer. Nat. Commun. 2017, 8, 14634. [Google Scholar] [CrossRef]
- Zhou, K.; Yao, Y.-L.; He, Z.-C.; Chen, C.; Zhang, X.-N.; Yang, K.-D.; Liu, Y.-Q.; Liu, Q.; Fu, W.-J.; Chen, Y.-P.; et al. VDAC2 interacts with PFKP to regulate glucose metabolism and phenotypic reprogramming of glioma stem cells. Cell Death Dis. 2018, 9, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.-H.; Lin, Y.-W.; Wu, T.-F.; Ko, J.-L.; Wang, P.-H. Clinical implication of voltage-dependent anion channel 1 in uterine cervical cancer and its action on cervical cancer cells. Oncotarget 2016, 7, 4210–4225. [Google Scholar] [CrossRef]
- White, C. The Regulation of Tumor Cell Invasion and Metastasis by Endoplasmic Reticulum-to-Mitochondrial Ca2+ Transfer. Front. Oncol. 2017, 7, 171. [Google Scholar] [CrossRef]
- Arif, T.; Krelin, Y.; Nakdimon, I.; Benharroch, D.; Paul, A.; Dadon-Klein, D.; Shoshan-Barmatz, V. VDAC1 is a molecular target in glioblastoma, with its depletion leading to reprogrammed metabolism and reversed oncogenic properties. Neuro-Oncol. 2017, 19, 951–964. [Google Scholar] [CrossRef] [PubMed]
- Yuan, S.; Fu, Y.; Wang, X.; Shi, H.; Huang, Y.; Song, X.; Li, L.; Song, N.; Luo, Y. Voltage-dependent anion channel 1 is involved in endostatin-induced endothelial cell apoptosis. FASEB J. 2008, 22, 2809–2820. [Google Scholar] [CrossRef]
- Chin, H.S.; Li, M.X.; Tan, I.K.L.; Ninnis, R.L.; Reljic, B.; Scicluna, K.; Dagley, L.F.; Sandow, J.J.; Kelly, G.L.; Samson, A.L.; et al. VDAC2 enables BAX to mediate apoptosis and limit tumor development. Nat. Commun. 2018, 9, 4976. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Townsend, D.M.; Morris, M.; Maldonado, E.N.; Jiang, Y.-L.; Broome, A.-M.; Bethard, J.R.; Ball, L.E.; Tew, K.D. Voltage-Dependent Anion Channels Influence Cytotoxicity of ME-344, a Therapeutic Isoflavone. J. Pharmacol. Exp. Ther. 2020, 374, 308–318. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Luo, M.; Zhang, K.; Zhang, J.; Gao, T.; Connell, D.O.; Yao, F.; Mu, C.; Cai, B.; Shang, Y.; et al. Nedd4 ubiquitylates VDAC2/3 to suppress erastin-induced ferroptosis in melanoma. Nat. Commun. 2020, 11, 1–14. [Google Scholar] [CrossRef]
- Arif, T.; Paul, A.; Krelin, Y.; Shteinfer-Kuzmine, A.; Shoshan-Barmatz, V. Mitochondrial VDAC1 Silencing Leads to Metabolic Rewiring and the Reprogramming of Tumour Cells into Advanced Differentiated States. Cancers 2018, 10, 499. [Google Scholar] [CrossRef] [PubMed]
- Qi, H.; Ning, X.; Yu, C.; Ji, X.; Jin, Y.; McNutt, M.A.; Yin, Y. Succinylation-dependent mitochondrial translocation of PKM2 promotes cell survival in response to nutritional stress. Cell Death Dis. 2019, 10, 1–16. [Google Scholar] [CrossRef]
- Pathak, T.; Gueguinou, M.; Walter, V.; Delierneux, C.; Johnson, M.T.; Zhang, X.; Xin, P.; Yoast, R.; Emrich, S.M.; Yochum, G.S.; et al. Dichotomous role of the human mitochondrial Na+/Ca2+/Li+ exchanger NCLX in colorectal cancer growth and metastasis. eLife 2020, 9. [Google Scholar] [CrossRef]
- Marchi, S.; Vitto, V.A.M.; Patergnani, S.; Pinton, P. High mitochondrial Ca2+ content increases cancer cell proliferation upon inhibition of mitochondrial permeability transition pore (mPTP). Cell Cycle 2019, 18, 914–916. [Google Scholar] [CrossRef]
- Don, A.; Kisker, O.; Dilda, P.; Donoghue, N.; Zhao, X.; Decollogne, S.; Creighton, B.; Flynn, E.; Folkman, J.; Hogg, P.J. A peptide trivalent arsenical inhibits tumor angiogenesis by perturbing mitochondrial function in angiogenic endothelial cells. Cancer Cell 2003, 3, 497–509. [Google Scholar] [CrossRef]
- Ling, X.; Zhou, Y.; Li, S.-W.; Yan, B.; Wen, L. Modulation of mitochondrial permeability transition pore affects multidrug resistance in human hepatocellular carcinoma cells. Int. J. Biol. Sci. 2010, 6, 773–783. [Google Scholar] [CrossRef][Green Version]
- Kaddour-Djebbar, I.; Lakshmikanthan, V.; Shirley, R.B.; Ismail, K.-D.; Lewis, R.W.; Kumar, M.V. Therapeutic advantage of combining calcium channel blockers and TRAIL in prostate cancer. Mol. Cancer Ther. 2006, 5, 1958–1966. [Google Scholar] [CrossRef][Green Version]
- Takata, N.; Ohshima, Y.; Suzuki-Karasaki, M.; Yoshida, Y.; Tokuhashi, Y. Mitochondrial Ca2+ removal amplifies TRAIL cytotoxicity toward apoptosis-resistant tumor cells via promotion of multiple cell death modalities. Int. J. Oncol. 2017, 51, 193–203. [Google Scholar] [CrossRef]
- Pang, G.; Xie, Q.; Yao, J. Mitofusin 2 inhibits bladder cancer cell proliferation and invasion via the Wnt/β-catenin pathway. Oncol. Lett. 2019, 18, 2434–2442. [Google Scholar] [CrossRef] [PubMed]
- Starenki, D.; Sosonkina, N.; Hong, S.-K.; Lloyd, R.V.; Park, J.-I. Mortalin (GRP75/HSPA9) Promotes Survival and Proliferation of Thyroid Carcinoma Cells. Int. J. Mol. Sci. 2019, 20, 2069. [Google Scholar] [CrossRef]
- Fu, R.; Yang, P.; Wu, H.-L.; Li, Z.-W.; Li, Z.-Y. GRP78 secreted by colon cancer cells facilitates cell proliferation via PI3K/Akt signaling. Asian Pac. J. Cancer Prev. 2014, 15, 7245–7249. [Google Scholar] [CrossRef]
- Crottès, D.; Guizouarn, H.; Martin, P.; Borgese, F.; Soriani, O. The sigma-1 receptor: A regulator of cancer cell electrical plasticity? Front. Physiol. 2013, 4, 175. [Google Scholar] [CrossRef]
- Moriya, C.; Taniguchi, H.; Nagatoishi, S.; Igarashi, H.; Tsumoto, K.; Imai, K. PRDM14 directly interacts with heat shock proteins HSP90α and glucose-regulated protein 78. Cancer Sci. 2017, 109, 373–383. [Google Scholar] [CrossRef] [PubMed]
- Kabakov, A.; Yakimova, A.; Matchuk, O. Molecular Chaperones in Cancer Stem Cells: Determinants of Stemness and Potential Targets for Antitumor Therapy. Cells 2020, 9, 892. [Google Scholar] [CrossRef]
- Xu, D.; Yi, W.; Chen, Y.; Ma, L.; Wang, J.; Yu, G. Overexpression of Sig1R is closely associated with tumor progression and poor outcome in patients with hilar cholangiocarcinoma. Med Oncol. 2014, 31, 261. [Google Scholar] [CrossRef]
- Liu, X.; Feng, C.; Wei, G.; Kong, W.; Meng, H.; Du, Y.; Li, J. Mitofusin1 Is a Major Mediator in Glucose-Induced Epithelial-to-Mesenchymal Transition in Lung Adenocarcinoma Cells. OncoTargets Ther. 2020, 13, 3511–3523. [Google Scholar] [CrossRef]
- You, M.-H.; Jeon, M.J.; Kim, S.R.; Lee, W.K.; Cheng, S.-Y.; Jang, G.; Kim, T.Y.; Kim, W.B.; Shong, Y.K. Mitofusin-2 modulates the epithelial to mesenchymal transition in thyroid cancer progression. Sci. Rep. 2021, 11, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Na, Y.; Kaul, S.; Ryu, J.; Lee, J.-S.; Ahn, H.M.; Kaul, Z.; Kalra, R.S.; Li, L.; Widodo, N.; Yun, C.-O.; et al. Stress Chaperone Mortalin Contributes to Epithelial-to-Mesenchymal Transition and Cancer Metastasis. Cancer Res. 2016, 76, 2754–2765. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.-J.; Chen, W.-Y.; Huang, C.-Y.; Liu, H.-H.; Wei, P.-L. Glucose-regulated protein 78 (GRP78) regulates colon cancer metastasis through EMT biomarkers and the NRF-2/HO-1 pathway. Tumor Biol. 2014, 36, 1859–1869. [Google Scholar] [CrossRef] [PubMed]
- Gueguinou, M.; Crottès, D.; Chantôme, A.; Rapetti-Mauss, R.; Potier-Cartereau, M.; Clarysse, L.; Girault, A.; Fourbon, Y.; Jézéquel, P.; Guérin-Charbonnel, C.; et al. The SigmaR1 chaperone drives breast and colorectal cancer cell migration by tuning SK3-dependent Ca2+ homeostasis. Oncogene 2017, 36, 3640–3647. [Google Scholar] [CrossRef] [PubMed]
- Yoo, J.Y.; Ryu, J.; Gao, R.; Yaguchi, T.; Kaul, S.C.; Wadhwa, R.; Yun, C.-O. Tumor suppression by apoptotic and anti-angiogenic effects of mortalin-targeting adeno-oncolytic virus. J. Gene Med. 2010, 12, 586–595. [Google Scholar] [CrossRef]
- Dong, D.; Stapleton, C.; Luo, B.; Xiong, S.; Ye, W.; Zhang, Y.; Jhaveri, N.; Zhu, G.; Ye, R.; Liu, Z.; et al. A Critical Role for GRP78/BiP in the Tumor Microenvironment for Neovascularization during Tumor Growth and Metastasis. Cancer Res. 2011, 71, 2848–2857. [Google Scholar] [CrossRef]
- Crottès, D.; Rapetti-Mauss, R.; Pérez, F.A.; Tichet, M.; Gariano, G.; Martial, S.; Guizouarn, H.; Pellissier, B.; Loubat, A.; Popa, A.M.; et al. SIGMAR1 Regulates Membrane Electrical Activity in Response to Extracellular Matrix Stimulation to Drive Cancer Cell Invasiveness. Cancer Res. 2016, 76, 607–618. [Google Scholar] [CrossRef]
- Wang, W.; Liu, X.; Guo, X.; Quan, H. Mitofusin-2 Triggers Cervical Carcinoma Cell Hela Apoptosis via Mitochondrial Pathway in Mouse Model. Cell. Physiol. Biochem. 2018, 46, 69–81. [Google Scholar] [CrossRef]
- Choudhary, V.; Kaddour-Djebbar, I.; Alaisami, R.; Kumar, M.V.; Bollag, W.B. Mitofusin 1 degradation is induced by a disruptor of mitochondrial calcium homeostasis, CGP37157: A role in apoptosis in prostate cancer cells. Int. J. Oncol. 2014, 44, 1767–1773. [Google Scholar] [CrossRef]
- Guo, W.; Yan, L.; Yang, L.; Liu, X.; E, Q.; Gao, P.; Ye, X.; Liu, W.; Zuo, J. Targeting GRP75 Improves HSP90 Inhibitor Efficacy by Enhancing p53-Mediated Apoptosis in Hepatocellular Carcinoma. PLoS ONE 2014, 9, e85766. [Google Scholar] [CrossRef]
- Wu, J.; Wu, Y.; Lian, X. Targeted inhibition of GRP78 by HA15 promotes apoptosis of lung cancer cells accompanied by ER stress and autophagy. Biol. Open 2020, 9, bio053298. [Google Scholar] [CrossRef]
- Do, W.; Herrera, C.; Mighty, J.; Shumskaya, M.; Redenti, S.M.; Sauane, M. Sigma 1 Receptor plays a prominent role in IL-24-induced cancer-specific apoptosis. Biochem. Biophys. Res. Commun. 2013, 439, 215–220. [Google Scholar] [CrossRef]
- Li, M.; Wang, L.; Wang, Y.; Zhang, S.; Zhou, G.; Lieshout, R.; Ma, B.; Liu, J.; Qu, C.; Verstegen, M.M.A.; et al. Mitochondrial Fusion Via OPA1 and MFN1 Supports Liver Tumor Cell Metabolism and Growth. Cells 2020, 9, 121. [Google Scholar] [CrossRef]
- Li, T.; Han, J.; Jia, L.; Hu, X.; Chen, L.; Wang, Y. PKM2 coordinates glycolysis with mitochondrial fusion and oxidative phosphorylation. Protein Cell 2019, 10, 583–594. [Google Scholar] [CrossRef]
- Li, Z.; Wang, Y.; Newton, I.P.; Zhang, L.; Ji, P.; Li, Z. GRP78 is implicated in the modulation of tumor aerobic glycolysis by promoting autophagic degradation of IKKβ. Cell. Signal. 2015, 27, 1237–1245. [Google Scholar] [CrossRef] [PubMed]
- Maher, C.M.; Thomas, J.D.; Haas, D.; Longen, C.G.; Oyer, H.M.; Tong, J.; Kim, F.J. Small-Molecule Sigma1 Modulator Induces Autophagic Degradation of PD-L1. Mol. Cancer Res. 2017, 16, 243–255. [Google Scholar] [CrossRef]
- Liu, Y.-N.; Yang, J.-F.; Huang, D.-J.; Ni, H.-H.; Zhang, C.-X.; Zhang, L.; He, J.; Gu, J.-M.; Chen, H.-X.; Mai, H.-Q.; et al. Hypoxia Induces Mitochondrial Defect That Promotes T Cell Exhaustion in Tumor Microenvironment Through MYC-Regulated Pathways. Front. Immunol. 2020, 11. [Google Scholar] [CrossRef]
- Lee, A.S. GRP78 Induction in Cancer: Therapeutic and Prognostic Implications. Cancer Res. 2007, 67, 3496–3499. [Google Scholar] [CrossRef]
- Zhu, L.X.; Sharma, S.; Gardner, B.; Escuadro, B.; Atianzar, K.; Tashkin, D.P.; Dubinett, S.M. IL-10 Mediates Sigma1 Receptor-Dependent Suppression of Antitumor Immunity. J. Immunol. 2003, 170, 3585–3591. [Google Scholar] [CrossRef]
- Pilzer, D.; Saar, M.; Koya, K.; Fishelson, Z. Mortalin inhibitors sensitize K562 leukemia cells to complement-dependent cytotoxicity. Int. J. Cancer 2009, 126, 1428–1435. [Google Scholar] [CrossRef]
- Faris, P.; Pellavio, G.; Ferulli, F.; Di Nezza, F.; Shekha, M.; Lim, D.; Maestri, M.; Guerra, G.; Ambrosone, L.; Pedrazzoli, P.; et al. Nicotinic acid adenine dinucleotide phosphate (NAADP) induces intracellular Ca2+ release through the two-pore channel TPC1 in metastatic colorectal cancer cells. Cancers 2019, 11, 542. [Google Scholar] [CrossRef]
- Müller, M.; Geisslinger, F.; Gerndt, S.; Schütz, R.; Chao, Y.-K.; Bracher, F.; Grimm, C.; Vollmar, A.; Bartel, K. Blocking Lysosomal Two-Pore Channel 2 Function Inhibits Proliferation of Multidrug Resistant Leukemia Cells and Sensitizes Them to Vincristine Treatment. Blood 2019, 134 (Suppl. 1), 2081. [Google Scholar] [CrossRef]
- Santoni, G.; Santoni, M.; Maggi, F.; Marinelli, O.; Morelli, M.B. Emerging Role of Mucolipins TRPML Channels in Cancer. Front. Oncol. 2020, 10. [Google Scholar] [CrossRef]
- Xu, M.; Almasi, S.; Yang, Y.; Yan, C.; Sterea, A.M.; Syeda, A.K.R.; Shen, B.; Derek, C.R.; Huang, P.; Gujar, S.; et al. The lysosomal TRPML1 channel regulates triple negative breast cancer development by promoting mTORC1 and purinergic signaling pathways. Cell Calcium 2019, 79, 80–88. [Google Scholar] [CrossRef]
- Pafumi, I.; Festa, M.; Papacci, F.; Lagostena, L.; Giunta, C.; Gutla, V.; Cornara, L.; Favia, A.; Palombi, F.; Gambale, F.; et al. Naringenin Impairs Two-Pore Channel 2 Activity And Inhibits VEGF-Induced Angiogenesis. Sci. Rep. 2017, 7, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Tang, T.; Yang, Z.-Y.; Wang, D.; Yang, X.-Y.; Wang, J.; Li, L.; Wen, Q.; Gao, L.; Bian, X.-W.; Yu, S.-C. The role of lysosomes in cancer development and progression. Cell Biosci. 2020, 10, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.; Yue, J. TPC2 mediates autophagy progression and extracellular vesicle secretion in cancer cells. Exp. Cell Res. 2018, 370, 478–489. [Google Scholar] [CrossRef]
- Almasi, S.; Kennedy, B.E.; Yoast, R.E.; Emrich, S.M.; Trebak, M.; Hiani, Y.E. The lysosomal TRPML1 channel promotes breast cancer survival by supporting mitochondrial function and cellular metabolism. BioRxiv 2020. [Google Scholar] [CrossRef]
- Xu, M.; Dong, X.-P. Endolysosomal TRPMLs in Cancer. Biomol. 2021, 11, 65. [Google Scholar] [CrossRef] [PubMed]
- Avnet, S.; Di Pompo, G.; Lemma, S.; Salerno, M.; Perut, F.; Bonuccelli, G.; Granchi, D.; Zini, N.; Baldini, N. V-ATPase is a candidate therapeutic target for Ewing sarcoma. Biochim. Biophys. Acta (BBA)—Mol. Basis Dis. 2013, 1832, 1105–1116. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Flinck, M.; Hagelund, S.; Gorbatenko, A.; Severin, M.; Pedraz-Cuesta, E.; Novak, I.; Stock, C.; Pedersen, S.F. The Vacuolar H+ ATPase α3 Subunit Negatively Regulates Migration and Invasion of Human Pancreatic Ductal Adenocarcinoma Cells. Cells 2020, 9, 465. [Google Scholar] [CrossRef] [PubMed]
- Di Cristofori, A.; Ferrero, S.; Bertolini, I.; Gaudioso, G.; Russo, M.V.; Berno, V.; Vanini, M.; Locatelli, M.; Zavanone, M.; Rampini, P.; et al. The vacuolar H+ ATPase is a novel therapeutic target for glioblastoma. Oncotarget 2015, 6, 17514–17531. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Chen, H.; Han, L.; Zou, X.; Shen, W. Expression and role of V1A subunit of V-ATPases in gastric cancer cells. Int. J. Clin. Oncol. 2015, 20, 725–735. [Google Scholar] [CrossRef] [PubMed]
- Sennoune, S.R.; E Bermudez, L.; Lees, J.C.; Hirsch, J.; Filleur, S.; Martínez-Zaguilán, R. Vacuolar H+-ATPase is down-regulated by the angiogenesis-inhibitory pigment epithelium-derived factor in metastatic prostate cancer cells. Cell. Mol. Boil. 2014, 60, 45–52. [Google Scholar]
- Lu, Q.; Lu, S.; Huang, L.; Wang, T.; Wan, Y.; Zhou, C.X.; Zhang, C.; Zhang, Z.; Li, X. The expression of V-ATPase is associated with drug resistance and pathology of non-small-cell lung cancer. Diagn. Pathol. 2013, 8, 145. [Google Scholar] [CrossRef]
- Stransky, L.; Cotter, K.; Forgac, M. The Function of V-ATPases in Cancer. Physiol. Rev. 2016, 96, 1071–1091. [Google Scholar] [CrossRef] [PubMed]
- Katara, G.K.; Jaiswal, M.K.; Kulshrestha, A.; Kolli, B.; Gilman-Sachs, A.; Beaman, K.D. Tumor-associated vacuolar ATPase subunit promotes tumorigenic characteristics in macrophages. Oncogene 2013, 33, 5649–5654. [Google Scholar] [CrossRef]
- Cui, C.; Yang, J.; Fu, L.; Wang, M.; Wang, X. Progress in understanding mitochondrial calcium uniporter complex-mediated calcium signalling: A potential target for cancer treatment. Br. J. Pharmacol. 2019, 176, 1190–1205. [Google Scholar] [CrossRef]
- Szabo, I.; Zoratti, M.; Biasutto, L. Targeting mitochondrial ion channels for cancer therapy. Redox Biol. 2021, 42, 101846. [Google Scholar] [CrossRef]
- Parrasia, S.; Mattarei, A.; Furlan, A.; Zoratti, M.; Biasutto, L. Small-Molecule Modulators of Mitochondrial Channels as Chemotherapeutic Agents. Cell. Physiol. Biochem. 2019, 53, 11–43. [Google Scholar] [CrossRef]
- Esuh, D.H.; Ekim, M.-K.; Ekim, H.S.; Echung, H.H.; Esong, Y.S. Mitochondrial permeability transition pore as a selective target for anti-cancer therapy. Front. Oncol. 2013, 3, 41. [Google Scholar] [CrossRef]
- Via, L.; Argaez, A.N.G.; Martínez-Vázquez, M.; Grancara, S.; Martinis, P.; Toninello, A. Mitochondrial Permeability Transition as Target of Anticancer Drugs. Curr. Pharm. Des. 2014, 20, 223–244. [Google Scholar] [CrossRef]
- Olivas-Aguirre, M.; Torres-López, L.; Valle-Reyes, J.S.; Hernández-Cruz, A.; Pottosin, I.; Dobrovinskaya, O. Cannabidiol directly targets mitochondria and disturbs calcium homeostasis in acute lymphoblastic leukemia. Cell Death Dis. 2019, 10, 779. [Google Scholar] [CrossRef]
- Ruiz, A.; Alberdi, E.; Matute, C. CGP37157, an inhibitor of the mitochondrial Na+/Ca2+ exchanger, protects neurons from excitotoxicity by blocking voltage-gated Ca2+ channels. Cell Death Dis. 2014, 5, e1156. [Google Scholar] [CrossRef] [PubMed]
- Mashayekhi, M.; Zhou, D.H. The advance of anti-cancer flexible heteroarotinoids compounds. J. Biochem. Mol. Biol. Res. 2020, 5, 230–241. [Google Scholar] [CrossRef]
- Yun, C.W.; Kim, H.J.; Lim, J.H.; Lee, S.H. Heat Shock Proteins: Agents of Cancer Development and Therapeutic Targets in Anti-Cancer Therapy. Cells 2019, 9, 60. [Google Scholar] [CrossRef]
- Bailly, C.; Waring, M.J. Pharmacological effectors of GRP78 chaperone in cancers. Biochem. Pharmacol. 2019, 163, 269–278. [Google Scholar] [CrossRef]
- Oyer, H.M.; Sanders, C.M.; Kim, F.J. Small-Molecule Modulators of Sigma1 and Sigma2/TMEM97 in the Context of Cancer: Foundational Concepts and Emerging Themes. Front. Pharmacol. 2019, 10, 1141. [Google Scholar] [CrossRef]
- Georgiadis, M.-O.; Karoutzou, O.; Foscolos, A.-S.; Papanastasiou, I. Sigma Receptor (σR) Ligands with Antiproliferative and Anticancer Activity. Mol. 2017, 22, 1408. [Google Scholar] [CrossRef]
- Alharbi, A.F.; Parrington, J. Endolysosomal Ca2+ Signaling in Cancer: The Role of TPC2, From Tumorigenesis to Metastasis. Front. Cell Dev. Biol. 2019, 7, 302. [Google Scholar] [CrossRef]
- Jin, X.; Zhang, Y.; Alharbi, A.; Hanbashi, A.; Alhoshani, A.; Parrington, J.; Alharbi, A.F. Targeting Two-Pore Channels: Current Progress and Future Challenges. Trends Pharmacol. Sci. 2020, 41, 582–594. [Google Scholar] [CrossRef]
- Huang, T.; Sun, Y.; Li, Y.; Wang, T.; Fu, Y.; Li, C.; Li, C. Growth Inhibition of a Novel Iron Chelator, DpdtC, against Hepatoma Carcinoma Cell Lines Partly Attributed to Ferritinophagy-Mediated Lysosomal ROS Generation. Oxidative Med. Cell. Longev. 2018, 2018, 1–13. [Google Scholar] [CrossRef]
- Zhang, X.; Chen, W.; Gao, Q.; Yang, J.; Yan, X.; Zhao, H.; Su, L.; Yang, M.; Gao, C.; Yao, Y.; et al. Rapamycin directly activates lysosomal mucolipin TRP channels independent of mTOR. PLoS Biol. 2019, 17, e3000252. [Google Scholar] [CrossRef]
- Chen, D.; Xie, J.; Fiskesund, R.; Dong, W.; Liang, X.; Lv, J.; Jin, X.; Liu, J.; Mo, S.; Zhang, T.; et al. Chloroquine modulates antitumor immune response by resetting tumor-associated macrophages toward M1 phenotype. Nat. Commun. 2018, 9, 873. [Google Scholar] [CrossRef]
- Pérez-Sayáns, M.; Martín, J.M.S.; Barros-Angueira, F.; Rey, J.M.G.; García-García, A. V-ATPase inhibitors and implication in cancer treatment. Cancer Treat. Rev. 2009, 35, 707–713. [Google Scholar] [CrossRef]
- Patil, R.; Powrozek, O.; Kumar, B.; Seibel, W.; Beaman, K.; Waris, G.; Sharma-Walia, N.; Patil, S. Bisbenzimidazoles: Anticancer vacuolar (H+)-atpase inhibitors. Chem. Appl. Benzimidazole Its Deriv. 2019. [Google Scholar] [CrossRef]










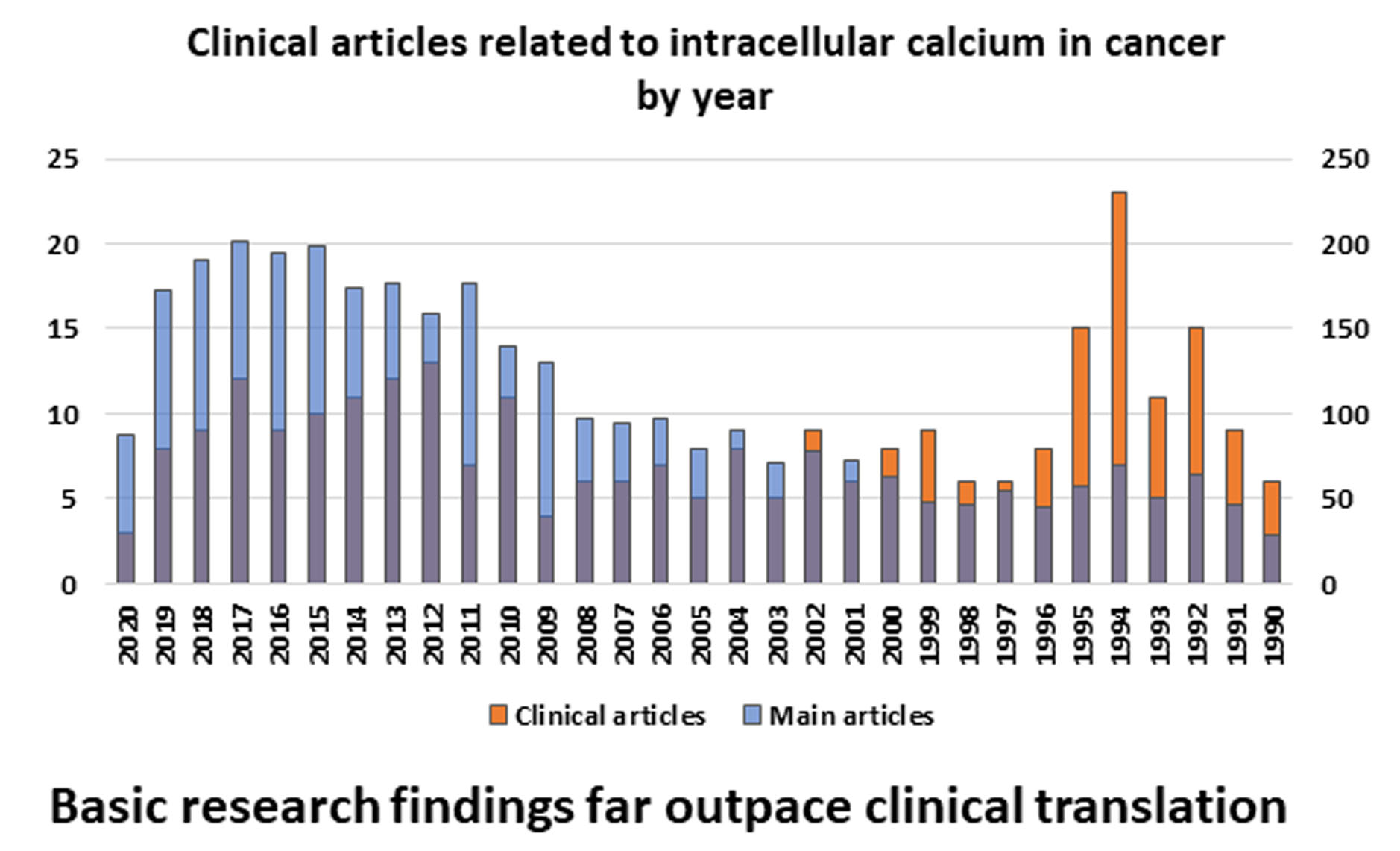{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
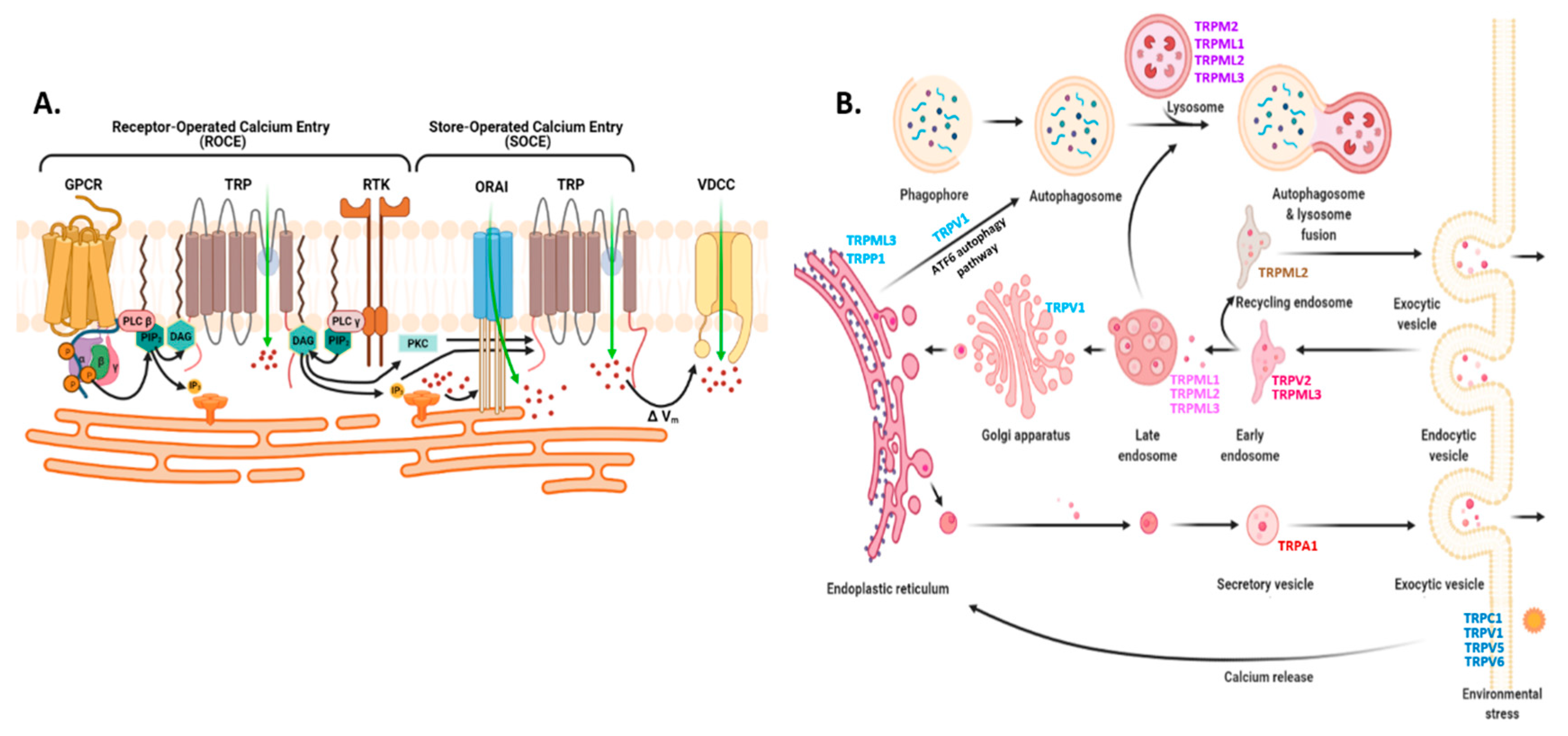{kind=link}
{kind=link}
{kind=link}
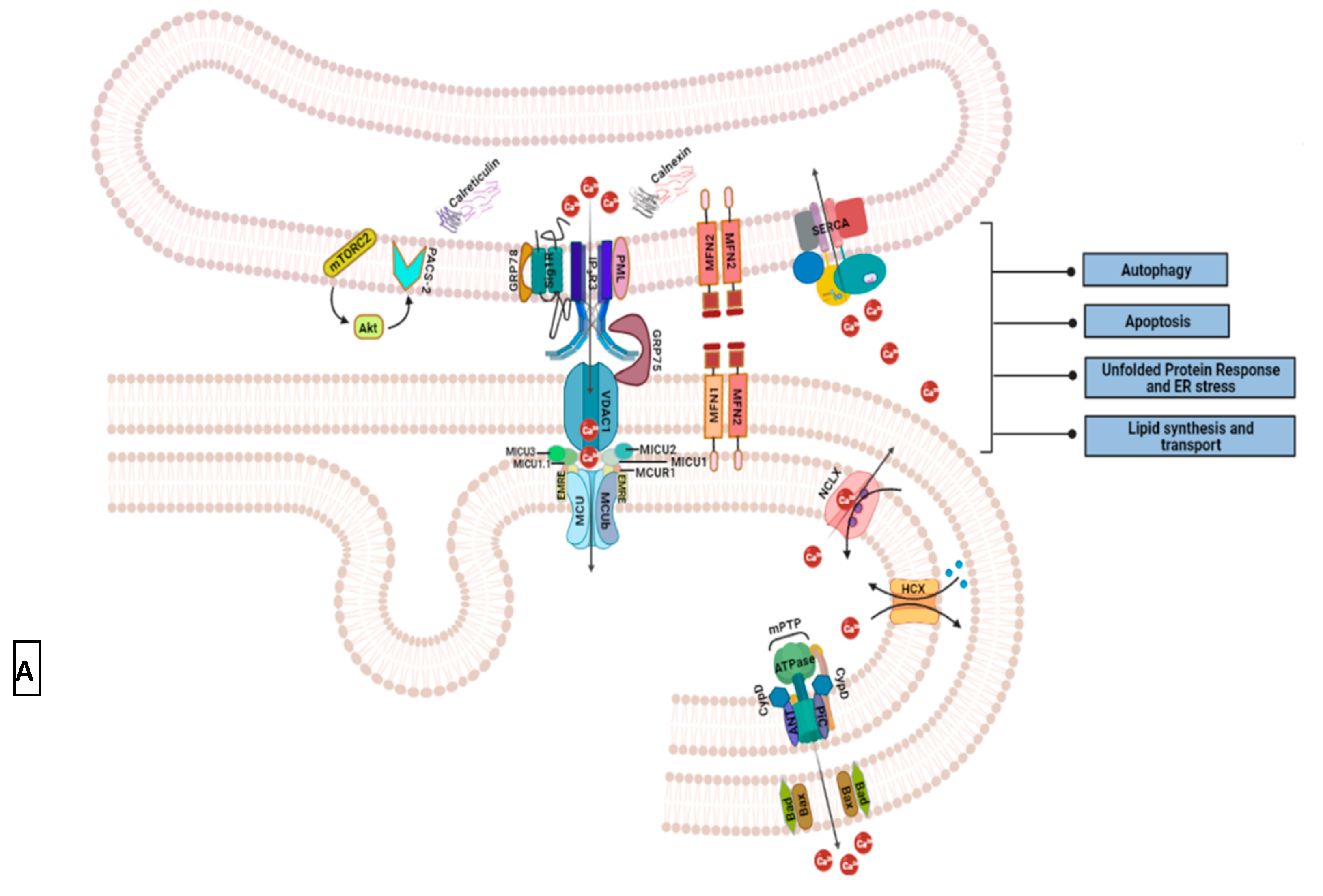{kind=link}
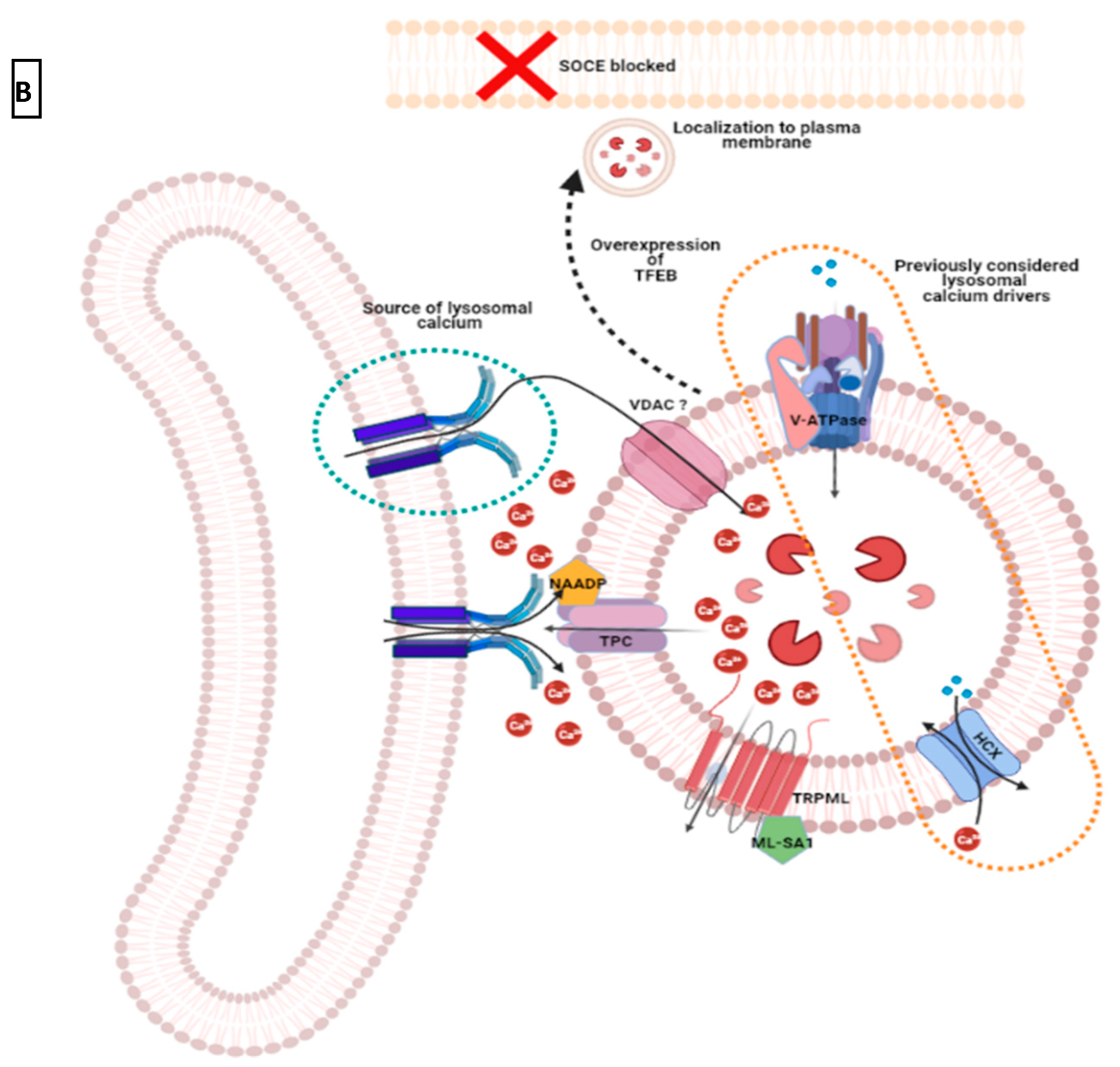{kind=link}
{kind=link}
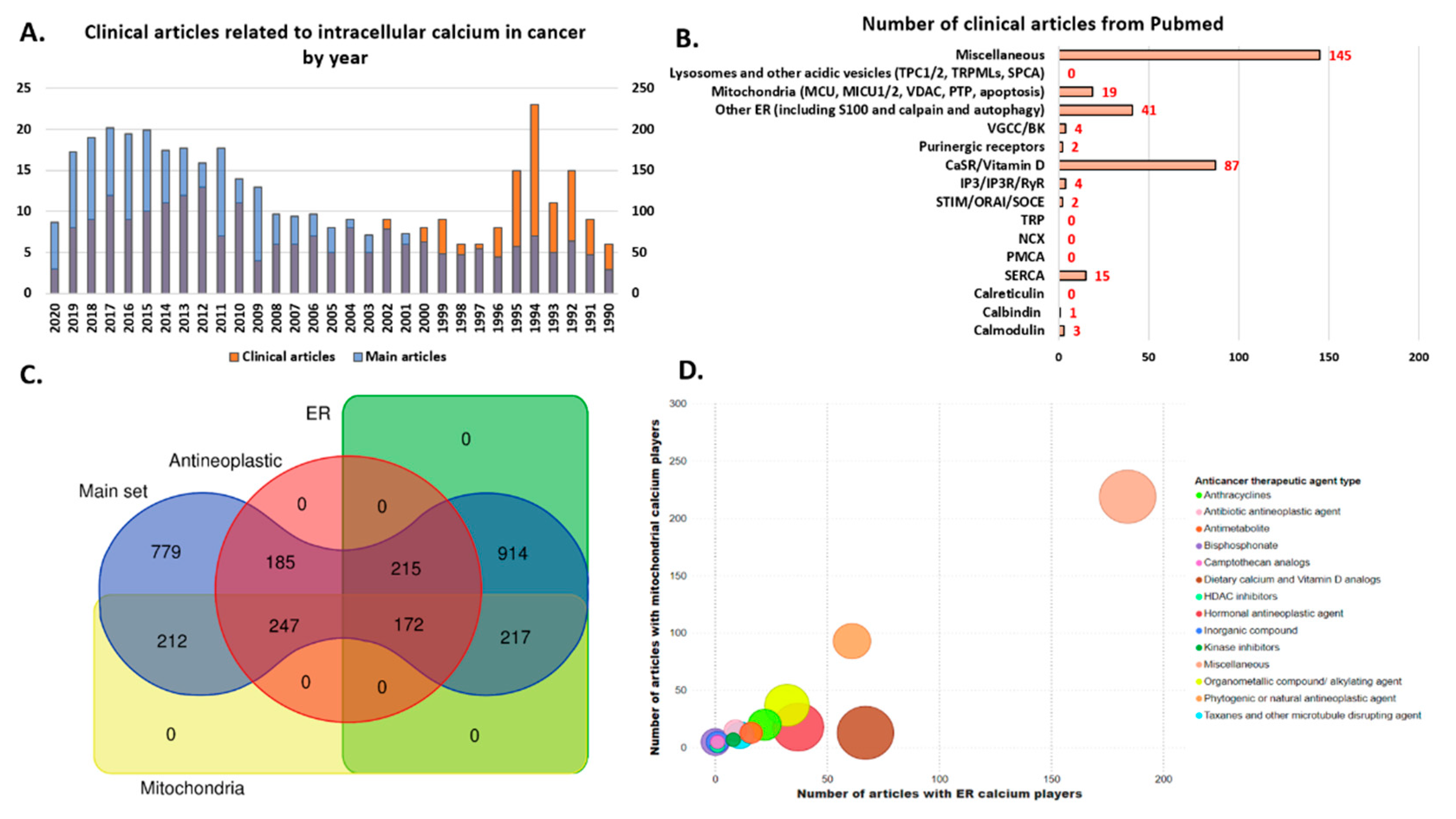{kind=link}
{kind=link}
{kind=link}
| SERCA Isoform | Tissue Localization | Inhibitor(s) |
|---|---|---|
| SERCA1 | Adult and neonatal skeletal muscles | Myoregulin (MLN), PLN, and SLN |
| SERCA2a | Cardiac muscles | PLN, SLN, miRNA-25 |
| SERCA2b | Non-muscle tissues | ALN, TM11 and its luminal extension |
| SERCA3 | Co-expressed with SERCA2b in endothelial, epithelial, and hematopoietic cells. | Endoregulin (ELN) |
| TRP Isoform | Protein Interactors |
|---|---|
| TRP A | PIP2, AKAP 79/150, Ca2+-CaM, TRPV1, Sig-1R |
| TRP C | CaBP1, CaM, Caveolin1, Homer, Immunophilins, IP3Rs, Junctate, Junctophilin, NCX1, PLCγ1 RhoA, Stathmin, VAMP, ZO-1, actin cytoskeleton (myosin, ERM proteins, NHERF, etc.), focal adhesion kinase, contactin, Src, STIM, ORAI, RyR, TRPV4, TRPP1, TRPM4 |
| TRP M | 14-3-3γ, 5HT1B, AKAP5/150, CaM, PKCα, PTPL1, Rac1, S100A10, Sig1R, TRPC3, RACK1, ENAC, Synaptotagmin1, α-actin, Myosin heavy chain, Annexin 1, Gαq |
| TRP ML | PDCD6, Cdc42, HSP40, HSP90, Rho1, Rac1, Rac2, RhoG, TPC1, TPC2, TRPV5 |
| TRP P | EGFR, eIF-2α, Filamin-A, HDAC6, IP3R1, IP3R3, PERK, α-actinin, PKD1, PKD1L1, RACK1, PLCγ, Troponin I, Tropomyosin, TRPC (1–4), TRPV4, RyR2, |
| TRP V | AKAP5/150, Calbindin D-28k, CaM, Caveolin1, Cyclophilin B, CAMKII, E-cadherin, EGFR, e-NOS, GABARAP, Fyn, F-actin, IP3R3, Klotho, Lck, Lyn, Myosin, α-integrin, α-tubulin, PKC, PPARα, S100A10, Src, TMEM16A, TRPA1, TRPC1, TRPML3, TRPP1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sharma, A.; Ramena, G.T.; Elble, R.C. Advances in Intracellular Calcium Signaling Reveal Untapped Targets for Cancer Therapy. Biomedicines 2021, 9, 1077. https://doi.org/10.3390/biomedicines9091077
Sharma A, Ramena GT, Elble RC. Advances in Intracellular Calcium Signaling Reveal Untapped Targets for Cancer Therapy. Biomedicines. 2021; 9(9):1077. https://doi.org/10.3390/biomedicines9091077
Chicago/Turabian StyleSharma, Aarushi, Grace T. Ramena, and Randolph C. Elble. 2021. "Advances in Intracellular Calcium Signaling Reveal Untapped Targets for Cancer Therapy" Biomedicines 9, no. 9: 1077. https://doi.org/10.3390/biomedicines9091077
APA StyleSharma, A., Ramena, G. T., & Elble, R. C. (2021). Advances in Intracellular Calcium Signaling Reveal Untapped Targets for Cancer Therapy. Biomedicines, 9(9), 1077. https://doi.org/10.3390/biomedicines9091077