
HMP-S7 Is a Novel Anti-Leukemic Peptide Discovered from Human Milk

 ,
,  , , , and
, , , and 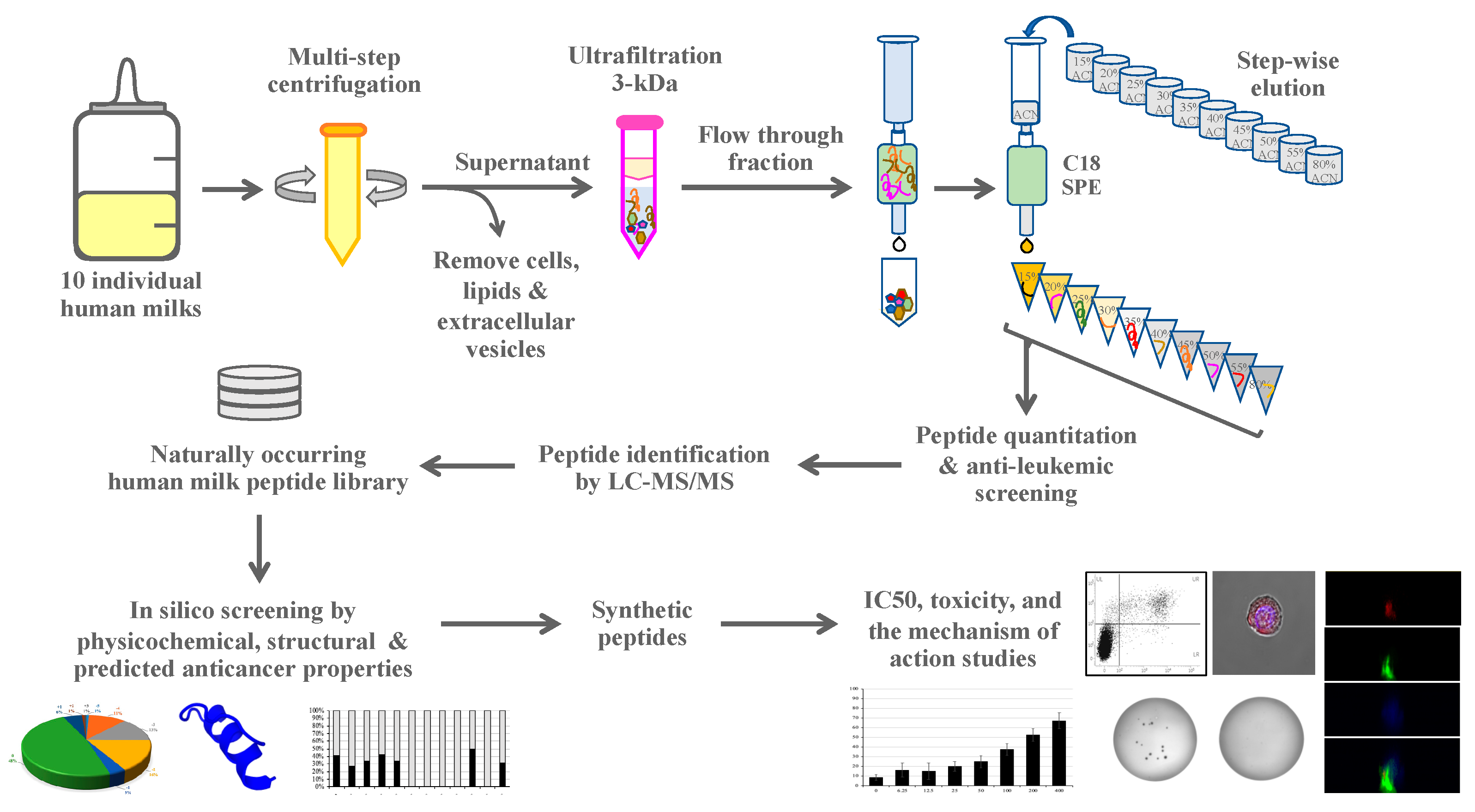{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
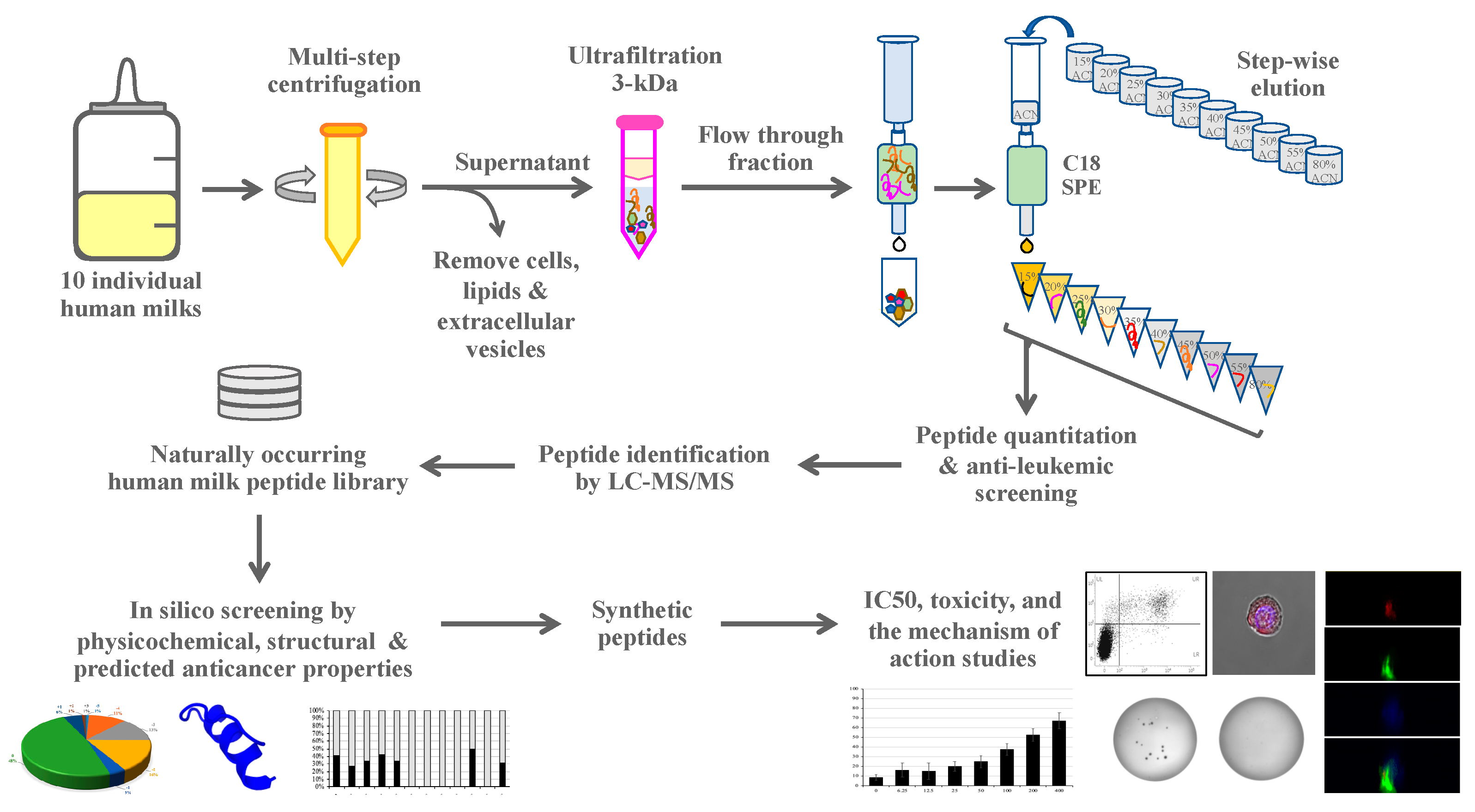
2.1. Experimental Design
2.2. Human Milk Collection
2.3. Peptide Isolation and Fractionation
2.4. Synthetic Peptides
2.5. Cell Culture
2.6. Human Milk Peptide Identification by Mass Spectrometry
2.7. In Silico Anticancer Peptide Screening
2.8. Cytotoxicity Assay
2.9. Half-Maximal Inhibitory Concentration (IC50) by Trypan Blue Assay
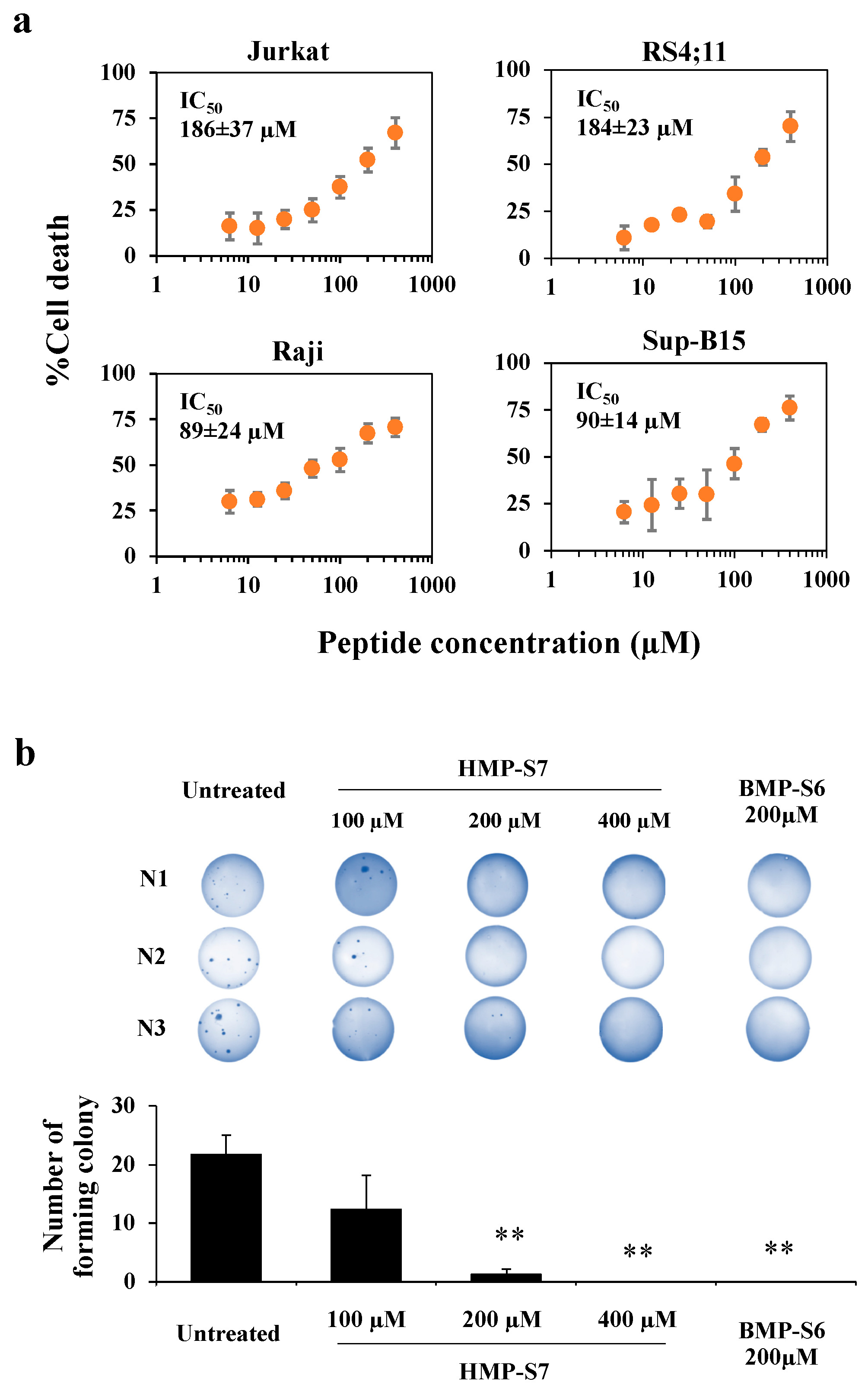
2.10. Soft Agar Assay for Colony Formation
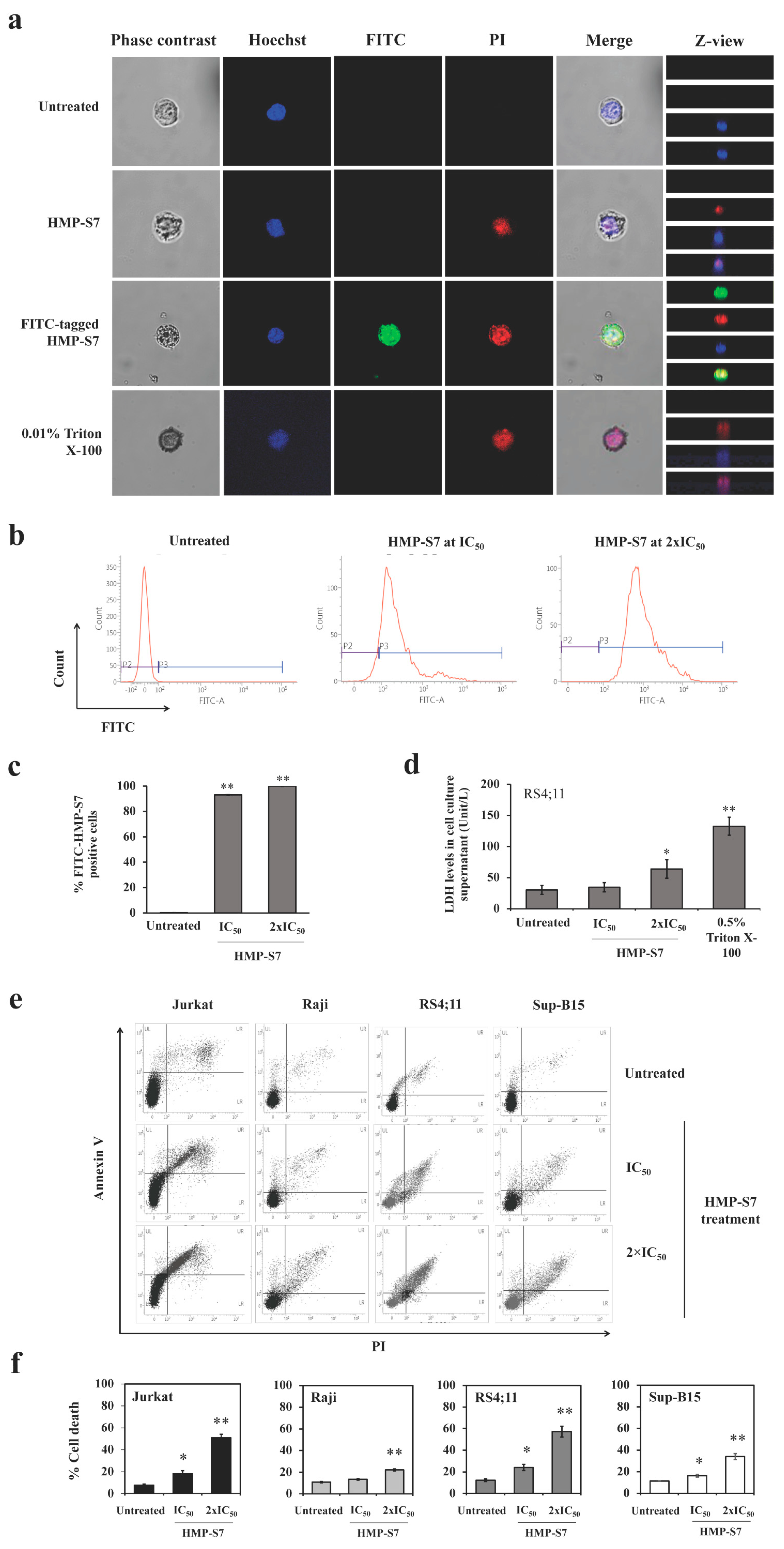
2.11. Flow Cytometric Cell Death Assay
2.12. Lactate Dehydrogenase (LDH) Release Assay
2.13. FITC Tagged HMP-S7 Internalized into Leukemic Cells
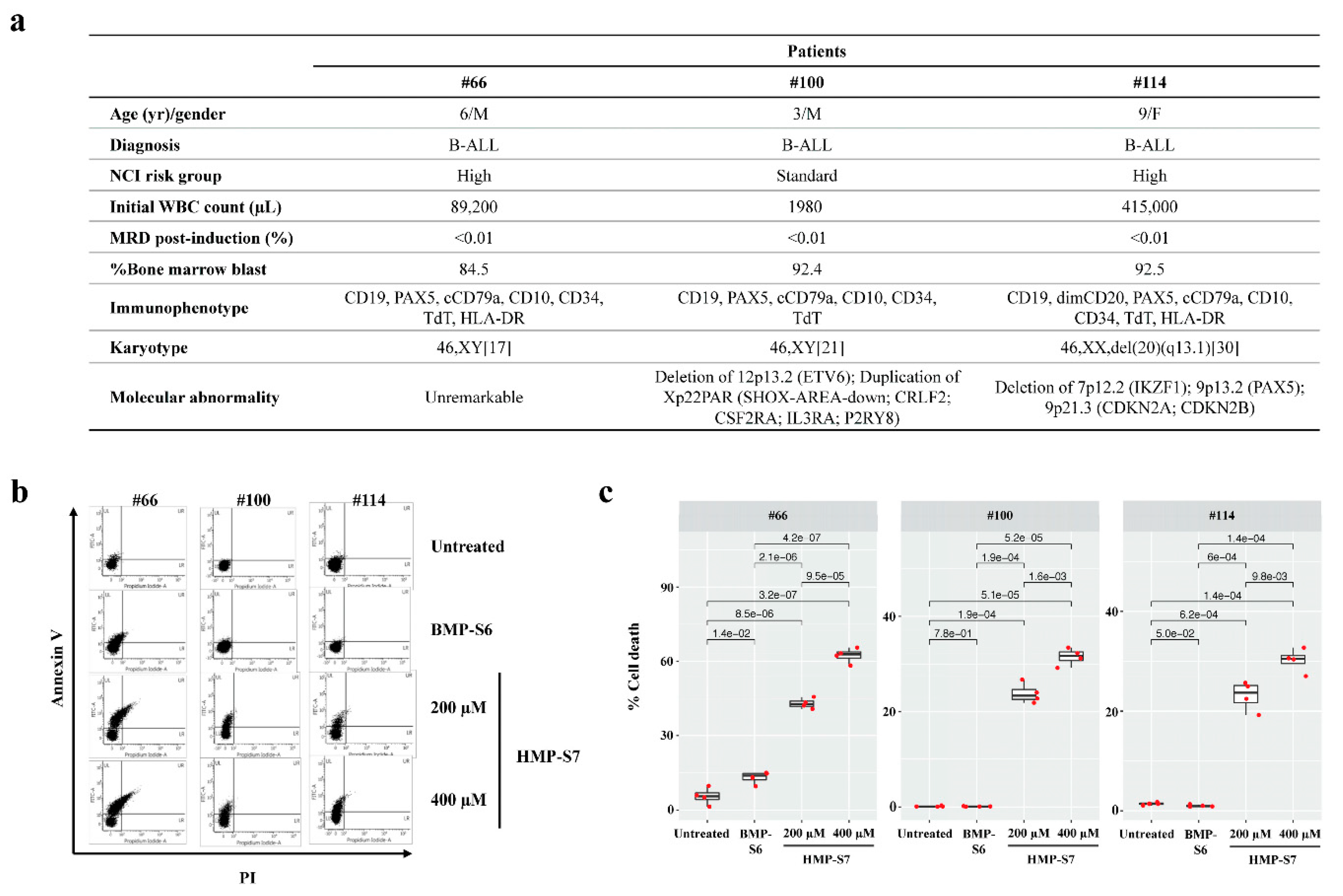
2.14. Ex Vivo Leukemic Cytotoxicity Study
2.15. Statistical Analysis
3. Results
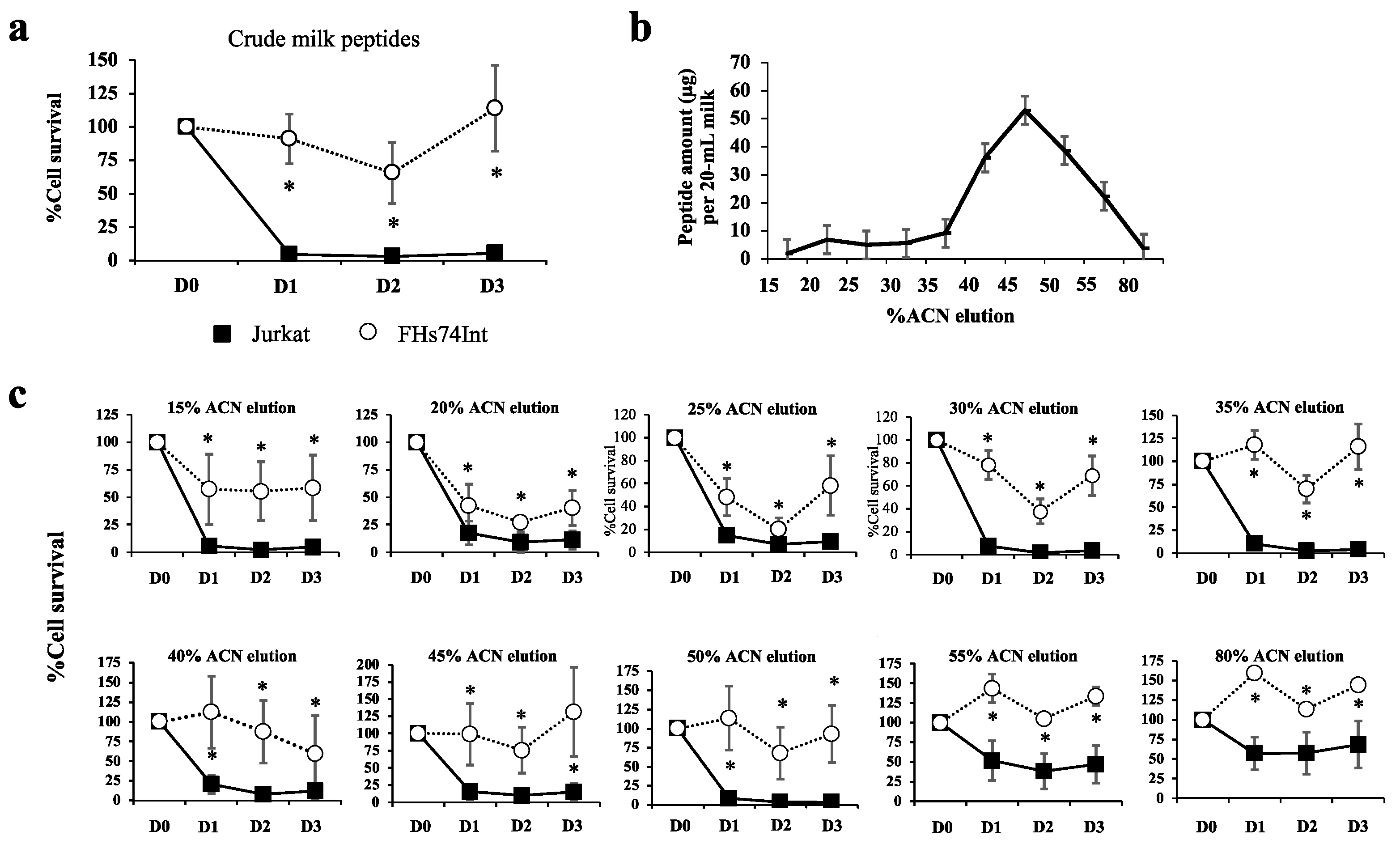
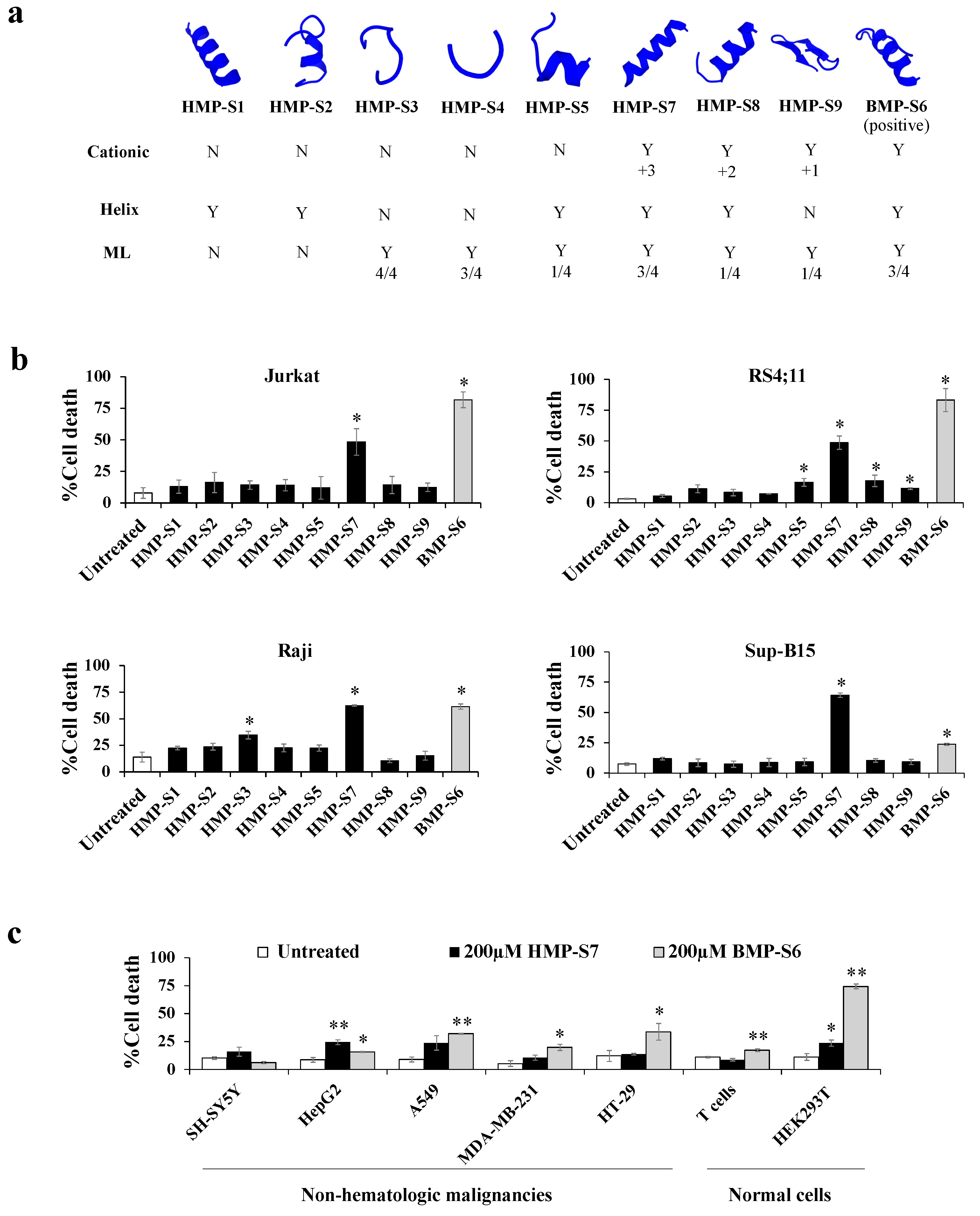
3.1. Most Human Milk Peptide Fractions Had Cytotoxic Effects on Leukemic and Normal Cell Lines
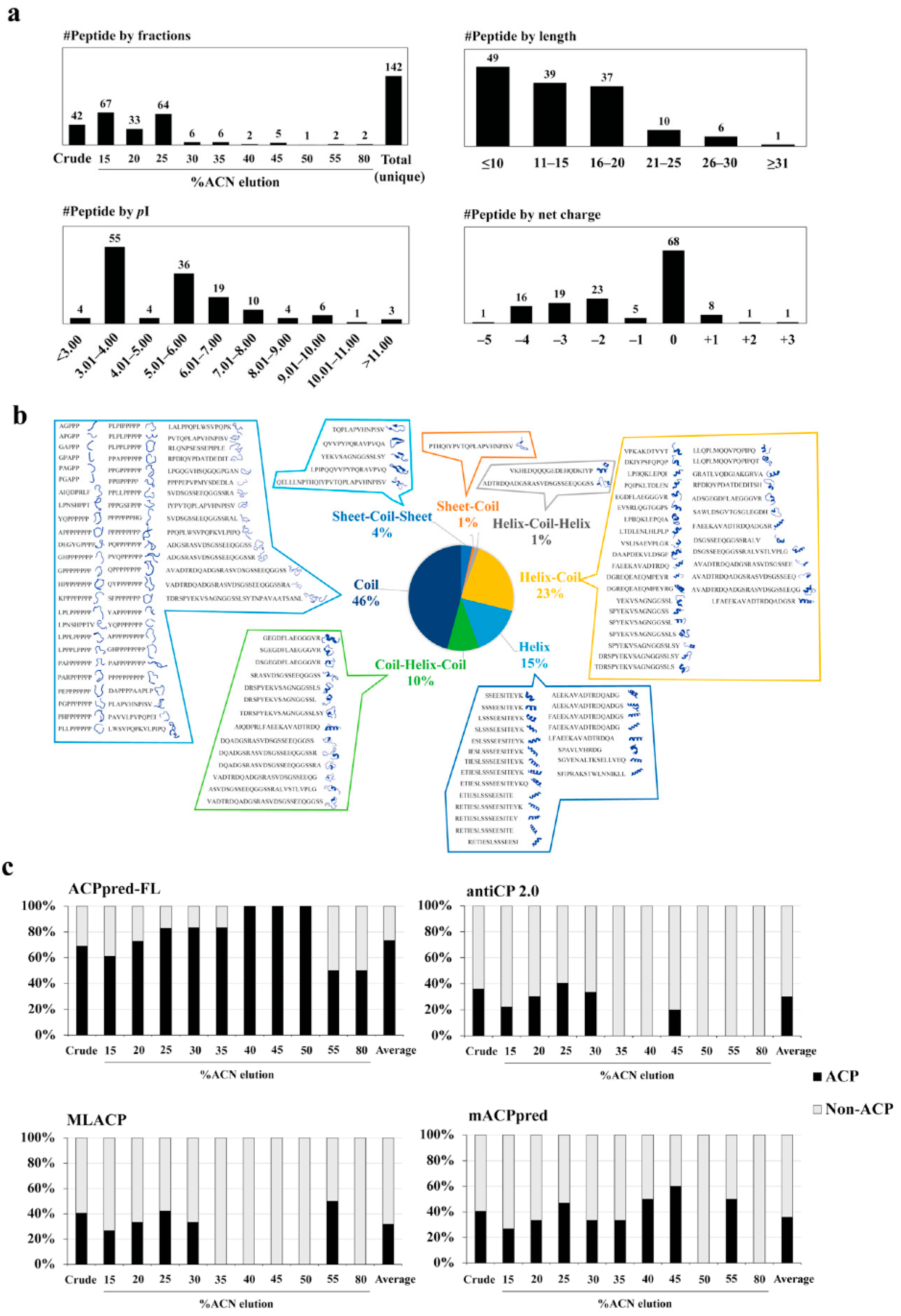
3.2. Peptide Identification, Library Construction and In Silico Anticancer Peptide Screening
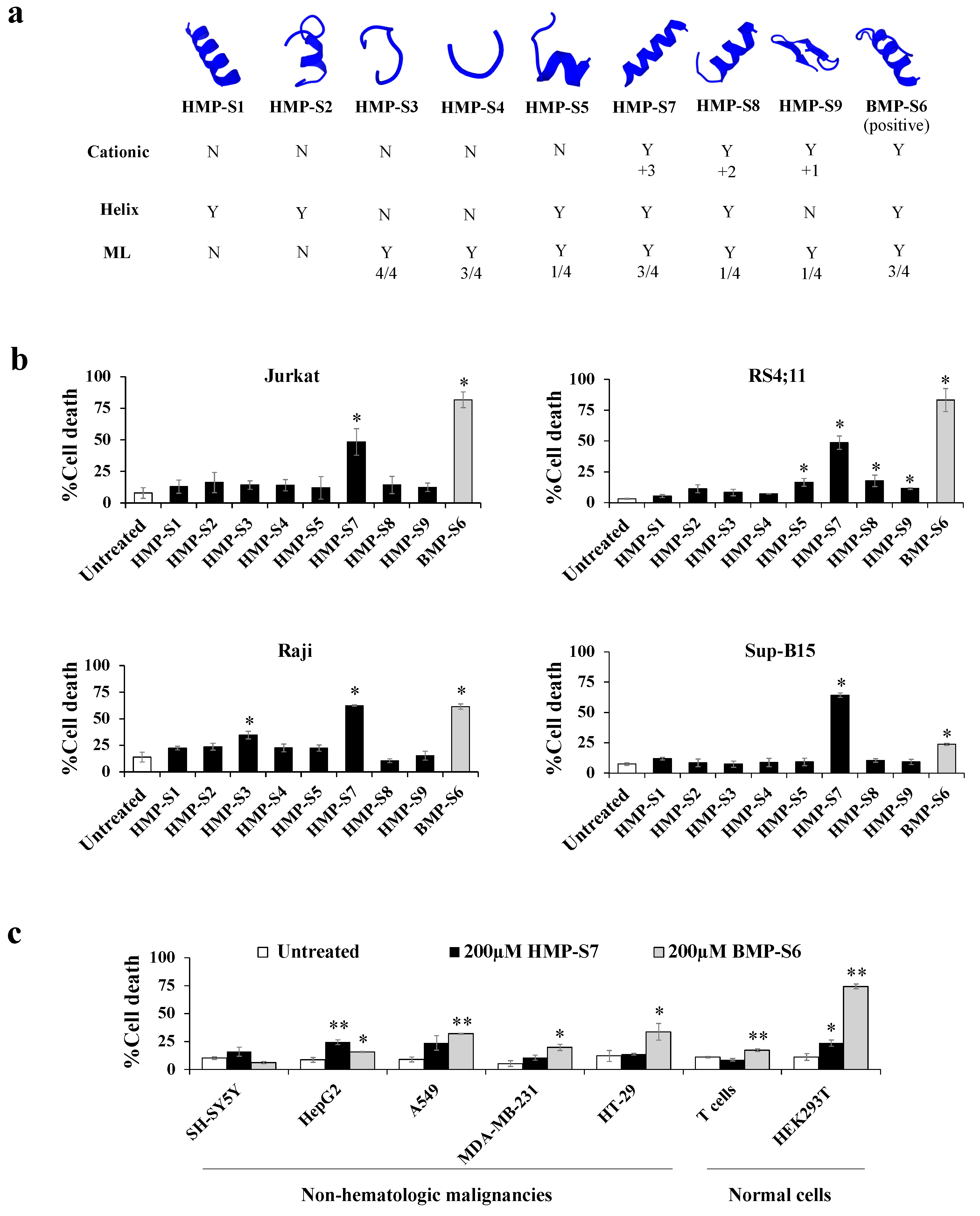
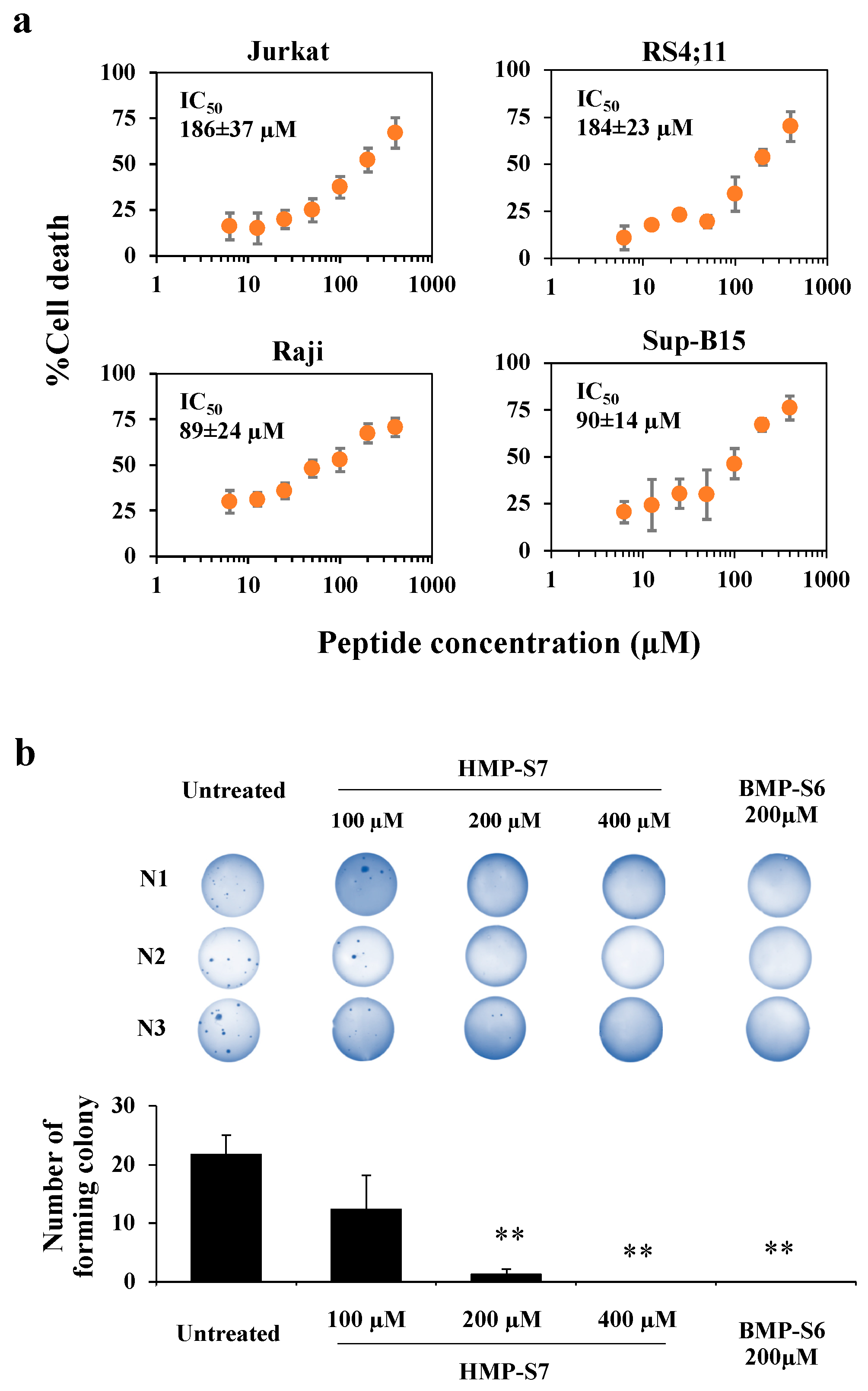
3.3. HMP-S7 Killed Leukemic Cells but Not Normal Cells or Solid Tumor Cell Lines
3.4. Mechanisms of HMP-S7-Induced Leukemic Cell Death
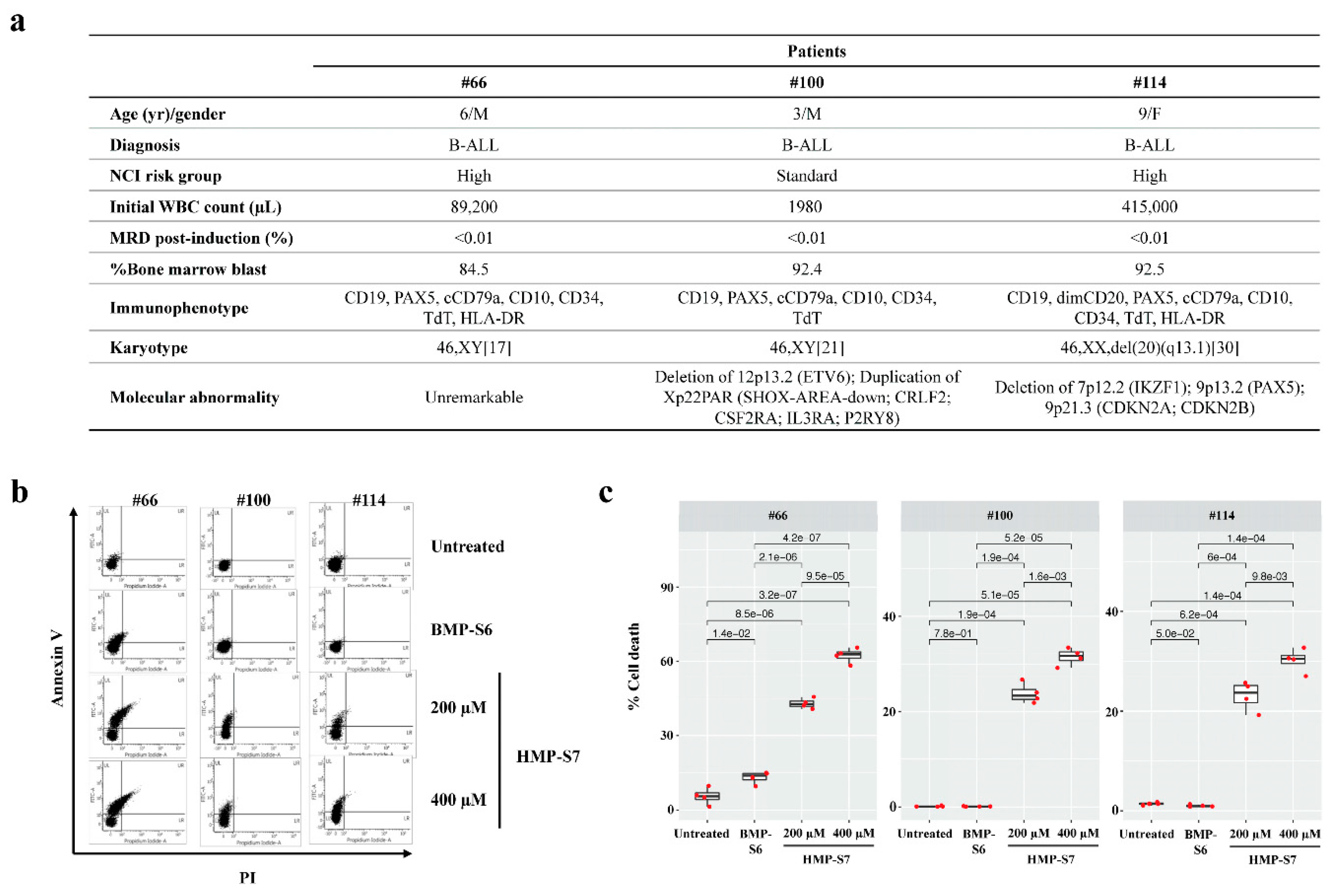
3.5. Ex Vivo Leukemic Cytotoxicity of HMP-S7
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Torre, L.A.; Siegel, R.L.; Ward, E.M.; Jemal, A. Global Cancer Incidence and Mortality Rates and Trends—An Update. Cancer Epidemiol. Biomark. Prev. 2016, 25, 16–27. [Google Scholar] [CrossRef] [Green Version]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2016. CA Cancer J. Clin. 2016, 66, 7–30. [Google Scholar] [CrossRef] [Green Version]
- Kantarjian, H.M.; Keating, M.J.; Freireich, E.J. Toward the potential cure of leukemias in the next decade. Cancer 2018, 124, 4301–4313. [Google Scholar] [CrossRef] [Green Version]
- Robison, L.L.; Bhatia, S. Late-effects among survivors of leukaemia and lymphoma during childhood and adolescence. Br. J. Haematol. 2003, 122, 345–359. [Google Scholar] [CrossRef] [PubMed]
- Mulrooney, D.A.; Hyun, G.; Ness, K.K.; Bhakta, N.; Pui, C.H.; Ehrhardt, M.J.; Krull, K.R.; Crom, D.B.; Chemaitilly, W.; Srivastava, D.K.; et al. The changing burden of long-term health outcomes in survivors of childhood acute lymphoblastic leukaemia: A retrospective analysis of the St Jude Lifetime Cohort Study. Lancet Haematol. 2019, 6, e306–e316. [Google Scholar] [CrossRef]
- Bhojwani, D.; Howard, S.C.; Pui, C.H. High-risk childhood acute lymphoblastic leukemia. Clin. Lymphoma Myeloma 2009, 9, S222–S230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harris, F.; Dennison, S.R.; Singh, J.; Phoenix, D.A. On the selectivity and efficacy of defense peptides with respect to cancer cells. Med. Res. Rev. 2013, 33, 190–234. [Google Scholar] [CrossRef]
- Papo, N.; Shai, Y. Host defense peptides as new weapons in cancer treatment. Cell. Mol. Life Sci. 2005, 62, 784–790. [Google Scholar] [CrossRef]
- Utsugi, T.; Schroit, A.J.; Connor, J.; Bucana, C.D.; Fidler, I.J. Elevated expression of phosphatidylserine in the outer membrane leaflet of human tumor cells and recognition by activated human blood monocytes. Cancer Res. 1991, 51, 3062–3066. [Google Scholar]
- Kozlowska, K.; Nowak, J.; Kwiatkowski, B.; Cichorek, M. ESR study of plasmatic membrane of the transplantable melanoma cells in relation to their biological properties. Exp. Toxicol. Pathol. 1999, 51, 89–92. [Google Scholar] [CrossRef]
- Sok, M.; Sentjurc, M.; Schara, M. Membrane fluidity characteristics of human lung cancer. Cancer Lett. 1999, 139, 215–220. [Google Scholar] [CrossRef]
- Schweizer, F. Cationic amphiphilic peptides with cancer-selective toxicity. Eur. J. Pharmacol. 2009, 625, 190–194. [Google Scholar] [CrossRef]
- Murota, I.; Taguchi, S.; Sato, N.; Park, E.Y.; Nakamura, Y.; Sato, K. Identification of antihyperuricemic peptides in the proteolytic digest of shark cartilage water extract using in vivo activity-guided fractionation. J. Agric. Food Chem. 2014, 62, 2392–2397. [Google Scholar] [CrossRef]
- Chen, X.; Zhang, L.; Ma, C.; Zhang, Y.; Xi, X.; Wang, L.; Zhou, M.; Burrows, J.F.; Chen, T. A novel antimicrobial peptide, Ranatuerin-2PLx, showing therapeutic potential in inhibiting proliferation of cancer cells. Biosci. Rep. 2018, 38. [Google Scholar] [CrossRef] [Green Version]
- Ma, R.; Wong, S.W.; Ge, L.; Shaw, C.; Siu, S.W.I.; Kwok, H.F. In Vitro and MD Simulation Study to Explore Physicochemical Parameters for Antibacterial Peptide to Become Potent Anticancer Peptide. Mol. Ther. Oncolytics 2020, 16, 7–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pihlanto, A.; Korhonen, H. Bioactive peptides and proteins. Adv. Food Nutr. Res. 2003, 47, 175–276. [Google Scholar] [PubMed]
- Mohanty, D.P.; Mohapatra, S.; Misra, S.; Sahu, P.S. Milk derived bioactive peptides and their impact on human health—A review. Saudi. J. Biol. Sci. 2016, 23, 577–583. [Google Scholar] [CrossRef] [Green Version]
- Fu, Y.; Ji, C.; Chen, X.; Cui, X.; Wang, X.; Feng, J.; Li, Y.; Qin, R.; Guo, X. Investigation into the antimicrobial action and mechanism of a novel endogenous peptide beta-casein 197 from human milk. AMB Express 2017, 7, 119. [Google Scholar] [CrossRef] [Green Version]
- Dallas, D.C.; Guerrero, A.; Khaldi, N.; Castillo, P.A.; Martin, W.F.; Smilowitz, J.T.; Bevins, C.L.; Barile, D.; German, J.B.; Lebrilla, C.B. Extensive in vivo human milk peptidomics reveals specific proteolysis yielding protective antimicrobial peptides. J. Proteome Res. 2013, 12, 2295–2304. [Google Scholar] [CrossRef] [Green Version]
- Politis, I.; Chronopoulou, R. Milk peptides and immune response in the neonate. Adv. Exp. Med. Biol. 2008, 606, 253–269. [Google Scholar] [CrossRef]
- Cui, X.; Li, Y.; Yang, L.; You, L.; Wang, X.; Shi, C.; Ji, C.; Guo, X. Peptidome analysis of human milk from women delivering macrosomic fetuses reveals multiple means of protection for infants. Oncotarget 2016, 7, 63514–63525. [Google Scholar] [CrossRef] [Green Version]
- Meisel, H.; FitzGerald, R.J. Biofunctional peptides from milk proteins: Mineral binding and cytomodulatory effects. Curr. Pharm. Des. 2003, 9, 1289–1295. [Google Scholar]
- Zhao, M.; Wei, C.; Yang, X.; Zhou, J.; Wang, J.; Gu, F.; Lei, T.; Qin, Y. The milk-derived hexapeptide PGPIPN inhibits the invasion and migration of human ovarian cancer cells by regulating the expression of MTA1 and NM23H1 genes. Int. J. Oncol. 2016, 48, 1721–1729. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; Zhao, M.; Tang, Y.; Wang, J.; Wei, C.; Gu, F.; Lei, T.; Chen, Z.; Qin, Y. The milk-derived fusion peptide, ACFP, suppresses the growth of primary human ovarian cancer cells by regulating apoptotic gene expression and signaling pathways. BMC Cancer 2016, 16, 246. [Google Scholar] [CrossRef] [Green Version]
- Smulevich, V.B.; Solionova, L.G.; Belyakova, S.V. Parental occupation and other factors and cancer risk in children: I. Study methodology and non-occupational factors. Int. J. Cancer 1999, 83, 712–717. [Google Scholar] [CrossRef]
- Amitay, E.L.; Keinan-Boker, L. Breastfeeding and Childhood Leukemia Incidence: A Meta-analysis and Systematic Review. JAMA Pediatr. 2015, 169, e151025. [Google Scholar] [CrossRef] [Green Version]
- Kanaprach, P.; Pongsakul, N.; Apiwattanakul, N.; Muanprasat, C.; Supapannachart, S.; Nuntnarumit, P.; Chutipongtanate, S. Evaluation of Fetal Intestinal Cell Growth and Antimicrobial Biofunctionalities of Donor Human Milk After Preparative Processes. Breastfeed Med. 2018, 13, 215–220. [Google Scholar] [CrossRef]
- Chutipongtanate, S.; Watcharatanyatip, K.; Homvises, T.; Jaturongkakul, K.; Thongboonkerd, V. Systematic comparisons of various spectrophotometric and colorimetric methods to measure concentrations of protein, peptide and amino acid: Detectable limits, linear dynamic ranges, interferences, practicality and unit costs. Talanta 2012, 98, 123–129. [Google Scholar] [CrossRef]
- Mader, J.S.; Richardson, A.; Salsman, J.; Top, D.; de Antueno, R.; Duncan, R.; Hoskin, D.W. Bovine lactoferricin causes apoptosis in Jurkat T-leukemia cells by sequential permeabilization of the cell membrane and targeting of mitochondria. Exp. Cell. Res. 2007, 313, 2634–2650. [Google Scholar] [CrossRef]
- Lamiable, A.; Thevenet, P.; Rey, J.; Vavrusa, M.; Derreumaux, P.; Tuffery, P. PEP-FOLD3: Faster de novo structure prediction for linear peptides in solution and in complex. Nucleic Acids Res. 2016, 44, W449–W454. [Google Scholar] [CrossRef] [Green Version]
- Shen, Y.; Maupetit, J.; Derreumaux, P.; Tuffery, P. Improved PEP-FOLD Approach for Peptide and Miniprotein Structure Prediction. J. Chem. Theory Comput. 2014, 10, 4745–4758. [Google Scholar] [CrossRef]
- Thevenet, P.; Shen, Y.; Maupetit, J.; Guyon, F.; Derreumaux, P.; Tuffery, P. PEP-FOLD: An updated de novo structure prediction server for both linear and disulfide bonded cyclic peptides. Nucleic Acids Res. 2012, 40, W288–W293. [Google Scholar] [CrossRef] [Green Version]
- Wei, L.; Zhou, C.; Chen, H.; Song, J.; Su, R. ACPred-FL: A sequence-based predictor using effective feature representation to improve the prediction of anti-cancer peptides. Bioinformatics 2018, 34, 4007–4016. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, P.; Bhagat, D.; Mahalwal, M.; Sharma, N.; Raghava, G.P.S. AntiCP 2.0: An updated model for predicting anticancer peptides. Brief. Bioinform. 2021, 22, bbaa153. [Google Scholar] [CrossRef]
- Manavalan, B.; Basith, S.; Shin, T.H.; Choi, S.; Kim, M.O.; Lee, G. MLACP: Machine-learning-based prediction of anticancer peptides. Oncotarget 2017, 8, 77121–77136. [Google Scholar] [CrossRef] [Green Version]
- Boopathi, V.; Subramaniyam, S.; Malik, A.; Lee, G.; Manavalan, B.; Yang, D.C. mACPpred: A Support Vector Machine-Based Meta-Predictor for Identification of Anticancer Peptides. Int. J. Mol. Sci. 2019, 20, 1964. [Google Scholar] [CrossRef] [Green Version]
- Kay, B.K.; Williamson, M.P.; Sudol, M. The importance of being proline: The interaction of proline-rich motifs in signaling proteins with their cognate domains. FASEB J. 2000, 14, 231–241. [Google Scholar] [CrossRef]
- Holfeld, L.; Knappe, D.; Hoffmann, R. Proline-rich antimicrobial peptides show a long-lasting post-antibiotic effect on Enterobacteriaceae and Pseudomonas aeruginosa. J. Antimicrob. Chemother. 2018, 73, 933–941. [Google Scholar] [CrossRef]
- Gaspar, D.; Veiga, A.S.; Castanho, M.A. From antimicrobial to anticancer peptides. A review. Front. Microbiol. 2013, 4, 294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Felicio, M.R.; Silva, O.N.; Goncalves, S.; Santos, N.C.; Franco, O.L. Peptides with Dual Antimicrobial and Anticancer Activities. Front. Chem. 2017, 5, 5. [Google Scholar] [CrossRef] [Green Version]
- Mahlapuu, M.; Hakansson, J.; Ringstad, L.; Bjorn, C. Antimicrobial Peptides: An Emerging Category of Therapeutic Agents. Front. Cell. Infect. Microbiol. 2016, 6, 194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dennison, S.R.; Whittaker, M.; Harris, F.; Phoenix, D.A. Anticancer alpha-helical peptides and structure/function relationships underpinning their interactions with tumour cell membranes. Curr. Protein Pept. Sci. 2006, 7, 487–499. [Google Scholar] [CrossRef] [PubMed]
- Svanborg, C.; Agerstam, H.; Aronson, A.; Bjerkvig, R.; Duringer, C.; Fischer, W.; Gustafsson, L.; Hallgren, O.; Leijonhuvud, I.; Linse, S.; et al. HAMLET kills tumor cells by an apoptosis-like mechanism—cellular, molecular, and therapeutic aspects. Adv. Cancer Res. 2003, 88, 1–29. [Google Scholar] [CrossRef] [PubMed]
- Hien, T.T.; Ambite, I.; Butler, D.; Wan, M.L.Y.; Tran, T.H.; Hoglund, U.; Babjuk, M.; Svanborg, C. Bladder cancer therapy without toxicity-A dose-escalation study of alpha1-oleate. Int. J. Cancer 2020, 147, 2479–2492. [Google Scholar] [CrossRef]
- Liu, Q.; Zhao, H.; Jiang, Y.; Wu, M.; Tian, Y.; Wang, D.; Lao, Y.; Xu, N.; Li, Z. Development of a lytic peptide derived from BH3-only proteins. Cell Death Discov. 2016, 2, 16008. [Google Scholar] [CrossRef] [Green Version]
- Hu, C.; Chen, X.; Zhao, W.; Chen, Y.; Huang, Y. Design and Modification of Anticancer Peptides. Drug Des. 2016, 5, 138. [Google Scholar] [CrossRef]
- Li, Z.J.; Cho, C.H. Peptides as targeting probes against tumor vasculature for diagnosis and drug delivery. J. Transl. Med. 2012, 10, S1. [Google Scholar] [CrossRef] [Green Version]
- Gupta, V.; Bhavanasi, S.; Quadir, M.; Singh, K.; Ghosh, G.; Vasamreddy, K.; Ghosh, A.; Siahaan, T.J.; Banerjee, S.; Banerjee, S.K. Protein PEGylation for cancer therapy: Bench to bedside. J. Cell Commun. Signal. 2019, 13, 319–330. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chiangjong, W.; Panachan, J.; Vanichapol, T.; Pongsakul, N.; Pongphitcha, P.; Siriboonpiputtana, T.; Lerksuthirat, T.; Nuntnarumit, P.; Supapannachart, S.; Srisomsap, C.; et al. HMP-S7 Is a Novel Anti-Leukemic Peptide Discovered from Human Milk. Biomedicines 2021, 9, 981. https://doi.org/10.3390/biomedicines9080981
Chiangjong W, Panachan J, Vanichapol T, Pongsakul N, Pongphitcha P, Siriboonpiputtana T, Lerksuthirat T, Nuntnarumit P, Supapannachart S, Srisomsap C, et al. HMP-S7 Is a Novel Anti-Leukemic Peptide Discovered from Human Milk. Biomedicines. 2021; 9(8):981. https://doi.org/10.3390/biomedicines9080981
Chicago/Turabian StyleChiangjong, Wararat, Jirawan Panachan, Thitinee Vanichapol, Nutkridta Pongsakul, Pongpak Pongphitcha, Teerapong Siriboonpiputtana, Tassanee Lerksuthirat, Pracha Nuntnarumit, Sarayut Supapannachart, Chantragan Srisomsap, and et al. 2021. "HMP-S7 Is a Novel Anti-Leukemic Peptide Discovered from Human Milk" Biomedicines 9, no. 8: 981. https://doi.org/10.3390/biomedicines9080981
APA StyleChiangjong, W., Panachan, J., Vanichapol, T., Pongsakul, N., Pongphitcha, P., Siriboonpiputtana, T., Lerksuthirat, T., Nuntnarumit, P., Supapannachart, S., Srisomsap, C., Svasti, J., Hongeng, S., & Chutipongtanate, S. (2021). HMP-S7 Is a Novel Anti-Leukemic Peptide Discovered from Human Milk. Biomedicines, 9(8), 981. https://doi.org/10.3390/biomedicines9080981