
Recognition of Oxidized Lipids by Macrophages and Its Role in Atherosclerosis Development


 ,
, 
Abstract
1. Introduction
2. Pathways of Lipid Oxidation and Their Relevance for Atherosclerosis
3. Receptor-Mediated Uptake and Effects of Oxidation-Specific Epitopes
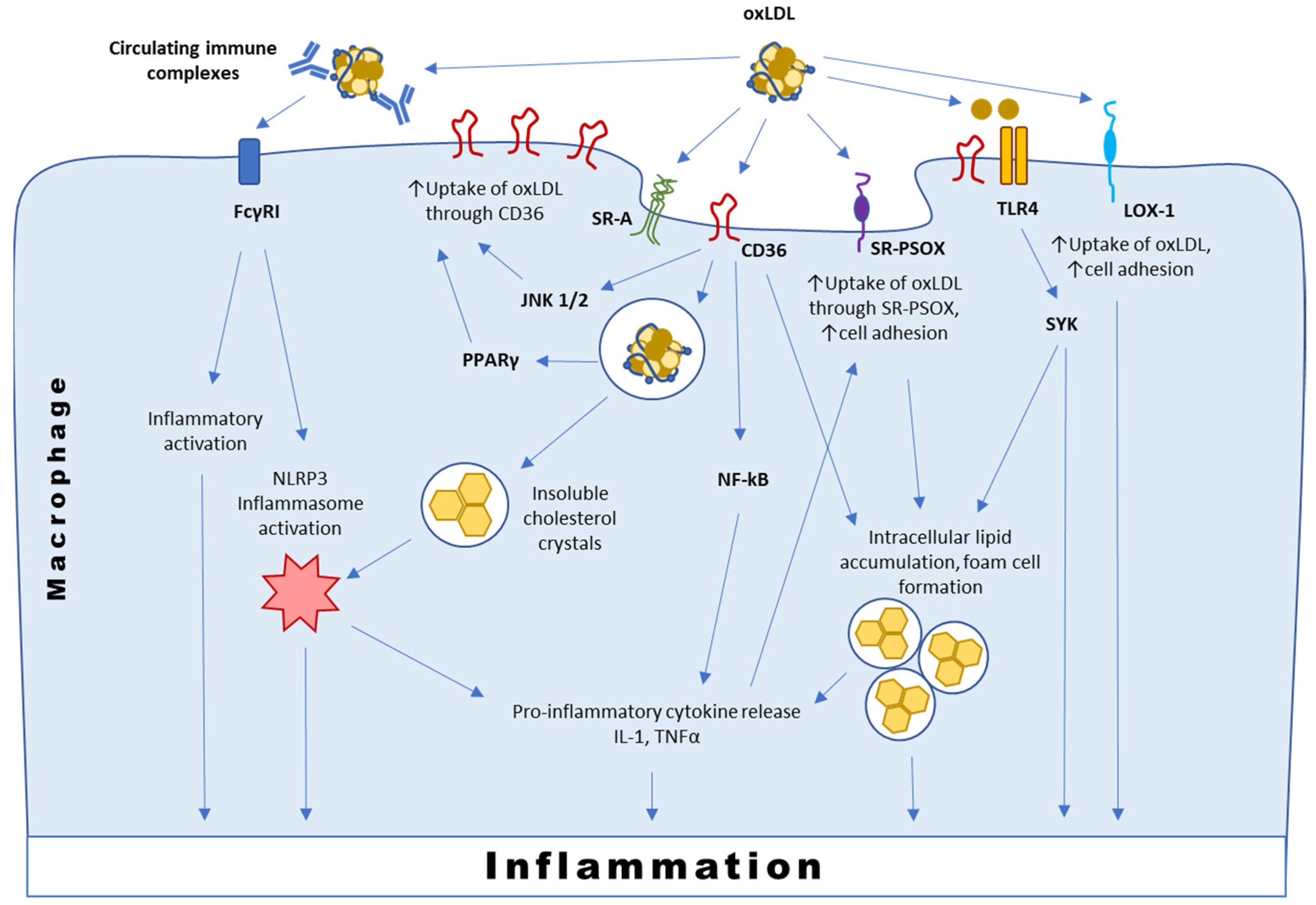
3.1. Interaction with CD36
3.2. Interaction with SR-PSOX
3.3. Interaction with Immunoglobulins and TLRs
3.4. Interaction with LOX-1 and Other Scavenger Receptors
3.5. Interaction with Soluble Factors and Chaperons
4. Anti-Inflammatory and Immunoregulatory Functions of oxLDL
5. OxLDL as a Macrophage Polarization Signal
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Cinoku, I.I.; Mavragani, C.P.; Moutsopoulos, H.M. Atherosclerosis: Beyond the lipid storage hypothesis. The role of autoimmunity. Eur. J. Clin. Investig. 2020, 50, e13195. [Google Scholar] [CrossRef]
- Zakiev, E.R.; Sukhorukov, V.N.; Melnichenko, A.A.; Sobenin, I.A.; Ivanova, E.A.; Orekhov, A.N. Lipid composition of circulating multiple-modified low density lipoprotein. Lipids Health Dis. 2016, 15, 134. [Google Scholar] [CrossRef]
- Orekhov, A.N.; Tertov, V.V.; Mukhin, D.N.; Mikhailenko, I.A. Modification of low density lipoprotein by desialylation causes lipid accumulation in cultured cells: Discovery of desialylated lipoprotein with altered cellular metabolism in the blood of atherosclerotic patients. Biochem. Biophys. Res. Commun. 1989, 162, 206–211. [Google Scholar] [CrossRef]
- Tertov, V.V.; Kaplun, V.V.; Sobenin, I.A.; Orekhov, A.N. Low-density lipoprotein modification occurring in human plasma: Possible mechanism of in vivo lipoprotein desialylation as a primary step of atherogenic modification. Atherosclerosis 1998, 138, 183–195. [Google Scholar] [CrossRef]
- Jaakkola, O.; Solakivi, T.; Tertov, V.V.; Orekhov, A.N.; Miettinen, T.A.; Nikkari, T. Characteristics of low-density lipoprotein subfractions from patients with coronary artery disease. Coron. Artery Dis. 1993, 4, 379–385. [Google Scholar] [CrossRef]
- Tertov, V.V.; Kaplun, V.V.; Sobenin, I.A.; Boytsova, E.Y.; Bovin, N.V.; Orekhov, A.N. Human plasma trans-sialidase causes atherogenic modification of low density lipoprotein. Atherosclerosis 2001, 159, 103–115. [Google Scholar] [CrossRef]
- Sukhorukov, V.N.; Karagodin, V.P.; Orekhov, A.N. Atherogenic modification of low-density lipoproteins. Biomed. Khim. 2016, 62, 391–402. [Google Scholar] [CrossRef]
- Tertov, V.V.; Sobenin, I.A.; Kaplun, V.V.; Orekhov, A.N. Antioxidant content in low density lipoprotein and lipoprotein oxidation in vivo and in vitro. Free Radic. Res. 1998, 29, 165–173. [Google Scholar] [CrossRef] [PubMed]
- Gianazza, E.; Brioschi, M.; Martinez Fernandez, A.; Casalnuovo, F.; Altomare, A.; Aldini, G.; Banfi, C. Lipid Peroxidation in Atherosclerotic Cardiovascular Diseases. Antioxid. Redox Signal. 2021, 34, 49–98. [Google Scholar] [CrossRef]
- Chinetti-Gbaguidi, G.; Colin, S.; Staels, B. Macrophage subsets in atherosclerosis. Nat. Rev. Cardiol. 2015, 12, 10–17. [Google Scholar] [CrossRef]
- Orekhov, A.N.; Nikiforov, N.G.; Sukhorukov, V.N.; Kubekina, M.V.; Sobenin, I.A.; Wu, W.-K.; Foxx, K.K.; Pintus, S.; Stegmaier, P.; Stelmashenko, D.; et al. Role of Phagocytosis in the Pro-Inflammatory Response in LDL-Induced Foam Cell Formation; a Transcriptome Analysis. Int. J. Mol. Sci. 2020, 21, 817. [Google Scholar] [CrossRef] [PubMed]
- Bhakdi, S.; Torzewski, M.; Klouche, M.; Hemmes, M. Complement and atherogenesis: Binding of CRP to degraded, nonoxidized LDL enhances complement activation. Arterioscler. Thromb. Vasc. Biol. 1999, 19, 2348–2354. [Google Scholar] [CrossRef] [PubMed]
- Martin-Ventura, J.L.; Martinez-Lopez, D.; Roldan-Montero, R.; Gomez-Guerrero, C.; Blanco-Colio, L.M. Role of complement system in pathological remodeling of the vascular wall. Mol. Immunol. 2019, 114, 207–215. [Google Scholar] [CrossRef]
- Hovland, A.; Jonasson, L.; Garred, P.; Yndestad, A.; Aukrust, P.; Lappegård, K.T.; Espevik, T.; Mollnes, T.E. The complement system and toll-like receptors as integrated players in the pathophysiology of atherosclerosis. Atherosclerosis 2015, 241, 480–494. [Google Scholar] [CrossRef] [PubMed]
- Quinn, M.T.; Parthasarathy, S.; Fong, L.G.; Steinberg, D. Oxidatively modified low density lipoproteins: A potential role in recruitment and retention of monocyte/macrophages during atherogenesis. Proc. Natl. Acad. Sci. USA 1987, 84, 2995–2998. [Google Scholar] [CrossRef]
- Palinski, W.; Hörkkö, S.; Miller, E.; Steinbrecher, U.P.; Powell, H.C.; Curtiss, L.K.; Witztum, J.L. Cloning of monoclonal autoantibodies to epitopes of oxidized lipoproteins from apolipoprotein E-deficient mice. Demonstration of epitopes of oxidized low density lipoprotein in human plasma. J. Clin. Investig. 1996, 98, 800–814. [Google Scholar] [CrossRef]
- Ito, F.; Sono, Y.; Ito, T. Measurement and Clinical Significance of Lipid Peroxidation as a Biomarker of Oxidative Stress: Oxidative Stress in Diabetes, Atherosclerosis, and Chronic Inflammation. Antioxidants 2019, 8, 72. [Google Scholar] [CrossRef]
- Nicholls, S.J.; Hazen, S.L. Myeloperoxidase, modified lipoproteins, and atherogenesis. J. Lipid Res. 2009, 50, S346–S351. [Google Scholar] [CrossRef] [PubMed]
- Meuwese, M.C.; Stroes, E.S.; Hazen, S.L.; van Miert, J.N.; Kuivenhoven, J.A.; Schaub, R.G.; Wareham, N.J.; Luben, R.; Kastelein, J.J.; Khaw, K.T.; et al. Serum myeloperoxidase levels are associated with the future risk of coronary artery disease in apparently healthy individuals: The EPIC-Norfolk Prospective Population Study. J. Am. Coll. Cardiol. 2007, 50, 159–165. [Google Scholar] [CrossRef] [PubMed]
- Binder, C.J.; Papac-Milicevic, N.; Witztum, J.L. Innate sensing of oxidation-specific epitopes in health and disease. Nat. Rev. Immunol. 2016, 16, 485–497. [Google Scholar] [CrossRef]
- Lee, S.; Birukov, K.G.; Romanoski, C.E.; Springstead, J.R.; Lusis, A.J.; Berliner, J.A. Role of phospholipid oxidation products in atherosclerosis. Circ. Res. 2012, 111, 778–799. [Google Scholar] [CrossRef]
- van Dijk, R.A.; Kolodgie, F.; Ravandi, A.; Leibundgut, G.; Hu, P.P.; Prasad, A.; Mahmud, E.; Dennis, E.; Curtiss, L.K.; Witztum, J.L.; et al. Differential expression of oxidation-specific epitopes and apolipoprotein(a) in progressing and ruptured human coronary and carotid atherosclerotic lesions. J. Lipid Res. 2012, 53, 2773–2790. [Google Scholar] [CrossRef]
- Alharby, H.; Abdelati, T.; Rizk, M.; Youssef, E.; Moghazy, K.; Gaber, N.; Yafei, S. Association of lipid peroxidation and interleukin-6 with carotid atherosclerosis in type 2 diabetes. Cardiovasc. Endocrinol. Metab. 2019, 8, 73–76. [Google Scholar] [CrossRef]
- Que, X.; Hung, M.Y.; Yeang, C.; Gonen, A.; Prohaska, T.A.; Sun, X.; Diehl, C.; Määttä, A.; Gaddis, D.E.; Bowden, K.; et al. Oxidized phospholipids are proinflammatory and proatherogenic in hypercholesterolaemic mice. Nature 2018, 558, 301–306. [Google Scholar] [CrossRef] [PubMed]
- Harkewicz, R.; Hartvigsen, K.; Almazan, F.; Dennis, E.A.; Witztum, J.L.; Miller, Y.I. Cholesteryl ester hydroperoxides are biologically active components of minimally oxidized low density lipoprotein. J. Biol. Chem. 2008, 283, 10241–10251. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.H.; Yin, H.; Ravandi, A.; Armando, A.; Dumlao, D.; Kim, J.; Almazan, F.; Taylor, A.M.; McNamara, C.A.; Tsimikas, S.; et al. Polyoxygenated cholesterol ester hydroperoxide activates TLR4 and SYK dependent signaling in macrophages. PLoS ONE 2013, 8, e83145. [Google Scholar] [CrossRef] [PubMed]
- Shibata, T.; Shimizu, K.; Hirano, K.; Nakashima, F.; Kikuchi, R.; Matsushita, T.; Uchida, K. Adductome-based identification of biomarkers for lipid peroxidation. J. Biol. Chem. 2017, 292, 8223–8235. [Google Scholar] [CrossRef] [PubMed]
- Uchida, K.; Shibata, T.; Toyokuni, S.; Daniel, B.; Zarkovic, K.; Zarkovic, N.; Sasson, S. Development of a novel monoclonal antibody against 4-hydroxy-2E,6Z-dodecadienal (4-HDDE)-protein adducts: Immunochemical application in quantitative and qualitative analyses of lipid peroxidation in vitro and ex vivo. Free Radic. Biol. Med. 2018, 124, 12–20. [Google Scholar] [CrossRef] [PubMed]
- Fredrikson, G.N.; Hedblad, B.; Berglund, G.; Alm, R.; Ares, M.; Cercek, B.; Chyu, K.Y.; Shah, P.K.; Nilsson, J. Identification of immune responses against aldehyde-modified peptide sequences in apoB associated with cardiovascular disease. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 872–878. [Google Scholar] [CrossRef]
- Duner, P.; To, F.; Alm, R.; Gonçalves, I.; Fredrikson, G.N.; Hedblad, B.; Berglund, G.; Nilsson, J.; Bengtsson, E. Immune responses against fibronectin modified by lipoprotein oxidation and their association with cardiovascular disease. J. Intern. Med. 2009, 265, 593–603. [Google Scholar] [CrossRef] [PubMed]
- Vallejo, J.; Dunér, P.; To, F.; Engelbertsen, D.; Gonçalves, I.; Nilsson, J.; Bengtsson, E. Activation of immune responses against the basement membrane component collagen type IV does not affect the development of atherosclerosis in ApoE-deficient mice. Sci. Rep. 2019, 9, 5964. [Google Scholar] [CrossRef] [PubMed]
- Vallejo, J.; Dunér, P.; Fredrikson, G.N.; Nilsson, J.; Bengtsson, E.J. Autoantibodies against aldehyde-modified collagen type IV are associated with risk of development of myocardial infarction. Intern. Med. 2017, 282, 496–507. [Google Scholar] [CrossRef] [PubMed]
- Leibundgut, G.; Witztum, J.L.; Tsimikas, S. Oxidation-specific epitopes and immunological responses: Translational biotheranostic implications for atherosclerosis. Curr. Opin. Pharmacol. 2013, 13, 168–179. [Google Scholar] [CrossRef]
- Kunjathoor, V.V.; Febbraio, M.; Podrez, E.A.; Moore, K.J.; Andersson, L.; Koehn, S.; Rhee, J.S.; Silverstein, R.; Hoff, H.F.; Freeman, M.W. Scavenger receptors class A-I/II and CD36 are the principal receptors responsible for the uptake of modified low density lipoprotein leading to lipid loading in macrophages. J. Biol. Chem. 2002, 277, 49982–49988. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.M. CD36, a scavenger receptor implicated in atherosclerosis. Exp. Mol. Med. 2014, 46, e99. [Google Scholar] [CrossRef]
- Kuchibhotla, S.; Vanegas, D.; Kennedy, D.J.; Guy, E.; Nimako, G.; Morton, R.E.; Febbraio, M. Absence of CD36 protects against atherosclerosis in ApoE knock-out mice with no additional protection provided by absence of scavenger receptor A I/II. Cardiovasc. Res. 2008, 78, 185–196. [Google Scholar] [CrossRef]
- Marleau, S.; Harb, D.; Bujold, K.; Avallone, R.; Iken, K.; Wang, Y.; Demers, A.; Sirois, M.G.; Febbraio, M.; Silverstein, R.L.; et al. EP 80317, a ligand of the CD36 scavenger receptor, protects apolipoprotein E-deficient mice from developing atherosclerotic lesions. FASEB J. 2005, 19, 1869–1871. [Google Scholar] [CrossRef]
- Bujold, K.; Mellal, K.; Zoccal, K.F.; Rhainds, D.; Brissette, L.; Febbraio, M.; Marleau, S.; Ong, H. EP 80317, a CD36 selective ligand, promotes reverse cholesterol transport in apolipoprotein E-deficient mice. Atherosclerosis 2013, 229, 408–414. [Google Scholar] [CrossRef]
- Sheedy, F.J.; Grebe, A.; Rayner, K.J.; Kalantari, P.; Ramkhelawon, B.; Carpenter, S.B.; Becker, C.E.; Ediriweera, H.N.; Mullick, A.E.; Golenbock, D.T.; et al. CD36 coordinates NLRP3 inflammasome activation by facilitating intracellular nucleation of soluble ligands into particulate ligands in sterile inflammation. Nature Immunol. 2013, 14, 812. [Google Scholar] [CrossRef]
- Paramel Varghese, G.; Folkersen, L.; Strawbridge, R.J.; Halvorsen, B.; Yndestad, A.; Ranheim, T.; Krohg-Sørensen, K.; Skjelland, M.; Espevik, T.; Aukrust, P.; et al. NLRP3 Inflammasome Expression and Activation in Human Atherosclerosis. J. Am. Heart Assoc. 2016, 5, e003031. [Google Scholar] [CrossRef]
- Duewell, P.; Kono, H.; Rayner, K.J.; Sirois, C.M.; Vladimer, G.; Bauernfeind, F.G.; Abela, G.S.; Franchi, L.; Nuñez, G.; Schnurr, M.; et al. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature 2010, 464, 1357–1361. [Google Scholar] [CrossRef] [PubMed]
- Grebe, A.; Hoss, F.; Latz, E. NLRP3 Inflammasome and the IL-1 Pathway in Atherosclerosis. Circ. Res. 2018, 122, 1722–1740. [Google Scholar] [CrossRef] [PubMed]
- Hoseini, Z.; Sepahvand, F.; Rashidi, B.; Sahebkar, A.; Masoudifar, A.; Mirzaei, H. NLRP3 inflammasome: Its regulation and involvement in atherosclerosis. J. Cell. Physiol. 2018, 233, 2116–2132. [Google Scholar] [CrossRef]
- Liaqat, A.; Asad, M.; Shoukat, F.; Khan, A.U. A Spotlight on the Underlying Activation Mechanisms of the NLRP3 Inflammasome and its Role in Atherosclerosis: A Review. Inflammation 2020, 43, 2011–2020. [Google Scholar] [CrossRef]
- Ridker, P.M.; Everett, B.M.; Thuren, T.; MacFadyen, J.G.; Chang, W.H.; Ballantyne, C.; Fonseca, F.; Nicolau, J.; Koenig, W.; Anker, S.D.; et al. Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease., CANTOS Trial Group. N. Engl. J. Med. 2017, 377, 1119–1131. [Google Scholar] [CrossRef]
- Tontonoz, P.; Nagy, L.; Alvarez, J.G.; Thomazy, V.A.; Evans, R.M. PPARgamma promotes monocyte/macrophage differentiation and uptake of oxidized LDL. Cell 1998, 93, 241–252. [Google Scholar] [CrossRef]
- Oppi, S.; Nusser-Stein, S.; Blyszczuk, P.; Wang, X.; Jomard, A.; Marzolla, V.; Yang, K.; Velagapudi, S.; Ward, L.J.; Yuan, X.M.; et al. Macrophage NCOR1 protects from atherosclerosis by repressing a proatherogenic PPARgamma signature. Eur. Heart J. 2020, 41, 995–1005. [Google Scholar] [PubMed]
- Hofnagel, O.; Engel, T.; Severs, N.J.; Robenek, H.; Buers, I. SR-PSOX at sites predisposed to atherosclerotic lesion formation mediates monocyte-endothelial cell adhesion. Atherosclerosis 2011, 217, 371–378. [Google Scholar] [CrossRef]
- Minami, M.; Kume, N.; Shimaoka, T.; Kataoka, H.; Hayashida, K.; Akiyama, Y.; Nagata, I.; Ando, K.; Nobuyoshi, M.; Hanyuu, M.; et al. Expression of SR-PSOX, a novel cell-surface scavenger receptor for phosphatidylserine and oxidized LDL in human atherosclerotic lesions. Arterioscler. Thromb. Vasc. Biol. 2001, 21, 1796–1800. [Google Scholar] [CrossRef]
- Zhang, L.; Liu, H.J.; Li, T.J.; Yang, Y.; Guo, X.L.; Wu, M.C.; Rui, Y.C.; Wei, L.X. Lentiviral vector-mediated siRNA knockdown of SR-PSOX inhibits foam cell formation in vitro. Acta Pharmacol. Sin. 2008, 29, 847–852. [Google Scholar] [CrossRef]
- Lundberg, G.A.; Kellin, A.; Samnegård, A.; Lundman, P.; Tornvall, P.; Dimmeler, S.; Zeihze, A.M.; Hamsten, A.; Hansson, G.K.; Eriksson, P. Severity of coronary artery stenosis is associated with a polymorphism in the CXCL16/SR-PSOX gene. J. Intern. Med. 2005, 257, 415–422. [Google Scholar] [CrossRef]
- Sun, Y.; Chang, Z.; Zhang, S. Increased serum CXCL16 level is a marker for acute coronary syndromes. Arch. Med. Res. 2008, 39, 332–337. [Google Scholar] [CrossRef] [PubMed]
- Tertov, V.V.; Sobenin, I.A.; Orekhov, A.N.; Jaakkola, O.; Solakivi, T.; Nikkari, T. Characteristics of low density lipoprotein isolated from circulating immune complexes. Atherosclerosis 1996, 122, 191–199. [Google Scholar] [CrossRef]
- Lopes-Virella, M.F.; McHenry, M.B.; Lipsitz, S.; Yim, E.; Wilson, P.F.; Lackland, D.T.; Lyons, T.; Jenkins, A.J.; Virella, G.; DCCT/EDIC Research Group. Immune complexes containing modified lipoproteins are related to the progression of internal carotid intima-media thickness in patients with type 1 diabetes. Atherosclerosis 2007, 190, 359–369. [Google Scholar] [CrossRef]
- Lopes-Virella, M.F.; Bebu, I.; Hunt, K.J.; Virella, G.; Baker, N.L.; Braffett, B.; Gao, X.; Lachin, J.M.; DCCT/EDIC Research Group. Immune Complexes and the Risk of CVD in Type 1 Diabetes. Diabetes 2019, 68, 1853–1860. [Google Scholar] [CrossRef]
- Imai, Y.; Kuba, K.; Neely, G.; Yaghubian-Malhami, R.; Perkmann, T.; van Loo, G.; Ermolaeva, M.; Veldhuizen, R.; Leung, Y.H.C.; Wang, H.; et al. Identification of oxidative stress and Toll-like receptor 4 signaling as a key pathway of acute lung injury. Cell 2008, 133, 235–249. [Google Scholar] [CrossRef]
- Kadl, A.; Sharma, P.R.; Chen, W.; Agrawal, R.; Meher, A.K.; Rudraiah, S.; Grubbs, N.; Sharma, R.; Leitinger, N. Oxidized phospholipid-induced inflammation is mediated by Toll-like receptor 2. Free Radic. Biol. Med. 2011, 51, 1903–1909. [Google Scholar] [CrossRef] [PubMed]
- Mendel, I.; Feige, E.; Yacov, N.; Salem, Y.; Levi, I.; Propheta-Meiran, O.; Shoham, A.; Ishai, E.; George, J.; Harats, D.; et al. VB-201, an oxidized phospholipid small molecule, inhibits CD14- and Toll-like receptor-2-dependent innate cell activation and constrains atherosclerosis. Clin. Exp. Immunol. 2014, 175, 126–137. [Google Scholar] [CrossRef]
- Hilgendorf, I.; Eisele, S.; Remer, I.; Schmitz, J.; Zeschky, K.; Colberg, C.; Stachon, P.; Wolf, D.; Willecke, F.; Buchner, M.; et al. The oral spleen tyrosine kinase inhibitor fostamatinib attenuates inflammation and atherogenesis in low-density lipoprotein receptor-deficient mice. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 1991–1999. [Google Scholar] [CrossRef] [PubMed]
- Lindau, A.; Härdtner, C.; Hergeth, S.P.; Blanz, K.D.; Dufner, B.; Hoppe, N.; Anto-Michel, N.; Kornemann, J.; Zou, J.; Gerhardt, L.M.; et al. Atheroprotection through SYK inhibition fails in established disease when local macrophage proliferation dominates lesion progression. Basic Res. Cardiol. 2016, 111, 20. [Google Scholar] [CrossRef] [PubMed]
- Mullick, A.E.; Soldau, K.; Kiosses, W.B.; Bell, T.A., III; Tobias, P.S.; Curtiss, L.K. Increased endothelial expression of Toll-like receptor 2 at sites of disturbed blood flow exacerbates early atherogenic events. J. Exp. Med. 2008, 205, 373–383. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.W.; Yakubenko, V.P.; West, X.Z.; Gugiu, G.B.; Renganathan, K.; Biswas, S.; Gao, D.; Crabb, J.W.; Salomon, R.G.; Podrez, E.A.; et al. Receptor-Mediated Mechanism Controlling Tissue Levels of Bioactive Lipid Oxidation Products. Circ. Res. 2015, 117, 321–332. [Google Scholar] [CrossRef] [PubMed]
- Balzan, S.; Lubrano, V. LOX-1 receptor: A potential link in atherosclerosis and cancer. Life Sci. 2018, 198, 79–86. [Google Scholar] [CrossRef]
- Chen, H.; Li, D.; Sawamura, T.; Inoue, K.; Mehta, J.L. Upregulation of LOX-1 expression in aorta of hypercholesterolemicrabbits: Modulation by losartan. Biochem. Biophys. Res. Commun. 2000, 276, 1100–1104. [Google Scholar] [CrossRef]
- Hansson, G.K.; Libby, P.; Tabas, I. Inflammation and plaque vulnerability. J. Intern. Med. 2015, 278, 483–493. [Google Scholar] [CrossRef]
- Zhu, X.; Ng, H.P.; Lai, Y.; Craigo, J.K.; Nagilla, P.S.; Raghani, P.; Nagarajan, S. Scavenger receptor function of mouse Fcgamma receptor III contributes to progression of atherosclerosis in apolipoprotein E hyperlipidemic mice. J. Immunol. 2014, 193, 2483–2495. [Google Scholar] [CrossRef] [PubMed]
- Ng, H.P.; Burris, R.L.; Nagarajan, S. Attenuated atherosclerotic lesions in apoE-Fcγ-chain-deficient hyperlipidemic mouse model is associated with inhibition of Th17 cells and promotion of regulatory T cells. J. Immunol. 2011, 187, 6082–6093. [Google Scholar] [CrossRef]
- Asare, Y.; Koehncke, J.; Selle, J.; Simsekyilmaz, S.; Jankowski, J.; Shagdarsuren, G.; Gessner, J.E.; Bernhagen, J.; Shagdarsuren, E. Differential Role for Activating FcγRIII in Neointima Formation After Arterial Injury and Diet-Induced Chronic Atherosclerosis in Apolipoprotein E-Deficient Mice. Front. Physiol. 2020, 11, 673. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Cai, Q.; Zhou, H.; He, C.; Chen, Y.; Zhang, P.; Wang, T.; Xu, L. OxLDL/beta2GPI/anti-beta2GPI Ab complex induces inflammatory activation via the TLR4/NF-kappaB pathway in HUVECs. J. Mol. Med. Rep. 2021, 23, 148. [Google Scholar] [CrossRef]
- Weismann, D.; Hartvigsen, K.; Lauer, N.; Bennett, K.L.; Scholl, H.P.; Charbel Issa, P.; Cano, M.; Brandstätter, H.; Tsimikas, S.; Skerka, C.; et al. Complement factor H binds malondialdehyde epitopes and protects from oxidative stress. Nature 2011, 478, 76–81. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, M.; Shur, A.; Tendler, Y. M1 Macrophages but Not M2 Macrophages Are Characterized by Upregulation of CRP Expression via Activation of NFkappaB: A Possible Role for Ox-LDL in Macrophage Polarization. Inflammation 2018, 41, 1477–1487. [Google Scholar] [CrossRef]
- Stancel, N.; Chen, C.C.; Ke, L.Y.; Chu, C.S.; Lu, J.; Sawamura, T.; Chen, C.H. Interplay between CRP, Atherogenic LDL, and LOX-1 and Its Potential Role in the Pathogenesis of Atherosclerosis. Clin. Chem. 2016, 62, 320–327. [Google Scholar] [CrossRef]
- Frostegard, J.; Kjellman, B.; Gidlund, M.; Andersson, B.; Jindal, S.; Kiessling, R. Induction of heat shock protein in monocytic cells by oxidized low density lipoprotein. Atherosclerosis 1996, 121, 93–103. [Google Scholar] [CrossRef]
- Shirsath, K.; Joshi, A.; Vohra, A.; Devkar, R. HSP60 knockdown exerts differential response in endothelial cells and monocyte derived macrophages during atherogenic transformation. Sci. Rep. 2021, 11, 1086. [Google Scholar] [CrossRef] [PubMed]
- Ayada, K.; Yokota, K.; Kobayashi, K.; Shoenfeld, Y.; Matsuura, E.; Oguma, K. Chronic infections and atherosclerosis. Clin. Rev. Allergy Immunol. 2009, 37, 44–48. [Google Scholar] [CrossRef]
- Almanzar, G.; Ollinger, R.; Leuenberger, J.; Onestingel, E.; Rather, B.; Zehm, S.; Cardini, B.; van der Zee, R.; Grundtman, C.; Wick, G. Autoreactive HSP60 epitope-specific T-cells in early human atherosclerotic lesions. J. Autoimmunol. 2012, 39, 441–450. [Google Scholar] [CrossRef] [PubMed]
- Liao, B.H.; Xu, Z.L.; Gao, F.; Zhang, S.H.; Liang, R.J.; Dong, S.H. The relationship between HSP60 gene polymorphisms and susceptibility to atherosclerosis. Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 2667–2673. [Google Scholar] [PubMed]
- Wick, C. Tolerization against atherosclerosis using heat shock protein 60. Cell Stress Chaperones 2016, 21, 201–211. [Google Scholar] [CrossRef]
- Mimura, J.; Itoh, K. Role of Nrf2 in the pathogenesis of atherosclerosis. Free Radic. Biol. Med. 2015, 88, 221–232. [Google Scholar] [CrossRef]
- Kadl, A.; Meher, A.K.; Sharma, P.R.; Lee, M.Y.; Doran, A.C.; Johnstone, S.R.; Elliott, M.R.; Gruber, F.; Han, J.; Chen, W.; et al. Identification of a novel macrophage phenotype that develops in response to atherogenic phospholipids via Nrf2. Circ. Res. 2010, 107, 737–746. [Google Scholar] [CrossRef]
- Feige, E.; Yacov, N.; Salem, Y.; Levi, I.; Mendel, I.; Propheta-Meiran, O.; Shoham, A.; Hait-Darshan, R.; Polonsky, O.; George, J.; et al. Inhibition of monocyte chemotaxis by VB-201, a small molecule lecinoxoid, hinders atherosclerosis development in ApoE−/− mice. Atherosclerosis 2013, 229, 430–439. [Google Scholar] [CrossRef][Green Version]
- Doran, A.C.; Yurdagul, A., Jr.; Tabas, I. Efferocytosis in health and disease. Nat. Rev. Immunol. 2020, 20, 254–267. [Google Scholar] [CrossRef]
- Martinet, W.; Kockx, M.M. Apoptosis in atherosclerosis: Focus on oxidized lipids and inflammation. Curr. Opin. Lipidol. 2001, 12, 535–541. [Google Scholar] [CrossRef]
- Xin, T.; Lu, C.; Zhang, J.; Wen, J.; Yan, S.; Li, C.; Zhang, F.; Zhang, J. Oxidized LDL Disrupts Metabolism and Inhibits Macrophage Survival by Activating a miR-9/Drp1/Mitochondrial Fission Signaling Pathway. Oxid. Med. Cell. Longev. 2020, 2020, 8848930. [Google Scholar] [CrossRef] [PubMed]
- Schrijvers, D.M.; De Meyer, G.R.; Kockx, M.M.; Herman, A.G.; Martinet, W. Phagocytosis of apoptotic cells by macrophages is impaired in atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 1256–1261. [Google Scholar] [CrossRef] [PubMed]
- Gordon, S.; Taylor, P.R. Monocyte and macrophage heterogeneity. Nat. Rev. Immunol. 2005, 5, 953–964. [Google Scholar] [CrossRef] [PubMed]
- Jinnouchi, H.; Guo, L.; Sakamoto, A.; Torii, S.; Sato, Y.; Cornelissen, A.; Kuntz, S.; Paek, K.H.; Fernandez, R.; Fuller, D.; et al. Diversity of macrophage phenotypes and responses in atherosclerosis. Cell. Mol. Life Sci. 2020, 77, 1919–1932. [Google Scholar] [CrossRef]
- Bi, Y.; Chen, J.; Hu, F.; Liu, J.; Li, M.; Zhao, L. M2 Macrophages as a Potential Target for Antiatherosclerosis Treatment. Neural Plast. 2019, 21, 6724903. [Google Scholar] [CrossRef]
- Han, X.; Ma, W.; Zhu, Y.; Sun, X.; Liu, N. Advanced glycation end products enhance macrophage polarization to the M1 phenotype via the HIF-1alpha/PDK4 pathway. Mol. Cell. Endocrinol. 2020, 514, 110878. [Google Scholar] [CrossRef] [PubMed]
- Ménégaut, L.; Thomas, C.; Jalil, A.; Julla, J.B.; Magnani, C.; Ceroi, A.; Basmaciyan, L.; Dumont, A.; Le Goff, W.; Mathew, M.J.; et al. Interplay between Liver X Receptor and Hypoxia Inducible Factor 1alpha Potentiates Interleukin-1beta Production in Human Macrophages. Cell Rep. 2020, 31, 107665. [Google Scholar] [CrossRef]
- Maguire, E.M.; Pearce, S.W.A.; Xiao, Q. Foam cell formation: A new target for fighting atherosclerosis and cardiovascular disease. Vasc. Pharmacol. 2019, 112, 54–71. [Google Scholar] [CrossRef]
- Podrez, E.A.; Poliakov, E.; Shen, Z.; Zhang, R.; Deng, Y.; Sun, M.; Finton, P.J.; Shan, L.; Gugiu, B.; Fox, P.L.; et al. Identification of a novel family of oxidized phospholipids that serve as ligands for the macrophage scavenger receptor CD36. J. Biol. Chem. 2002, 277, 38503–38516. [Google Scholar] [CrossRef] [PubMed]
- Serbulea, V.; DeWeese, D.; Leitinger, N. The effect of oxidized phospholipids on phenotypic polarization and function of macrophages. Free Radic. Biol. Med. 2017, 111, 156–168. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Gupta, P.; Rana, M.; Chandra, T.; Dikshit, M.; Barthwal, M.K. Role of pyruvate kinase M2 in oxidized LDL-induced macrophage foam cell formation and inflammation. J. Lipid Res. 2020, 61, 351–364. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| PRR | OSE | Effect | Cells |
|---|---|---|---|
| Scavenger receptors | |||
| SR-A1,2 | MDA | Uptake | Macrophages, mast, dendritic, endothelial, smooth muscle cells |
| SR-B1 | PC-OxPL | Uptake | Monocytes/macrophages, hepatocytes, adipocytes |
| SRECI/II | OxLDL | Uptake | Endothelial cells, macrophages, CD8+ cells |
| SR-PSOX | Ox-PS | Uptake Foam cell formation | Macrophages, smooth muscle, dendritic, and endothelial cells, and B-cells and T cells |
| LOX-1 | MDA | Monocyte adhesion Uptake Inflammation | Endothelial and smooth muscle cells, macrophages, platelets |
| 4-HNE | |||
| CD36 | PC-OxPL | Uptake Inflammation | Macrophages, platelets, adipocytes, epithelial and endothelial cells |
| OxPS | Uptake Inflammation | ||
| CEP | Uptake Inflammation | ||
| TLRs | |||
| TLRs 4-6 | PC-OxPL | Inflammation | Monocytes/macrophages, dendritic cells, mast cells, B cells |
| TLR4 | OxCE | Inflammation Foam cell formation | Monocytes/macrophages, dendritic cells, mast cells, B cells |
| OxPE | Inflammation Foam cell formation | ||
| 4-HNE | Inflammation | ||
| TLRs 2-6 | CEP | Inflammation Thrombosis | Monocytes/macrophages, dendritic cells, mast cells, B cells, platelets |
| OxPL | Angiogenesis ER stress | ||
| TLR9 | CEP | Promotion of platelet hyperreactivity and thrombosis | Platelets |
| Complement | |||
| CFH | MDA | Neutralization Opsonization | |
| C3a | MDA | Complement activation | |
| CRP | PC-OxPL | Enhanced efferocytosis | |
| Other PRRs | |||
| MFG-E8 | OxPS | Enhanced efferocytosis | |
| OxPE | Enhanced efferocytosis | ||
| Annexin A5 | OxCL | Neutralization | |
| CD16 | MDA | Inflammation | Macrophages |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mushenkova, N.V.; Bezsonov, E.E.; Orekhova, V.A.; Popkova, T.V.; Starodubova, A.V.; Orekhov, A.N. Recognition of Oxidized Lipids by Macrophages and Its Role in Atherosclerosis Development. Biomedicines 2021, 9, 915. https://doi.org/10.3390/biomedicines9080915
Mushenkova NV, Bezsonov EE, Orekhova VA, Popkova TV, Starodubova AV, Orekhov AN. Recognition of Oxidized Lipids by Macrophages and Its Role in Atherosclerosis Development. Biomedicines. 2021; 9(8):915. https://doi.org/10.3390/biomedicines9080915
Chicago/Turabian StyleMushenkova, Nataliya V., Evgeny E. Bezsonov, Varvara A. Orekhova, Tatyana V. Popkova, Antonina V. Starodubova, and Alexander N. Orekhov. 2021. "Recognition of Oxidized Lipids by Macrophages and Its Role in Atherosclerosis Development" Biomedicines 9, no. 8: 915. https://doi.org/10.3390/biomedicines9080915
APA StyleMushenkova, N. V., Bezsonov, E. E., Orekhova, V. A., Popkova, T. V., Starodubova, A. V., & Orekhov, A. N. (2021). Recognition of Oxidized Lipids by Macrophages and Its Role in Atherosclerosis Development. Biomedicines, 9(8), 915. https://doi.org/10.3390/biomedicines9080915