
Predictive Antibiotic Susceptibility Testing by Next-Generation Sequencing for Periprosthetic Joint Infections: Potential and Limitations

 , , , ,
, , , ,
Abstract
:1. Introduction
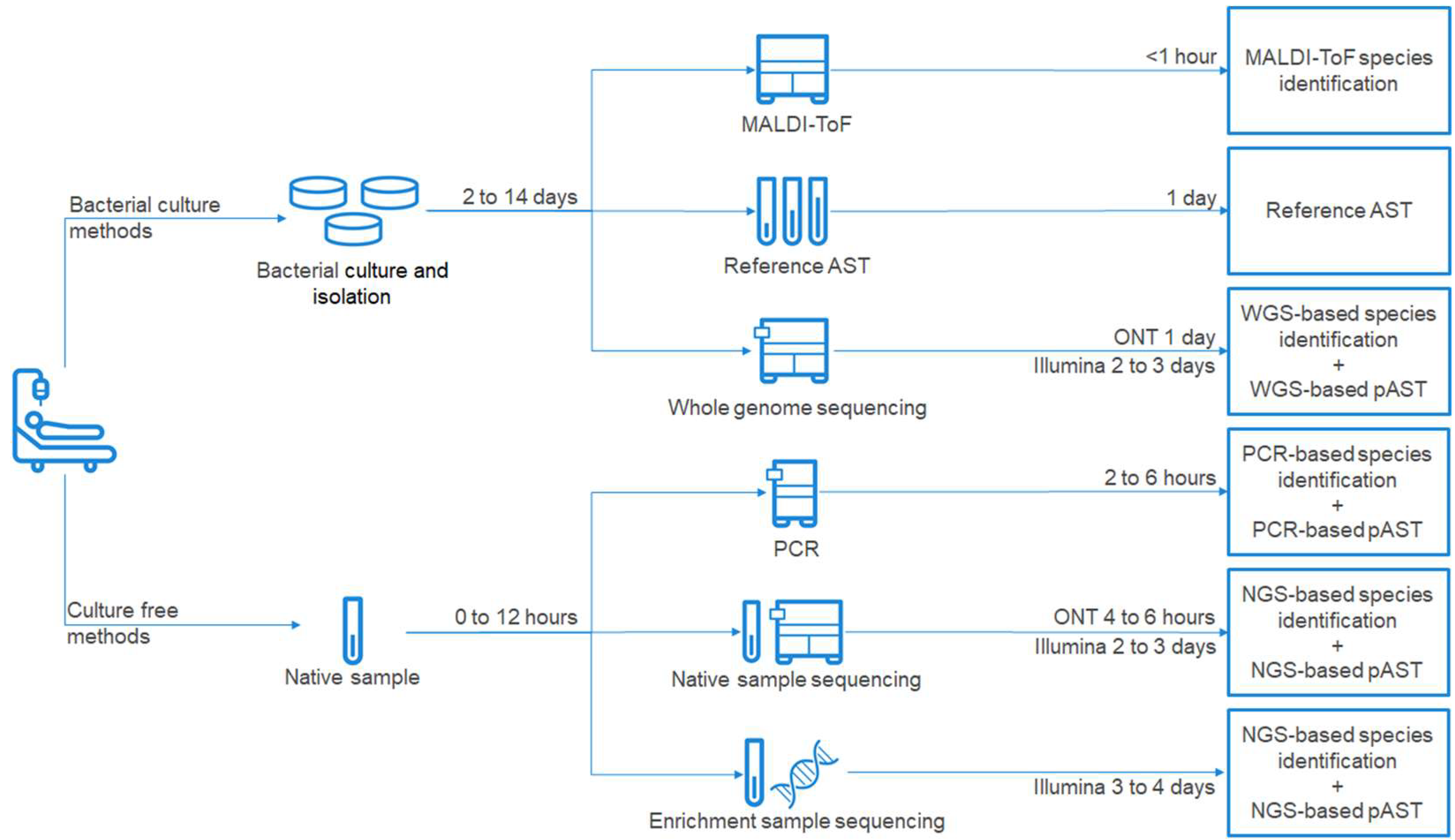
2. Standard of Care Diagnostic Methods for PJI Management
3. Next-Generation Sequencing—Diagnostic Opportunities in PJI
4. Predictive Antimicrobial Susceptibility Testing
4.1. Method Grouping by Decision Algorithm
4.2. Method Grouping by Internal Representation of NGS Data
5. Advances in Predictive AST for Common PJI Pathogens
5.1. Staphylococcus aureus and Staphylococcus spp.
5.2. Enterobacterales and Non-Fermentative Gram-Negative Bacteria

{kind=link}
| Organism Group | Organism | Input Data Type | Decision Algorithm Type | Data Input to Decision Algorithm | Decision Algorithm | Validation Strategy | Validation Dataset Sizes | Accuracy on Validation Datasets [# Compounds] | Year | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|
| Staphylococci | S. aureus | Gene | Rule | AMR markers in reads or assemblies | ResFinder DB lookup | Multiple independent validation datasets | ~80 | 96% and 97% [8] | 2020 | [28] |
| Gene | Rule | AMR markers in reads or assemblies | Custom AMR DB lookup | Independent validation dataset | 470 | (Sensitivity/specificity of 99.1%/99.6%) [12] | 2015 | [29] | ||
| Sequence | ML | DNA k-mer counts from assemblies | AdaBoost | 10× cross-validation | 11 | 99.5% [1] | 2016 | [43] | ||
| Sequence | ML | DNA k-mer counts from reads or assemblies | Set Covering Machine | 80%/20% data split | ~330 | 95%–99% [10] | 2019 | [51] | ||
| Gram-negatives | K. pneumoniae | Sequence | ML | DNA k-mer counts from assemblies | XGBoost | 10× cross-validation | ~160 | (VME/ME of 4%/7%) [21] | 2018 | [35] |
| E. coli | Gene | Rule | AMR markers in reads or assemblies | ResFinder DB lookup | Multiple independent validation datasets | 95, 99, 390 | 97%, 98% and 95% [16] | 2020 | [28] | |
| Gene | ML | gene content, isolation year, population structure | Multiple | 80%/20% data split | 387 | 91% [11] | 2018 | [60] | ||
| Sequence | ML | DNA k-mer counts from reads or assemblies | Set Covering Machine | 80%/20% data split | ~300 | 80%–98% [16] | 2019 | [51] | ||
| P. aeruginosa | Gene | ML | AMR markers, gene content, gene expression data | Support Vector Machine | 80%/20% data split | ~80 | (Sensitivity of 81%–91%) [4] | 2020 | [61] | |
| Sequence | ML | DNA k-mer counts from reads or assemblies | Set Covering Machine | 80%/20% data split | ~100 | 73% and 95% [4] | 2019 | [51] | ||
| Sequence | ML | DNA k-mer presence/absence patterns | Regression | 75%/25% data split | 48 | 88% [1] | 2018 | [63] | ||
| Enterococci | E. faecium | Sequence | ML | DNA k-mer counts from reads or assemblies | Set Covering Machine | 80%/20% data split | 27 | 100% [1] | 2019 | [51] |
| Gene | Rule | AMR markers in reads or assemblies | ResFinder DB lookup | Multiple independent validation datasets | 50 and 56 | 93% and 96% [8] | 2020 | [28] | ||
| E. faecalis | Gene | Rule | AMR markers in reads or assemblies | ResFinder DB lookup | Independent validation dataset | 50 | 97% [5] | 2020 | [28] |
5.3. Streptococci
5.4. Enterococci
6. Current Limitations and Perspectives
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Sloan, M.; Premkumar, A.; Sheth, N.P. Projected Volume of Primary Total Joint Arthroplasty in the U.S., 2014 to 2030. J. Bone Jt. Surg. 2018, 100, 1455–1460. [Google Scholar] [CrossRef]
- Klug, A.; Gramlich, Y.; Rudert, M.; Drees, P.; Hoffmann, R.; Weißenberger, M.; Kutzner, K.P. The projected volume of primary and revision total knee arthroplasty will place an immense burden on future heath care systems over the next 30 years. Knee Surg. Sport. Traumatol. Arthrosc. 2020, 1–12. [Google Scholar] [CrossRef]
- Alp, E.; Cevahir, F.; Ersoy, S.; Guney, A. Incidence and economic burden of prosthetic joint infections in a university hospital: A report from a middle-income country. J. Infect. Public Health 2016, 9, 494–498. [Google Scholar] [CrossRef] [Green Version]
- Kurtz, S.M.; Lau, E.; Watson, H.; Schmier, J.K.; Parvizi, J. Economic burden of periprosthetic joint infection in the United States. J. Arthroplasty 2012, 27, 61–65.e1. [Google Scholar] [CrossRef] [PubMed]
- Tande, A.J.; Patel, R. Prosthetic joint infection. Clin. Microbiol. Rev. 2014, 27, 302–345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Izakovicova, P.; Borens, O.; Trampuz, A. Periprosthetic joint infection: Current concepts and outlook. EFORT Open Rev. 2019, 4, 482–494. [Google Scholar] [CrossRef]
- Motro, Y.; Moran-Gilad, J. Next-generation sequencing applications in clinical bacteriology. Biomol. Detect. Quantif. 2017, 14, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Qin, D. Next-generation sequencing and its clinical application. Cancer Biol. Med. 2019, 16, 4–10. [Google Scholar] [CrossRef]
- Hendriksen, R.S.; Bortolaia, V.; Tate, H.; Tyson, G.H.; Aarestrup, F.M.; McDermott, P.F. Using Genomics to Track Global Antimicrobial Resistance. Front. Public Health 2019, 7, 242. [Google Scholar] [CrossRef] [Green Version]
- Vegyari, C.; Underwood, A.; Kekre, M.; Argimon, S.; Muddyman, D.; Abrudan, M.; Carlos, C.; Donado-Godoy, P.; Okeke, I.N.; Ravikumar, K.L.; et al. Whole-genome sequencing as part of national and international surveillance programmes for antimicrobial resistance: A roadmap. BMJ Glob. Health 2020, 5, e002244. [Google Scholar] [CrossRef]
- World Health Organization. The Use of Next-Generation Sequencing Technologies for the Detection of Mutations Associated with Drug Resistance in Mycobacterium Tuberculosis Complex: Technical Guide 2018; WHO: Geneva, Switzerland, 2018. [Google Scholar]
- Rychert, J. Benefits and Limitations of MALDI-TOF Mass Spectrometry for the Identification of Microorganisms. J. Infect. 2019, 2, 1–5. [Google Scholar] [CrossRef]
- Jorgensen, J.H.; Ferraro, M.J. Antimicrobial Susceptibility Testing: A Review of General Principles and Contemporary Practices. Clin. Infect. Dis. 2009, 49, 1749–1755. [Google Scholar] [CrossRef] [PubMed]
- Kalbian, I.; Park, J.W.; Goswami, K.; Lee, Y.-K.; Parvizi, J.; Koo, K.-H. Culture-negative periprosthetic joint infection: Prevalence, aetiology, evaluation, recommendations, and treatment. Int. Orthop. 2020, 44, 1255–1261. [Google Scholar] [CrossRef] [PubMed]
- Klein, M.; Bacher, J.; Barth, S.; Atrzadeh, F.; Siebenhaller, K.; Ferreira, I.; Beisken, S.; Posch, A.E.; Carroll, K.C.; Wunderink, R.G.; et al. Multicenter Evaluation of the Unyvero Platform for Testing Bronchoalveolar Lavage Fluid. J. Clin. Microbiol. 2020. [Google Scholar] [CrossRef]
- Lee, B.R.; Hassan, F.; Jackson, M.A.; Selvarangan, R. Impact of multiplex molecular assay turn-around-time on antibiotic utilization and clinical management of hospitalized children with acute respiratory tract infections. J. Clin. Virol. 2019, 110, 11–16. [Google Scholar] [CrossRef]
- Lausmann, C.; Kolle, K.N.; Citak, M.; Abdelaziz, H.; Schulmeyer, J.; Delgado, G.D.; Gehrke, T.; Gebauer, M.; Zahar, A. How reliable is the next generation of multiplex-PCR for diagnosing prosthetic joint infection compared to the MSIS criteria? Still missing the ideal test. HIP Int. 2020, 30, 72–77. [Google Scholar] [CrossRef]
- Graue, C.; Schmitt, B.H.; Waggoner, A.; Laurent, F.; Abad, L.; Bauer, T.; Mazariegos, I.; Balada-Llasat, J.-M.; Horn, J.; Wolk, D.; et al. 322. Evaluation of the BioFire® Bone and Joint Infection (BJI) Panel for the Detection of Microorganisms and Antimicrobial Resistance Genes in Synovial Fluid Specimens. Open Forum Infect. Dis. 2020, 7, S233–S234. [Google Scholar] [CrossRef]
- ORTHOPEDICS|Orthopedic & Periprosthetic Joint Infection|MicroGen Diagnostics. Available online: https://microgendx.com/orthopedic-joint-infections/ (accessed on 7 June 2021).
- Wragg, P.; Randall, L.; Whatmore, A.M. Comparison of Biolog GEN III MicroStation semi-automated bacterial identification system with matrix-assisted laser desorption ionization-time of flight mass spectrometry and 16S ribosomal RNA gene sequencing for the identification of bacteria of veterinary interest. J. Microbiol. Methods 2014, 105, 16–21. [Google Scholar] [CrossRef] [Green Version]
- Garza-González, E.; Bocanegra-Ibarias, P.; Dinh, A.; Morfín-Otero, R.; Camacho-Ortiz, A.; Rojas-Larios, F.; Rodríguez-Zulueta, P.; Arias, C.A. Species identification of Enterococcus spp: Whole genome sequencing compared to three biochemical test-based systems and two Matrix-Assisted Laser Desorption/Ionization Time-of-Flight Mass Spectrometry (MALDI-TOF MS) systems. J. Clin. Lab. Anal. 2020, 34, e23348. [Google Scholar] [CrossRef]
- Hong, E.; Bakhalek, Y.; Taha, M.K. Identification of Neisseria meningitidis by MALDI-TOF MS may not be reliable. Clin. Microbiol. Infect. 2019, 25, 717–722. [Google Scholar] [CrossRef] [PubMed]
- MacFadden, D.R.; Melano, R.G.; Coburn, B.; Tijet, N.; Hanage, W.P.; Daneman, N. Comparing Patient Risk Factor-, Sequence Type-, and Resistance Locus Identification-Based Approaches for Predicting Antibiotic Resistance in Escherichia coli Bloodstream Infections. J. Clin. Microbiol. 2019, 57, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Břinda, K.; Callendrello, A.; Ma, K.C.; MacFadden, D.R.; Charalampous, T.; Lee, R.S.; Cowley, L.; Wadsworth, C.B.; Grad, Y.H.; Kucherov, G.; et al. Rapid inference of antibiotic resistance and susceptibility by genomic neighbour typing. Nat. Microbiol. 2020, 5, 455–464. [Google Scholar] [CrossRef] [Green Version]
- Ivy, M.I.; Thoendel, M.J.; Jeraldo, P.R.; Greenwood-Quaintance, K.E.; Hanssen, A.D.; Abdel, M.P.; Chia, N.; Yao, J.Z.; Tande, A.J.; Mandrekar, J.N.; et al. Direct detection and identification of prosthetic joint infection pathogens in synovial fluid by metagenomic shotgun sequencing. J. Clin. Microbiol. 2018, 56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kozarewa, I.; Armisen, J.; Gardner, A.F.; Slatko, B.E.; Hendrickson, C.L. Overview of target enrichment strategies. Curr. Protoc. Mol. Biol. 2015. [CrossRef]
- Allicock, O.M.; Guo, C.; Uhlemann, A.C.; Whittier, S.; Chauhan, L.V.; Garcia, J.; Price, A.; Morse, S.S.; Mishra, N.; Briese, T.; et al. Baccapseq: A platform for diagnosis and characterization of bacterial infections. mBio 2018, 9, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bortolaia, V.; Kaas, R.S.; Ruppe, E.; Roberts, M.C.; Schwarz, S.; Philippon, A.; Allesoe, R.L.; Rebelo, A.R.; Florensa, A.F.; Cattoir, V.; et al. ResFinder 4.0 for predictions of phenotypes from genotypes. J. Antimicrob. Chemother. 2020. [Google Scholar] [CrossRef] [PubMed]
- Bradley, P.; Gordon, N.C.; Walker, T.M.; Dunn, L.; Heys, S.; Huang, B.; Earle, S.; Pankhurst, L.J.; Anson, L.; De Cesare, M.; et al. Rapid antibiotic-resistance predictions from genome sequence data for Staphylococcus aureus and Mycobacterium tuberculosis. Nat. Commun. 2015, 6. [Google Scholar] [CrossRef] [Green Version]
- Ruppert, D. The Elements of Statistical Learning: Data Mining, Inference, and Prediction. J. Am. Stat. Assoc. 2004, 99, 567. [Google Scholar] [CrossRef]
- Cortes, C.; Vapnik, V. Support-Vector Networks. Mach. Learn. 1995, 20, 273–297. [Google Scholar] [CrossRef]
- Freund, Y.; Schapire, R.E. A Decision-Theoretic Generalization of On-Line Learning and an Application to Boosting. J. Comput. Syst. Sci. 1997, 55, 119–139. [Google Scholar] [CrossRef] [Green Version]
- Marchand, M.; Shawe-Taylor, J.; Brodley, C.E.; Danyluk, A. The Set Covering Machine. J. Mach. Learn. Res. 2002, 3, 723–746. [Google Scholar] [CrossRef]
- Wayne, P. Performance Standards for Antimicrobial Susceptibility Testing, 29th ed.; CLSI supplement M100; Clinical and Laboratory Standards Institute: Wayne, PA, USA, 2019. [Google Scholar]
- Nguyen, M.; Brettin, T.; Long, S.W.; Musser, J.M.; Olsen, R.J.; Olson, R.; Shukla, M.; Stevens, R.L.; Xia, F.; Yoo, H.; et al. Developing an in silico minimum inhibitory concentration panel test for Klebsiella pneumoniae. Sci. Rep. 2018, 8, 421. [Google Scholar] [CrossRef] [Green Version]
- Antimicrobial Susceptibility Test (AST) Systems—Class II Special Controls Guidance for Industry and FDA | FDA. Available online: https://www.fda.gov/medical-devices/guidance-documents-medical-devices-and-radiation-emitting-products/antimicrobial-susceptibility-test-ast-systems-class-ii-special-controls-guidance-industry-and-fda (accessed on 14 May 2021).
- European Committee on Antimicrobial Susceptibility Testing MIC Distributions and the Setting of Epidemiological Cut-Off (ECOFF) Values. EUCAST: Basel, Switzerland, 2019.
- Zankari, E.; Hasman, H.; Kaas, R.S.; Seyfarth, A.M.; Agersø, Y.; Lund, O.; Larsen, M.V.; Aarestrup, F.M. Genotyping using whole-genome sequencing is a realistic alternative to surveillance based on phenotypic antimicrobial susceptibility testing. J. Antimicrob. Chemother. 2013, 68, 771–777. [Google Scholar] [CrossRef]
- Clausen, P.T.L.C.; Zankari, E.; Aarestrup, F.M.; Lund, O. Benchmarking of methods for identification of antimicrobial resistance genes in bacterial whole genome data. J. Antimicrob. Chemother. 2016, 71, 2484–2488. [Google Scholar] [CrossRef] [Green Version]
- Hunt, M.; Mather, A.E.; Sánchez-Busó, L.; Page, A.J.; Parkhill, J.; Keane, J.A.; Harris, S.R. ARIBA: Rapid antimicrobial resistance genotyping directly from sequencing reads. Microb. Genomics 2017, 3, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Anjum, M.F.; Zankari, E.; Hasman, H. Molecular Methods for Detection of Antimicrobial Resistance. Antimicrob. Resist. Bact. Livest. Companion Anim. 2017, 33–50. [Google Scholar] [CrossRef] [Green Version]
- Drouin, A.; Giguère, S.; Déraspe, M.; Marchand, M.; Tyers, M.; Loo, V.G.; Bourgault, A.-M.M.; Laviolette, F.; Corbeil, J. Predictive computational phenotyping and biomarker discovery using reference-free genome comparisons. BMC Genomics 2016, 17, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Davis, J.J.; Boisvert, S.; Brettin, T.; Kenyon, R.W.; Mao, C.; Olson, R.; Overbeek, R.; Santerre, J.; Shukla, M.; Wattam, A.R.; et al. Antimicrobial Resistance Prediction in PATRIC and RAST. Sci. Rep. 2016, 6, 1–12. [Google Scholar] [CrossRef]
- Valizadehaslani, T.; Zhao, Z.; Sokhansanj, B.A.; Rosen, G.L. Amino acid K-mer feature extraction for quantitative antimicrobial resistance (AMR) prediction by machine learning and model interpretation for biological insights. Biology 2020, 9, 365. [Google Scholar] [CrossRef]
- Hicks, A.L.; Wheeler, N.; Sánchez-Busó, L.; Rakeman, J.L.; Harris, S.R.; Grad, Y.H. Evaluation of parameters affecting performance and reliability of machine learning-based antibiotic susceptibility testing from whole genome sequencing data. PLoS Comput. Biol. 2019, 15, e1007349. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, M.; Wesley Long, S.; McDermott, P.F.; Olsen, R.J.; Olson, R.; Stevens, R.L.; Tyson, G.H.; Zhao, S.; Davisa, J.J. Using machine learning to predict antimicrobial MICs and associated genomic features for nontyphoidal Salmonella. J. Clin. Microbiol. 2019, 57. [Google Scholar] [CrossRef] [Green Version]
- Chua, K.Y.L.; Stinear, T.P.; Howden, B.P. Functional genomics of staphylococcus aureus. Brief. Funct. Genom. 2013, 12, 305–315. [Google Scholar] [CrossRef] [PubMed]
- Foster, T.J. Antibiotic resistance in Staphylococcus aureus. Current status and future prospects. FEMS Microbiol. Rev. 2017, 41, 430–449. [Google Scholar] [CrossRef]
- Gordon, N.C.; Price, J.R.; Cole, K.; Everitt, R.; Morgan, M.; Finney, J.; Kearns, A.M.; Pichon, B.; Young, B.; Wilson, D.J.; et al. Prediction of staphylococcus aureus antimicrobial resistance by whole-genome sequencing. J. Clin. Microbiol. 2014, 52, 1182–1191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Drouin, A.; Letarte, G.; Raymond, F.; Marchand, M.; Corbeil, J.; Laviolette, F. Interpretable genotype-to-phenotype classifiers with performance guarantees. Sci. Rep. 2019, 9. [Google Scholar] [CrossRef] [Green Version]
- Becker, K.; Heilmann, C.; Peters, G. Coagulase-negative staphylococci. Clin. Microbiol. Rev. 2014, 27, 870–926. [Google Scholar] [CrossRef] [Green Version]
- Wielders, C.L.C.; Vriens, M.R.; Brisse, S.; De Graaf-Miltenburg, L.A.M.; Troelstra, A.; Fleer, A.; Schmitz, F.J.; Verhoef, J.; Fluit, A.C. Evidence for in-vivo transfer of mecA DNA between strains of Staphylococcus aureus. Lancet 2001, 357, 1674–1675. [Google Scholar] [CrossRef]
- Heilmann, C.; Ziebuhr, W.; Becker, K. Are coagulase-negative staphylococci virulent? Clin. Microbiol. Infect. 2019, 25, 1071–1080. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, P.H.; Lee, M.S.; Hsu, K.Y.; Chang, Y.H.; Shin, H.N.; Ueng, S.W. Gram-negative prosthetic joint infections: Risk factors and outcome of treatment. Clin. Infect. Dis. 2009, 49, 1036–1043. [Google Scholar] [CrossRef] [PubMed]
- Cerioli, M.; Batailler, C.; Conrad, A.; Roux, S.; Perpoint, T.; Becker, A.; Triffault-Fillit, C.; Lustig, S.; Fessy, M.-H.; Laurent, F.; et al. Pseudomonas aeruginosa Implant-Associated Bone and Joint Infections: Experience in a Regional Reference Center in France. Front. Med. 2020, 7, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Shah, N.B.; Osmon, D.R.; Steckelberg, J.M.; Sierra, R.J.; Walker, R.C.; Tande, A.J.; Berbari, E.F. Pseudomonas Prosthetic Joint Infections: A Review of 102 Episodes. J. Bone Jt. Infect. 2016, 1, 25–30. [Google Scholar] [CrossRef]
- Stoesser, N.; Batty, E.M.; Eyre, D.W.; Morgan, M.; Wyllie, D.H.; Del Ojo Elias, C.; Johnson, J.R.; Walker, A.S.; Peto, T.E.A.; Crook, D.W. Predicting antimicrobial susceptibilities for Escherichia coli and Klebsiella pneumoniae isolates using whole genomic sequence data. J. Antimicrob. Chemother. 2013, 68, 2234–2244. [Google Scholar] [CrossRef]
- Pesesky, M.W.; Hussain, T.; Wallace, M.; Patel, S.; Andleeb, S.; Burnham, C.A.D.; Dantas, G. Evaluation of machine learning and rules-based approaches for predicting antimicrobial resistance profiles in gram-negative bacilli from whole genome sequence data. Front. Microbiol. 2016, 7, 1887. [Google Scholar] [CrossRef]
- Moradigaravand, D.; Palm, M.; Farewell, A.; Mustonen, V.; Warringer, J.; Parts, L. Prediction of antibiotic resistance in Escherichia coli from large-scale pan-genome data. PLoS Comput. Biol. 2018, 14. [Google Scholar] [CrossRef] [Green Version]
- Khaledi, A.; Weimann, A.; Schniederjans, M.; Asgari, E.; Kuo, T.; Oliver, A.; Cabot, G.; Kola, A.; Gastmeier, P.; Hogardt, M.; et al. Predicting antimicrobial resistance in Pseudomonas aeruginosa with machine learning-enabled molecular diagnostics. EMBO Mol. Med. 2020, 12, 1–19. [Google Scholar] [CrossRef]
- Jeukens, J.; Freschi, L.; Kukavica-Ibrulj, I.; Emond-Rheault, J.G.; Tucker, N.P.; Levesque, R.C. Genomics of antibiotic-resistance prediction in pseudomonas aeruginosa. Ann. N. Y. Acad. Sci. 2019, 1435, 5–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aun, E.; Brauer, A.; Kisand, V.; Tenson, T.; Remm, M. A k-mer-based method for the identification of phenotype-associated genomic biomarkers and predicting phenotypes of sequenced bacteria. PLoS Comput. Biol. 2018, 14. [Google Scholar] [CrossRef] [Green Version]
- Jacoby, G.A. Mechanisms of Resistance to Quinolones. Clin. Infect. Dis. 2005, 41, S120–S126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pang, Z.; Raudonis, R.; Glick, B.R.; Lin, T.J.; Cheng, Z. Antibiotic resistance in Pseudomonas aeruginosa: Mechanisms and alternative therapeutic strategies. Biotechnol. Adv. 2019, 37, 177–192. [Google Scholar] [CrossRef] [PubMed]
- Akgün, D.; Trampuz, A.; Perka, C.; Renz, N. High failure rates in treatment of streptococcal periprosthetic joint infection RESULTS FROM A SEVEN-YEAR RETROSPECTIVE COHORT STUDY. Bone Joint J. 2017, 99, 653–659. [Google Scholar] [CrossRef]
- Li, Y.; Metcalf, B.J.; Chochua, S.; Li, Z.; Gertz, R.E.; Walker, H.; Hawkins, P.A.; Tran, T.; Whitney, C.G.; McGee, L.; et al. Penicillin-binding protein transpeptidase signatures for tracking and predicting β-lactam resistance levels in Streptococcus pneumoniae. mBio 2016, 7, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, R.; Gallagher, J.C. Vancomycin-Resistant Enterococcal Bacteremia Pharmacotherapy. Ann. Pharmacother. 2015, 49, 69–85. [Google Scholar] [CrossRef]
- Clarke, R.; Ressom, H.W.; Wang, A.; Xuan, J.; Liu, M.C.; Gehan, E.A.; Wang, Y. The properties of high-dimensional data spaces: Implications for exploring gene and protein expression data. Nat. Rev. Cancer 2008, 8, 37–49. [Google Scholar] [CrossRef] [Green Version]
- Libbrecht, M.W.; Noble, W.S. Machine learning applications in genetics and genomics. Nat. Rev. Genet. 2015, 16, 321–332. [Google Scholar] [CrossRef] [Green Version]
- Lüftinger, L.; Májek, P.; Beisken, S.; Rattei, T.; Posch, A.E. Learning from Limited Data: Towards Best Practice Techniques for Antimicrobial Resistance Prediction From Whole Genome Sequencing Data. Front. Cell. Infect. Microbiol. 2021, 11, 1. [Google Scholar] [CrossRef] [PubMed]
- Consultation on Report from the EUCAST Subcommittee on the Role of Whole Genome Sequencing (WGS) in Antimicrobial Susceptibility Testing of Bacteria Report from the EUCAST Subcommittee on the Role of Whole Genome Sequencing (WGS) in Bacteria Antimicro; EUCAST: Geneva, Switzerland, 2016; pp. 1–72.
- Mahfouz, N.; Ferreira, I.; Beisken, S.; von Haeseler, A.; Posch, A.E. Large-scale assessment of antimicrobial resistance marker databases for genetic phenotype prediction: A systematic review. J. Antimicrob. Chemother. 2020, 75, 3099–3108. [Google Scholar] [CrossRef] [PubMed]
- Sczyrba, A.; Hofmann, P.; Belmann, P.; Koslicki, D.; Janssen, S.; Dröge, J.; Gregor, I.; Majda, S.; Fiedler, J.; Dahms, E.; et al. Critical Assessment of Metagenome Interpretation—A benchmark of metagenomics software. Nat. Methods 2017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruppé, E.; Lazarevic, V.; Girard, M.; Mouton, W.; Ferry, T.; Laurent, F.; Schrenzel, J. Clinical metagenomics of bone and joint infections: A proof of concept study. Sci. Rep. 2017, 7, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Payne, A.; Holmes, N.; Clarke, T.; Munro, R.; Debebe, B.; Loose, M. Nanopore adaptive sequencing for mixed samples, whole exome capture and targeted panels. bioRxiv 2020. [Google Scholar] [CrossRef] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lüftinger, L.; Ferreira, I.; Frank, B.J.H.; Beisken, S.; Weinberger, J.; von Haeseler, A.; Rattei, T.; Hofstaetter, J.G.; Posch, A.E.; Materna, A. Predictive Antibiotic Susceptibility Testing by Next-Generation Sequencing for Periprosthetic Joint Infections: Potential and Limitations. Biomedicines 2021, 9, 910. https://doi.org/10.3390/biomedicines9080910
Lüftinger L, Ferreira I, Frank BJH, Beisken S, Weinberger J, von Haeseler A, Rattei T, Hofstaetter JG, Posch AE, Materna A. Predictive Antibiotic Susceptibility Testing by Next-Generation Sequencing for Periprosthetic Joint Infections: Potential and Limitations. Biomedicines. 2021; 9(8):910. https://doi.org/10.3390/biomedicines9080910
Chicago/Turabian StyleLüftinger, Lukas, Ines Ferreira, Bernhard J. H. Frank, Stephan Beisken, Johannes Weinberger, Arndt von Haeseler, Thomas Rattei, Jochen G. Hofstaetter, Andreas E. Posch, and Arne Materna. 2021. "Predictive Antibiotic Susceptibility Testing by Next-Generation Sequencing for Periprosthetic Joint Infections: Potential and Limitations" Biomedicines 9, no. 8: 910. https://doi.org/10.3390/biomedicines9080910
APA StyleLüftinger, L., Ferreira, I., Frank, B. J. H., Beisken, S., Weinberger, J., von Haeseler, A., Rattei, T., Hofstaetter, J. G., Posch, A. E., & Materna, A. (2021). Predictive Antibiotic Susceptibility Testing by Next-Generation Sequencing for Periprosthetic Joint Infections: Potential and Limitations. Biomedicines, 9(8), 910. https://doi.org/10.3390/biomedicines9080910