
Structural and Functional Alterations in Mitochondria-Associated Membranes (MAMs) and in Mitochondria Activate Stress Response Mechanisms in an In Vitro Model of Alzheimer’s Disease

 , ,
, ,  , ,
, ,  and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Subcellular Fractionation
2.3. Protein Analysis by Western Blot
2.4. Mitochondria Morphology Analysis by Transmission Electron Microscopy (TEM)
2.5. Mitochondria Morphology Analysis by Confocal Microscopy Using MitoTracker Green
2.6. Fluorometric Analysis of Calcium and Mitochondrial Membrane Potential
2.7. Activity of Mitochondrial Electron Transport Chain and Glycolysis Measured by the Seahorse XFe24 Analyzer
2.8. Statistical Analysis
3. Results
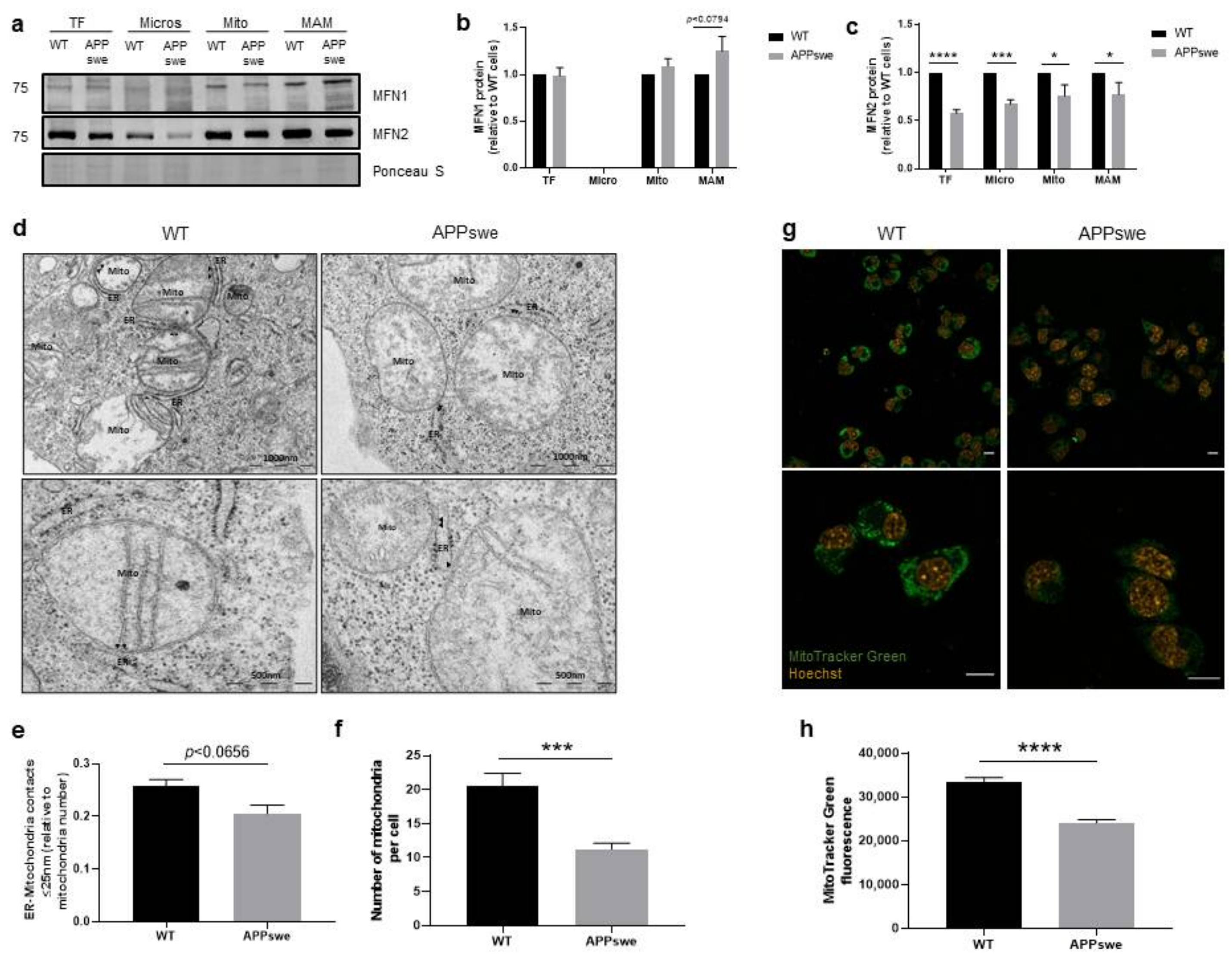
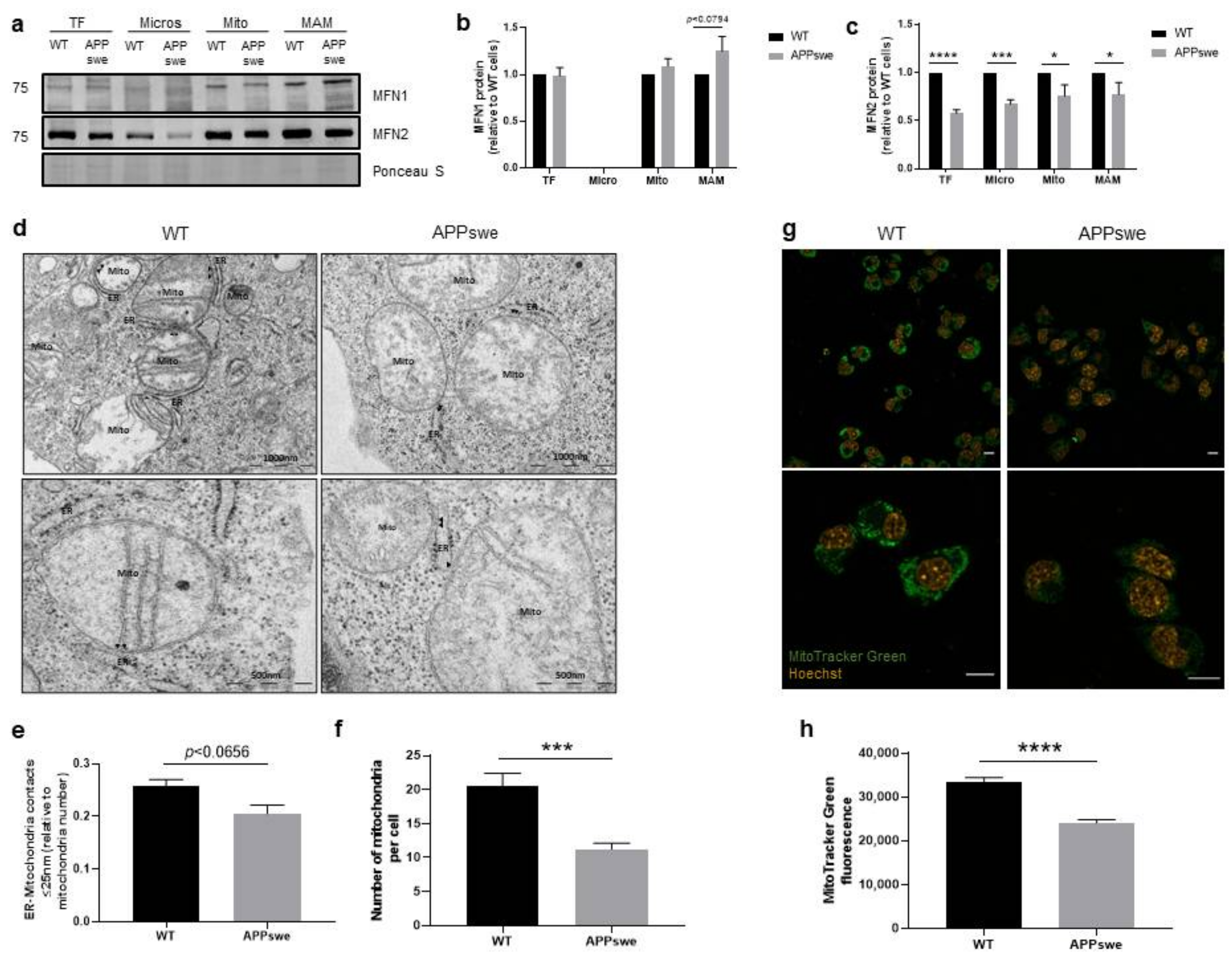
3.1. Alterations of ER-Mitochondria Contacts in APPswe Cells
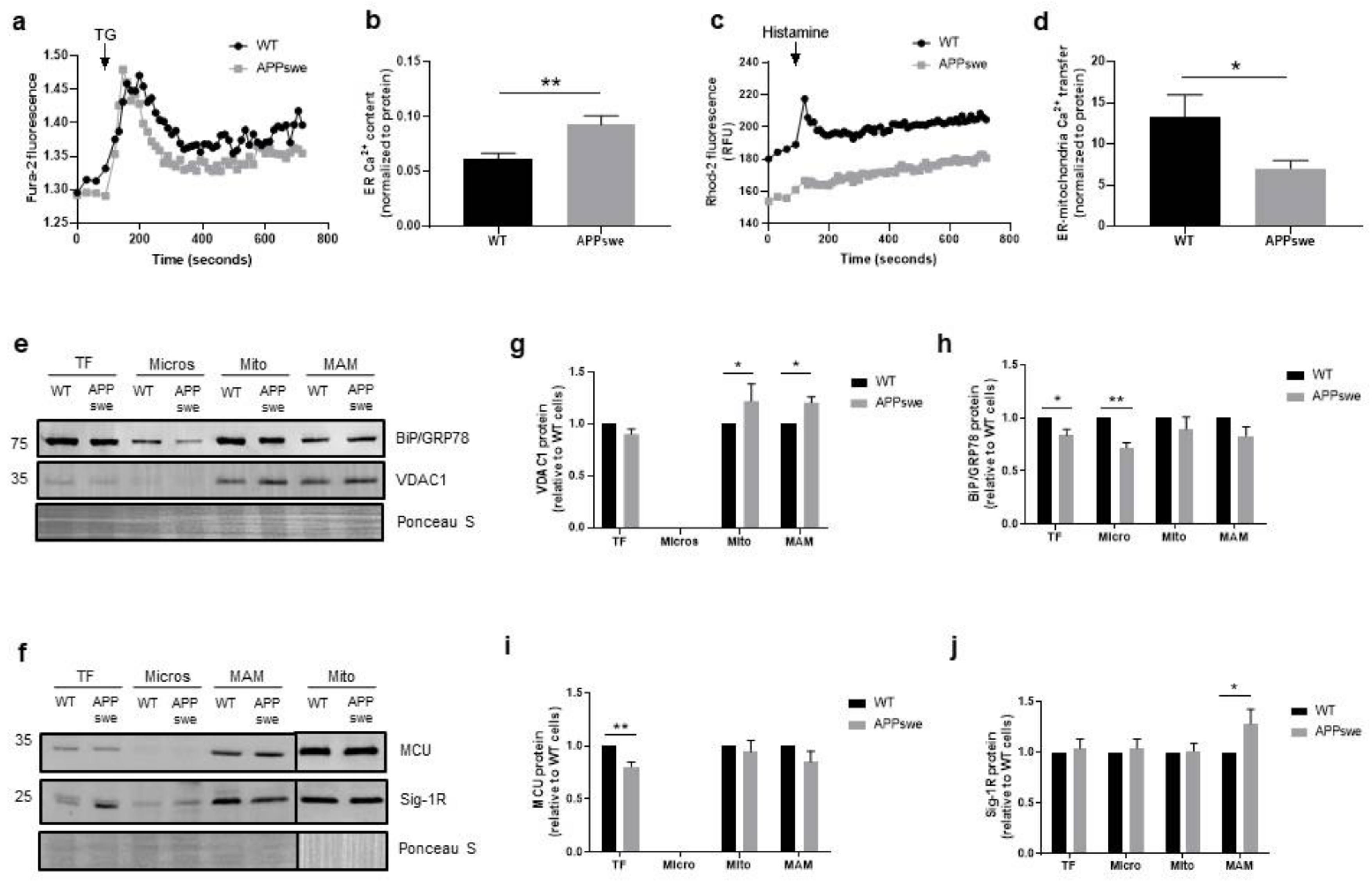
3.2. Impairment of Ca2+ Transfer from ER to Mitochondria in APPswe Cells
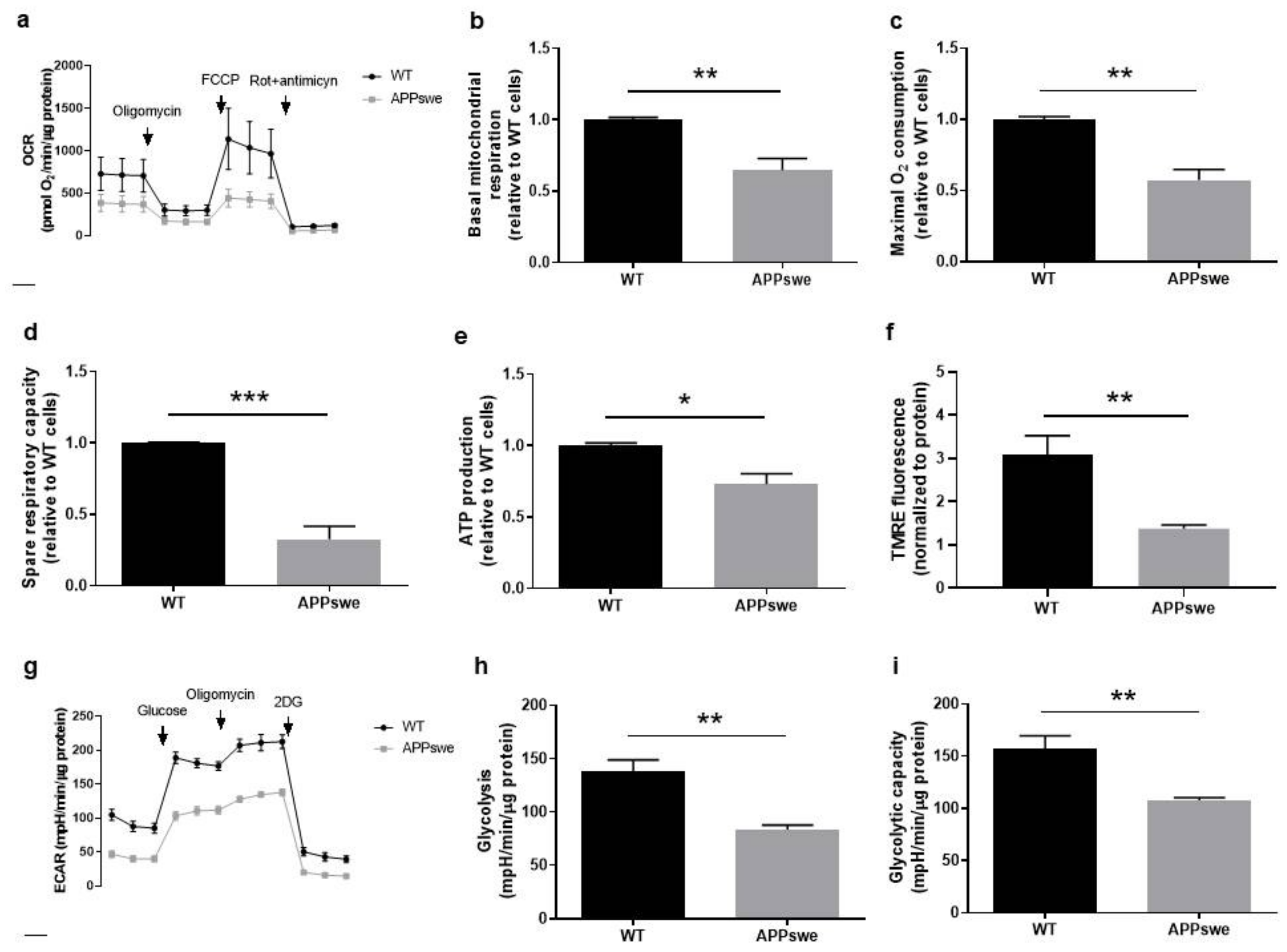
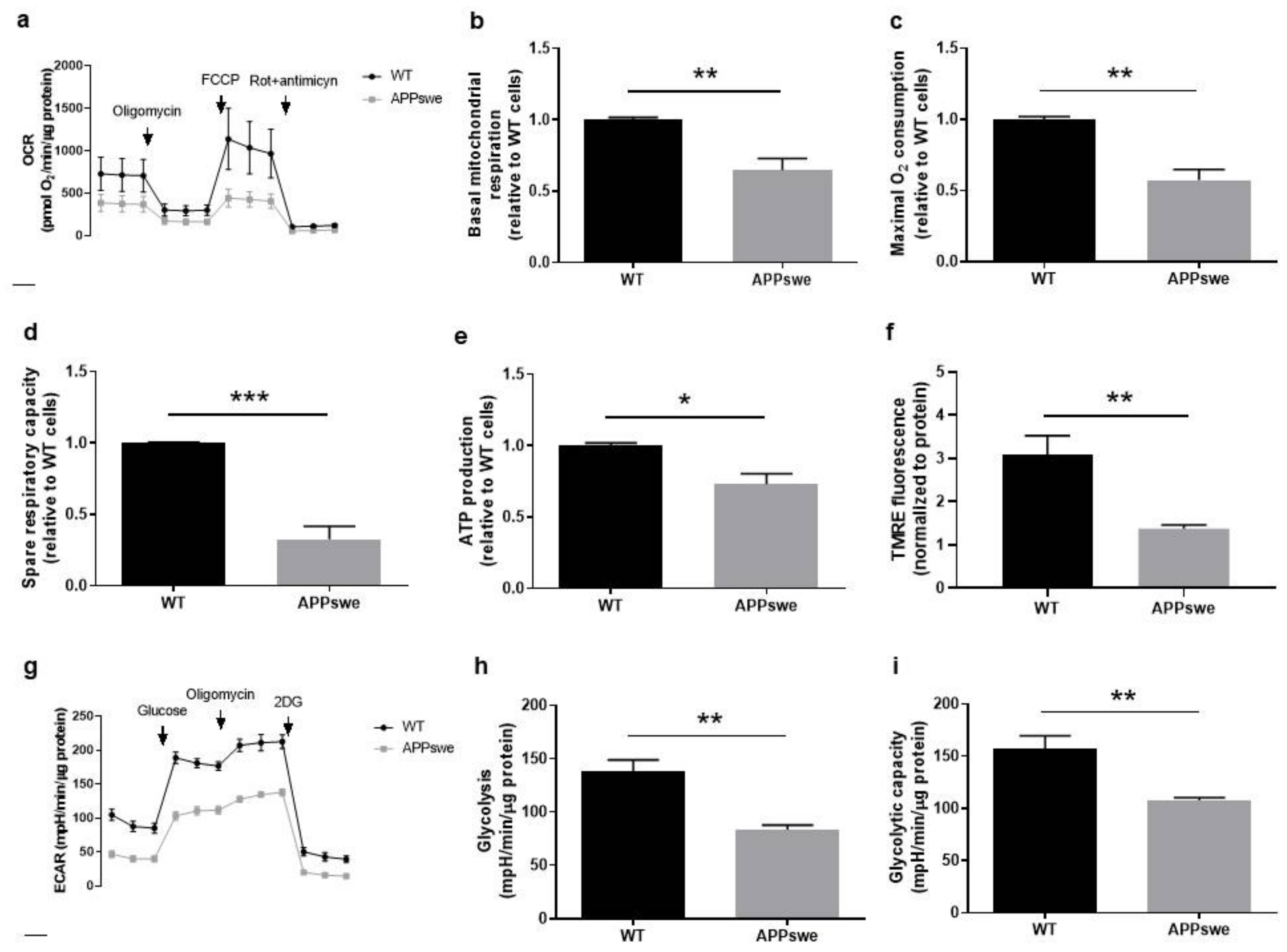
3.3. Oxygen and Glucose Fluxes in APPswe Cells
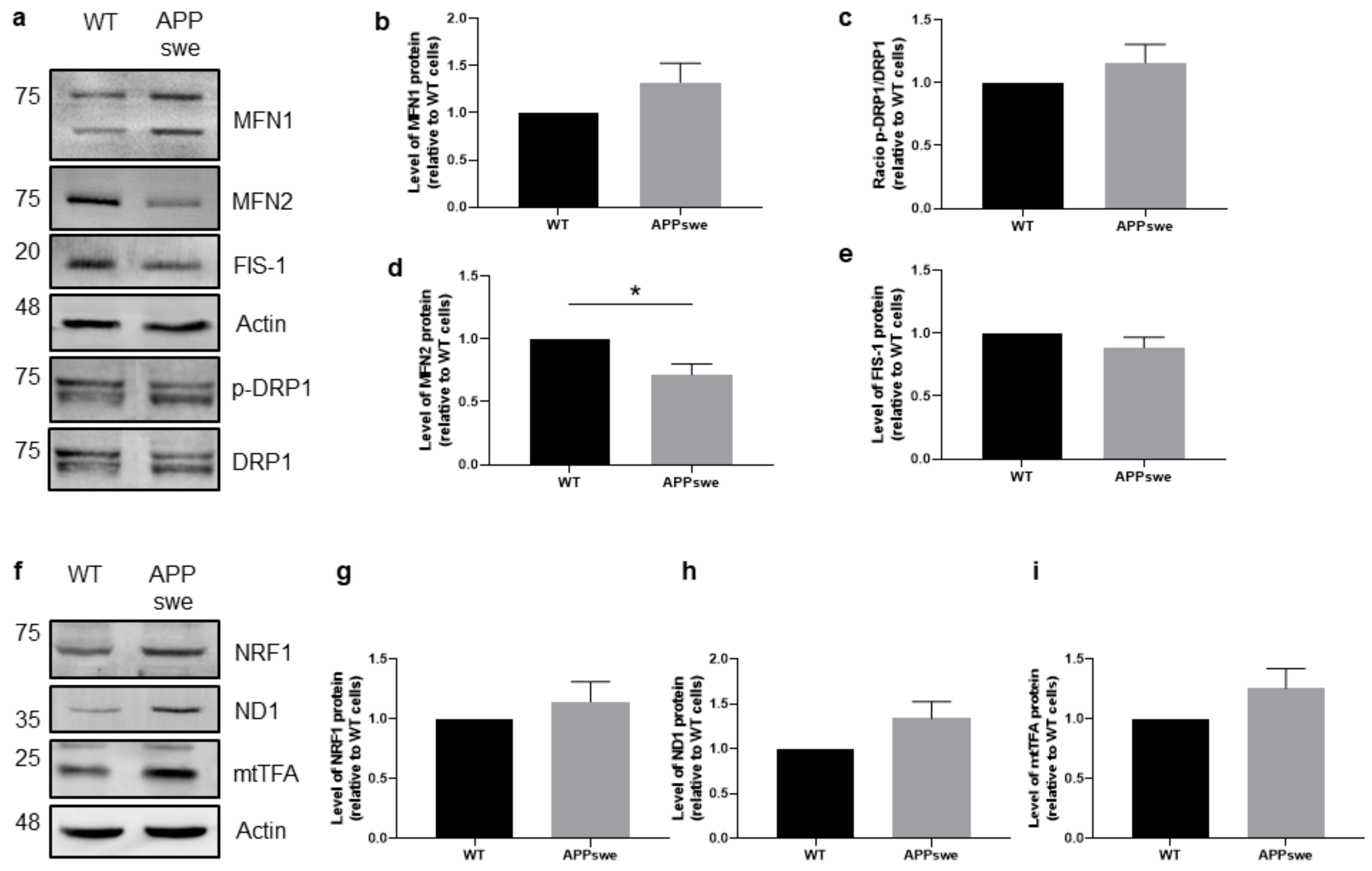
3.4. Impaired Mitochondrial Dynamics and Biogenesis in APPswe Cells
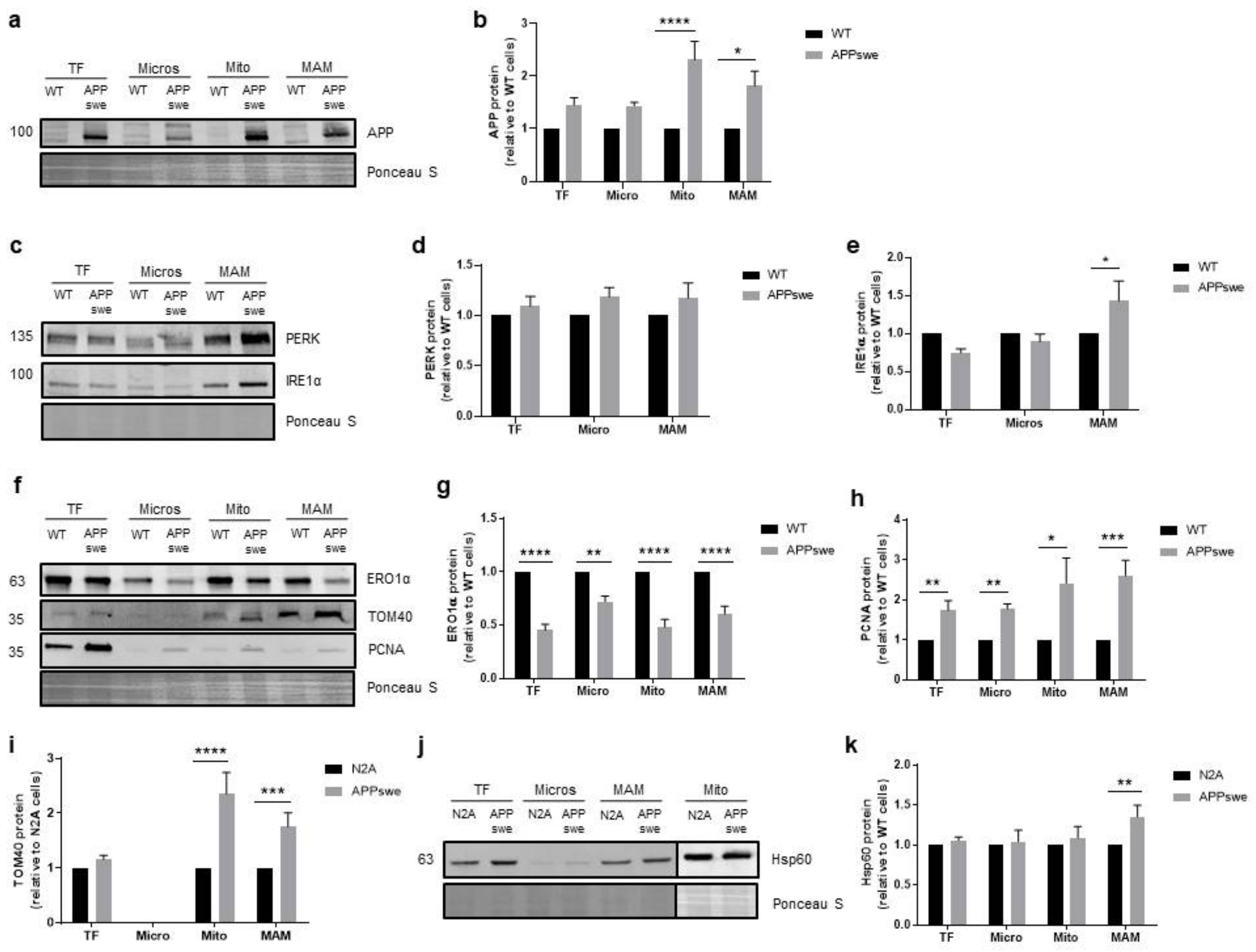
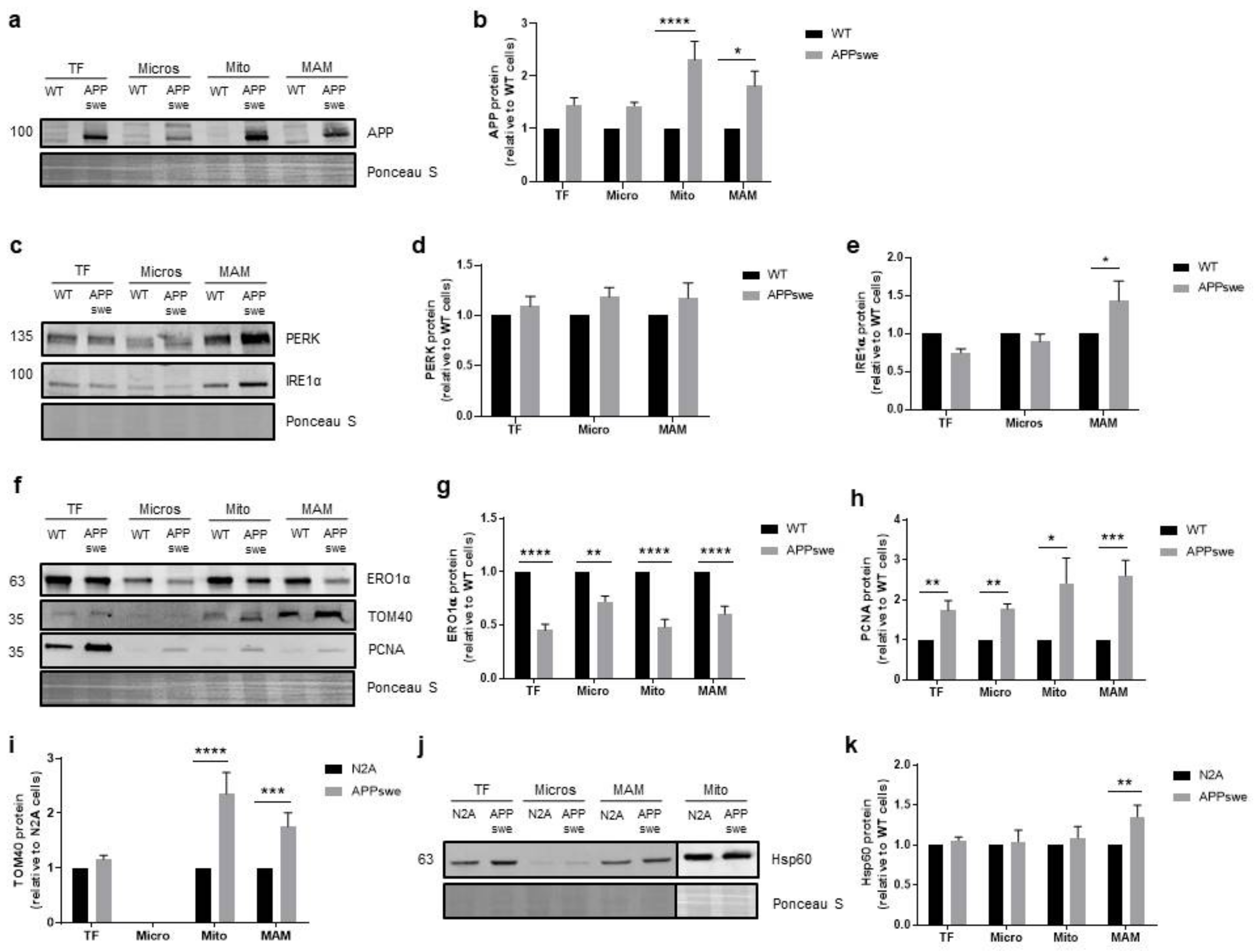
3.5. Cellular Stress Responses Are Affected in APPswe Cells
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Poirier, Y.; Grimm, A.; Schmitt, K.; Eckert, A. Link between the unfolded protein response and dysregulation of mitochondrial bioenergetics in Alzheimer’s disease. Cell Mol. Life Sci. 2019, 76, 1419–1431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winblad, B.; Amouyel, P.; Andrieu, S.; Ballard, C.; Brayne, C.; Brodaty, H.; Cedazo-Minguez, A.; Dubois, B.; Edvardsson, D.; Feldman, H.; et al. Defeating Alzheimer’s disease and other dementias: A priority for European science and society. Lancet Neurol. 2016, 15, 455–532. [Google Scholar] [CrossRef] [Green Version]
- Nelson, P.T.; Abner, E.L.; Schmitt, F.A.; Kryscio, R.J.; Jicha, G.A.; Santacruz, K.; Smith, C.D.; Patel, E.; Markesbery, W.R. Brains with medial temporal lobe neurofibrillary tangles but no neuritic amyloid plaques are a diagnostic dilemma but may have pathogenetic aspects distinct from Alzheimer disease. J. Neuropathol. Exp. Neurol. 2009, 68, 774–784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Area-Gomez, E.; Schon, E.A. Mitochondria-associated ER membranes and Alzheimer disease. Curr. Opin. Genet. Dev. 2016, 38, 90–96. [Google Scholar] [CrossRef] [Green Version]
- Area-Gomez, E.; Schon, E.A. On the pathogenesis of Alzheimer’s disease: The MAM hypothesis. FASEB J. 2017, 31, 864–867. [Google Scholar] [CrossRef] [Green Version]
- Area-Gomez, E.; de Groof, A.; Bonilla, E.; Montesinos, J.; Tanji, K.; Boldogh, I.; Pon, L.; Schon, E.A. A key role for MAM in mediating mitochondrial dysfunction in Alzheimer disease. Cell Death Dis. 2018, 9, 335. [Google Scholar] [CrossRef] [Green Version]
- Ferreiro, E.; Baldeiras, I.; Ferreira, I.L.; Costa, R.O.; Rego, A.C.; Pereira, C.F.; Oliveira, C.R. Mitochondrial- and endoplasmic reticulum-associated oxidative stress in Alzheimer’s disease: From pathogenesis to biomarkers. Int. J. Cell Biol. 2012, 2012, 1–23. [Google Scholar] [CrossRef]
- Cardoso, S.; Carvalho, C.; Correia, S.C.; Seiça, R.M.; Moreira, P.I. Alzheimer’s disease: From mitochondrial perturbations to mitochondrial medicine. Brain Pathol. 2016, 26, 632–647. [Google Scholar] [CrossRef] [PubMed]
- Simmen, T.; Herrera-Cruz, M.S. Plastic mitochondria-endoplasmic reticulum (ER) contacts use chaperones and tethers to mould their structure and signaling. Curr. Opin. Cell Biol. 2018, 53, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Giacomello, M.; Pellegrini, L. The coming of age of the mitochondria–ER contact: A matter of thickness. Cell Death Differ. 2016, 23, 1417–1427. [Google Scholar] [CrossRef] [PubMed]
- Wilson, E.L.; Metzakopian, E. ER-mitochondria contact sites in neurodegeneration: Genetic screening approaches to investigate novel disease mechanisms. Cell Death Differ. 2021, 28, 1804–1821. [Google Scholar] [CrossRef]
- Bernard-Marissal, N.; Chrast, R.; Schneider, B.L. Endoplasmic reticulum and mitochondria in diseases of motor and sensory neurons: A broken relationship? Cell Death Dis. 2018, 9, 333. [Google Scholar] [CrossRef]
- Gordaliza-Alaguero, I.; Cantó, C.; Zorzano, A. Metabolic implications of organelle–mitochondria communication. EMBO Rep. 2019, 20, 1–27. [Google Scholar] [CrossRef] [PubMed]
- Van Vliet, A.R.; Verfaillie, T.; Agostinis, P. New functions of mitochondria associated membranes in cellular signaling. Biochim. Biophys. Acta Mol. Cell Res. 2014, 1843, 2253–2262. [Google Scholar] [CrossRef] [Green Version]
- Rossi, A.; Pizzo, P.; Filadi, R. Calcium, mitochondria and cell metabolism: A functional triangle in bioenergetics. Biochim. Biophys. Acta Mol. Cell Res. 2019, 1866, 1068–1078. [Google Scholar] [CrossRef]
- Brunetti, D.; Catania, A.; Viscomi, C.; Deleidi, M.; Bindoff, L.A.; Ghezzi, D.; Zeviani, M. Role of PITRM1 in mitochondrial dysfunction and neurodegeneration. Biomedicines 2021, 9, 833. [Google Scholar] [CrossRef]
- Moreira, P.I.; Carvalho, C.; Zhu, X.; Smith, M.A.; Perry, G. Mitochondrial dysfunction is a trigger of Alzheimer’s disease pathophysiology. Biochim. Biophys. Acta Mol. Basis Dis. 2010, 1802, 2–10. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Zhao, F.; Ma, X.; Perry, G.; Zhu, X. Mitochondria dysfunction in the pathogenesis of Alzheimer’s disease: Recent advances. Mol. Neurodegener. 2020, 15, 30. [Google Scholar] [CrossRef] [PubMed]
- Cunnane, S.C.; Trushina, E.; Morland, C.; Prigione, A.; Casadesus, G.; Andrews, Z.B.; Beal, M.F.; Bergersen, L.H.; Brinton, R.D.; de la Monte, S.; et al. Brain energy rescue: An emerging therapeutic concept for neurodegenerative disorders of ageing. Nat. Rev. Drug Discov. 2020, 19, 609–633. [Google Scholar] [CrossRef]
- Thinakaran, G.; Teplow, D.B.; Siman, R.; Greenberg, B.; Sisodia, S.S. Metabolism of the “Swedish” amyloid precursor protein variant in Neuro2a (N2a) cells. J. Biol. Chem. 1996, 271, 9390–9397. [Google Scholar] [CrossRef] [Green Version]
- Cagnin, M.; Ozzano, M.; Bellio, N.; Fiorentino, I.; Follo, C.; Isidoro, C. Dopamine induces apoptosis in APPswe-expressing Neuro2A cells following Pepstatin-sensitive proteolysis of APP in acid compartments. Brain Res. 2012, 1471, 102–117. [Google Scholar] [CrossRef]
- Resende, R.; Ferreira-Marques, M.; Moreira, P.; Coimbra, J.R.M.; Baptista, S.J.; Isidoro, C.; Salvador, J.A.R.; Dinis, T.C.P.; Pereira, C.F.; Santos, A.E. New BACE1 chimeric peptide inhibitors selectively prevent AβPP-β cleavage decreasing amyloid-β production and accumulation in Alzheimer’s disease models. J. Alzheimer’s Dis. 2020, 76, 1317–1337. [Google Scholar] [CrossRef]
- Wieckowski, M.R.M.R.; Giorgi, C.; Lebiedzinska, M.; Duszynski, J.; Pinton, P. Isolation of mitochondria-associated membranes and mitochondria from animal tissues and cells. Nat. Protoc. 2009, 4, 1582–1590. [Google Scholar] [CrossRef]
- Williamson, C.D.; Wong, D.S.; Bozidis, P.; Zhang, A.; Colberg-Poley, A.M. Isolation of endoplasmic reticulum, mitochondria, and mitochondria-associated membrane and detergent resistant membrane fractions from transfected cells and from human cytomegalovirus-infected primary fibroblasts. In Current Protocols in Cell Biology; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2015; Volume 118, pp. 3–27. ISBN 0471143030. [Google Scholar]
- Prinz, W.A.; Toulmay, A.; Balla, T. The functional universe of membrane contact sites. Nat. Rev. Mol. Cell Biol. 2020, 21, 7–24. [Google Scholar] [CrossRef] [PubMed]
- Giorgi, C.; Missiroli, S.; Patergnani, S.; Duszynski, J.; Wieckowski, M.R.; Pinton, P. Mitochondria-associated membranes: Composition, molecular mechanisms, and physiopathological implications. Antioxid. Redox Signal. 2015, 22, 995–1019. [Google Scholar] [CrossRef]
- Herrera-Cruz, M.S.; Simmen, T. Over six decades of discovery and characterization of the architecture at mitochondria-associated membranes (MAMs). Adv. Exp. Med. Biol. 2017, 997, 13–31. [Google Scholar] [CrossRef] [PubMed]
- Raturi, A.; Simmen, T. Where the endoplasmic reticulum and the mitochondrion tie the knot: The mitochondria-associated membrane (MAM). Biochim. Biophys. Acta Mol. Cell Res. 2013, 1833, 213–224. [Google Scholar] [CrossRef] [Green Version]
- Manczak, M.; Calkins, M.J.; Reddy, P.H. Impaired mitochondrial dynamics and abnormal interaction of amyloid beta with mitochondrial protein Drp1 in neurons from patients with Alzheimer’s disease: Implications for neuronal damage. Hum. Mol. Genet. 2011, 20, 2495–2509. [Google Scholar] [CrossRef]
- Manczak, M.; Kandimalla, R.; Yin, X.; Reddy, P.H. Hippocampal mutant APP and amyloid beta-induced cognitive decline, dendritic spine loss, defective autophagy, mitophagy and mitochondrial abnormalities in a mouse model of Alzheimer’s disease. Hum. Mol. Genet. 2018, 27, 1332–1342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calvo-Rodriguez, M.; Hou, S.S.; Snyder, A.C.; Kharitonova, E.K.; Russ, A.N.; Das, S.; Fan, Z.; Muzikansky, A.; Garcia-Alloza, M.; Serrano-Pozo, A.; et al. Increased mitochondrial calcium levels associated with neuronal death in a mouse model of Alzheimer’s disease. Nat. Commun. 2020, 11, 2146. [Google Scholar] [CrossRef]
- Jadiya, P.; Kolmetzky, D.W.; Tomar, D.; Di Meco, A.; Lombardi, A.A.; Lambert, J.P.; Luongo, T.S.; Ludtmann, M.H.; Praticò, D.; Elrod, J.W. Impaired mitochondrial calcium efflux contributes to disease progression in models of Alzheimer’s disease. Nat. Commun. 2019, 10, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Marchi, S.; Patergnani, S.; Missiroli, S.; Morciano, G.; Rimessi, A.; Wieckowski, M.R.; Giorgi, C.; Pinton, P. Mitochondrial and endoplasmic reticulum calcium homeostasis and cell death. Cell Calcium 2018, 69, 62–72. [Google Scholar] [CrossRef]
- Patergnani, S.; Suski, J.M.; Agnoletto, C.; Bononi, A.; Bonora, M.; De Marchi, E.; Giorgi, C.; Marchi, S.; Missiroli, S.; Poletti, F.; et al. Calcium signaling around mitochondria associated membranes (MAMs). Cell Commun. Signal. 2011, 9, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weng, T.Y.; Tsai, S.Y.A.; Su, T.P. Roles of Sigma-1 receptors on mitochondrial functions relevant to neurodegenerative diseases. J. Biomed. Sci. 2017, 24, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Picone, P.; Nuzzo, D.; Caruana, L.; Scafidi, V.; Di Carlo, M. Mitochondrial dysfunction: Different routes to Alzheimer’s disease therapy. Oxid. Med. Cell Longev. 2014, 2014, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Carvalho, C.; Cardoso, S.; Correia, S.C.; Santos, R.X.; Santos, M.S.; Baldeiras, I.; Oliveira, C.R.; Moreira, P.I. Metabolic alterations induced by sucrose Intake and Alzheimer’s disease promote similar brain mitochondrial abnormalities. Diabetes 2012, 61, 1234–1242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Correia, C.S.; Santos, X.R.; Santos, S.M.; Casadesus, G.; LaManna, C.J.; Perry, G.; Smith, A.M.; Moreira, I.P. Mitochondrial abnormalities in a streptozotocin-induced rat model of sporadic Alzheimer’s disease. Curr. Alzheimer Res. 2013, 10, 406–419. [Google Scholar] [CrossRef]
- Tilokani, L.; Nagashima, S.; Paupe, V.; Prudent, J. Mitochondrial dynamics: Overview of molecular mechanisms. Essays Biochem. 2018, 62, 341–360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Onyango, I.G.; Lu, J.; Rodova, M.; Lezi, E.; Crafter, A.B.; Swerdlow, R.H. Regulation of neuron mitochondrial biogenesis and relevance to brain health. Biochim. Biophys. Acta Mol. Basis Dis. 2010, 1802, 228–234. [Google Scholar] [CrossRef] [Green Version]
- Gerakis, Y.; Hetz, C. Emerging roles of ER stress in the etiology and pathogenesis of Alzheimer’s disease. FEBS J. 2018, 285, 995–1011. [Google Scholar] [CrossRef] [Green Version]
- Hashimoto, S.; Saido, T.C. Critical review: Involvement of endoplasmic reticulum stress in the aetiology of Alzheimer’s disease. Open Biol. 2018, 8, 180024. [Google Scholar] [CrossRef] [PubMed]
- Anelli, T.; Bergamelli, L.; Margittai, E.; Rimessi, A.; Fagioli, C.; Malgaroli, A.; Pinton, P.; Ripamonti, M.; Rizzuto, R.; Sitia, R. Ero1α regulates Ca2+ fluxes at the endoplasmic reticulum–mitochondria interface (MAM). Antioxid. Redox Signal. 2012, 16, 1077–1087. [Google Scholar] [CrossRef]
- Seervi, M.; Sobhan, P.K.; Joseph, J.; Ann Mathew, K.; Santhoshkumar, T.R. ERO1α-dependent endoplasmic reticulum-mitochondrial calcium flux contributes to ER stress and mitochondrial permeabilization by procaspase-activating compound-1 (PAC-1). Cell Death Dis. 2013, 4, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Silva, A.; Pereira, M.; Carrascal, M.A.; Brites, G.; Neves, B.; Moreira, P.; Resende, R.; Silva, M.M.; Santos, A.E.; Pereira, C.; et al. Calcium modulation, anti-oxidant and anti-inflammatory effect of skin allergens targeting the Nrf2 signaling pathway in Alzheimer’s disease cellular models. Int. J. Mol. Sci. 2020, 21, 7791. [Google Scholar] [CrossRef]
- Costa, R.O.; Ferreiro, E.; Martins, I.; Santana, I.; Cardoso, S.M.; Oliveira, C.R.; Pereira, C.M.F. Amyloid β-induced ER stress is enhanced under mitochondrial dysfunction conditions. Neurobiol. Aging 2012, 33, 824.e5–824.e16. [Google Scholar] [CrossRef]
- Fonseca, A.C.R.G.; Ferreiro, E.; Oliveira, C.R.; Cardoso, S.M.; Pereira, C.F. Activation of the endoplasmic reticulum stress response by the amyloid-beta 1–40 peptide in brain endothelial cells. Biochim. Biophys. Acta Mol. Basis Dis. 2013, 1832, 2191–2203. [Google Scholar] [CrossRef] [Green Version]
- Witko-Sarsat, V.; Mocek, J.; Bouayad, D.; Tamassia, N.; Ribeil, J.-A.; Candalh, C.; Davezac, N.; Reuter, N.; Mouthon, L.; Hermine, O.; et al. Proliferating cell nuclear antigen acts as a cytoplasmic platform controlling human neutrophil survival. J. Exp. Med. 2010, 207, 2631–2645. [Google Scholar] [CrossRef] [PubMed]
- Gottschalk, W.K.; Lutz, M.W.; He, Y.T.; Saunders, A.M.; Burns, D.K.; Roses, A.D.; Chiba-Falek, O. The broad impact of TOM40 on neurodegenerative diseases in aging. J. Park. Dis. Alzheimer’s Dis. 2014, 1, 1–25. [Google Scholar] [CrossRef]
- Campanella, C.; Pace, A.; Bavisotto, C.C.; Marzullo, P.; Gammazza, A.M.; Buscemi, S.; Piccionello, A.P. Heat shock proteins in Alzheimer’s disease: Role and targeting. Int. J. Mol. Sci. 2018, 19, 2603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Zhu, X. Endoplasmic reticulum-mitochondria tethering in neurodegenerative diseases. Transl. Neurodegener. 2017, 6, 21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Völgyi, K.; Badics, K.; Sialana, F.J.; Gulyássy, P.; Udvari, E.B.; Kis, V.; Drahos, L.; Lubec, G.; Kékesi, K.A.; Juhász, G. Early presymptomatic changes in the proteome of mitochondria-associated membrane in the APP/PS1 mouse model of Alzheimer’s disease. Mol. Neurobiol. 2018, 55, 7839–7857. [Google Scholar] [CrossRef]
- Jiang, S.; Nandy, P.; Wang, W.; Ma, X.; Hsia, J.; Wang, C.; Wang, Z.; Niu, M.; Siedlak, S.L.; Torres, S.; et al. Mfn2 ablation causes an oxidative stress response and eventual neuronal death in the hippocampus and cortex. Mol. Neurodegener. 2018, 13, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, S.; Nandy, P.; Austria, Q.; Siedlak, S.L.; Torres, S.; Fujioka, H.; Wang, W.; Zhu, X. Mfn2 ablation in the adult mouse hippocampus and cortex causes neuronal death. Cells 2020, 9, 116. [Google Scholar] [CrossRef] [Green Version]
- Park, J.; Choi, H.; Min, J.S.; Kim, B.; Lee, S.R.; Yun, J.W.; Choi, M.S.; Chang, K.T.; Lee, D.S. Loss of mitofusin 2 links beta-amyloid-mediated mitochondrial fragmentation and Cdk5-induced oxidative stress in neuron cells. J. Neurochem. 2015, 132, 687–702. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Su, B.; Zheng, L.; Perry, G.; Smith, M.A.; Zhu, X. The role of abnormal mitochondrial dynamics in the pathogenesis of Alzheimer’s disease. J. Neurochem. 2009, 109, 153–159. [Google Scholar] [CrossRef] [Green Version]
- De Brito, O.M.; Scorrano, L. Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature 2008, 456, 605–610. [Google Scholar] [CrossRef]
- Naon, D.; Zaninello, M.; Giacomello, M.; Varanita, T.; Grespi, F.; Lakshminaranayan, S.; Serafini, A.; Semenzato, M.; Herkenne, S.; Hernández-Alvarez, M.I.; et al. Critical reappraisal confirms that mitofusin 2 is an endoplasmic reticulum–mitochondria tether. Proc. Natl. Acad. Sci. USA 2016, 113, 11249–11254. [Google Scholar] [CrossRef] [Green Version]
- Janikiewicz, J.; Szymański, J.; Malinska, D.; Patalas-Krawczyk, P.; Michalska, B.; Duszyński, J.; Giorgi, C.; Bonora, M.; Dobrzyn, A.; Wieckowski, M.R. Mitochondria-associated membranes in aging and senescence: Structure, function, and dynamics. Cell Death Dis. 2018, 9, 332. [Google Scholar] [CrossRef] [PubMed]
- Esteras, N.; Abramov, A.Y. Mitochondrial calcium deregulation in the mechanism of beta-amyloid and tau pathology. Cells 2020, 9, 2135. [Google Scholar] [CrossRef] [PubMed]
- Sanz-Blasco, S.; Valero, R.A.; Rodríguez-Crespo, I.; Villalobos, C.; Núñez, L. Mitochondrial Ca2+ overload underlies Aβ oligomers neurotoxicity providing an unexpected mechanism of neuroprotection by NSAIDs. PLoS ONE 2008, 3, e2718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mallilankaraman, K.; Cárdenas, C.; Doonan, P.J.; Chandramoorthy, H.C.; Irrinki, K.M.; Golenár, T.; Csordás, G.; Madireddi, P.; Yang, J.; Müller, M.; et al. MCUR1 is an essential component of mitochondrial Ca2+ uptake that regulates cellular metabolism. Nat. Cell Biol. 2012, 14, 1336–1343. [Google Scholar] [CrossRef] [Green Version]
- Filadi, R.; Greotti, E.; Turacchio, G.; Luini, A.; Pozzan, T.; Pizzo, P. Mitofusin 2 ablation increases endoplasmic reticulum–mitochondria coupling. Proc. Natl. Acad. Sci. USA 2015, 112, E2174–E2181. [Google Scholar] [CrossRef] [Green Version]
- Leal, N.S.; Schreiner, B.; Pinho, C.M.; Filadi, R.; Wiehager, B.; Karlström, H.; Pizzo, P.; Ankarcrona, M. Mitofusin-2 knockdown increases ER-mitochondria contact and decreases amyloid β-peptide production. J. Cell. Mol. Med. 2016, 20, 1686–1695. [Google Scholar] [CrossRef] [PubMed]
- Finkel, T.; Menazza, S.; Holmström, K.M.; Parks, R.J.; Liu, J.; Sun, J.; Liu, J.; Pan, X.; Murphy, E. The ins and outs of mitochondrial calcium. Circ. Res. 2015, 116, 1810–1819. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shoshan-Barmatz, V.; Nahon-Crystal, E.; Shteinfer-Kuzmine, A.; Gupta, R. VDAC1, mitochondrial dysfunction, and Alzheimer’s disease. Pharmacol. Res. 2018, 131, 87–101. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, T.; Su, T.-P. Sigma-1 receptor chaperones at the ER- mitochondrion interface regulate Ca2+ signaling and cell survival. Cell 2007, 131, 596–610. [Google Scholar] [CrossRef] [Green Version]
- Hedskog, L.; Pinho, C.M.; Filadi, R.; Ronnback, A.; Hertwig, L.; Wiehager, B.; Larssen, P.; Gellhaar, S.; Sandebring, A.; Westerlund, M.; et al. Modulation of the endoplasmic reticulum-mitochondria interface in Alzheimer’s disease and related models. Proc. Natl. Acad. Sci. USA 2013, 110, 7916–7921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitsuda, T.; Omi, T.; Tanimukai, H.; Sakagami, Y.; Tagami, S.; Okochi, M.; Kudo, T.; Takeda, M. Sigma-1Rs are upregulated via PERK/eIF2α/ATF4 pathway and execute protective function in ER stress. Biochem. Biophys. Res. Commun. 2011, 415, 519–525. [Google Scholar] [CrossRef]
- Sun, F.C.; Wei, S.; Li, C.W.; Chang, Y.S.; Chao, C.C.; Lai, Y.K. Localization of GRP78 to mitochondria under the unfolded protein response. Biochem. J. 2006, 396, 31–39. [Google Scholar] [CrossRef] [Green Version]
- Paz Gavilán, M.; Vela, J.; Castaño, A.; Ramos, B.; del Río, J.C.; Vitorica, J.; Ruano, D. Cellular environment facilitates protein accumulation in aged rat hippocampus. Neurobiol. Aging 2006, 27, 973–982. [Google Scholar] [CrossRef] [PubMed]
- Naidoo, N.; Ferber, M.; Master, M.; Zhu, Y.; Pack, A.I. Aging impairs the unfolded protein response to sleep deprivation and leads to proapoptotic signaling. J. Neurosci. 2008, 28, 6539–6548. [Google Scholar] [CrossRef] [PubMed]
- Katayama, T.; Imaizumi, K.; Sato, N.; Miyoshi, K.; Kudo, T.; Hitomi, J.; Morihara, T.; Yoneda, T.; Gomi, F.; Mori, Y.; et al. Presenilin-1 mutations downregulate the signalling pathway of the unfolded-protein response. Nat. Cell Biol. 1999, 1, 479–485. [Google Scholar] [CrossRef]
- Hoozemans, J.J.M.; Veerhuis, R.; Van Haastert, E.S.; Rozemuller, J.M.; Baas, F.; Eikelenboom, P.; Scheper, W. The unfolded protein response is activated in Alzheimer’s disease. Acta Neuropathol. 2005, 110, 165–172. [Google Scholar] [CrossRef]
- Soejima, N.; Ohyagi, Y.; Nakamura, N.; Himeno, E.; Iinuma, K.M.; Sakae, N.; Yamasaki, R.; Tabira, T.; Murakami, K.; Irie, K.; et al. Intracellular accumulation of toxic turn amyloid-β is associated with endoplasmic reticulum stress in Alzheimer’s disease. Curr. Alzheimer Res. 2013, 10, 11–20. [Google Scholar] [CrossRef]
- Costa, R.O.; Lacor, P.N.; Ferreira, I.L.; Resende, R.; Auberson, Y.P.; Klein, W.L.; Oliveira, C.R.; Rego, A.C.; Pereira, C.M.F. Endoplasmic reticulum stress occurs downstream of GluN2B subunit of N -methyl-D-aspartate receptor in mature hippocampal cultures treated with amyloid-β oligomers. Aging Cell 2012, 11, 823–833. [Google Scholar] [CrossRef] [PubMed]
- Kudo, T.; Okumura, M.; Imaizumi, K.; Araki, W.; Morihara, T.; Tanimukai, H.; Kamagata, E.; Tabuchi, N.; Kimura, R.; Kanayama, D.; et al. Altered localization of amyloid precursor protein under endoplasmic reticulum stress. Biochem. Biophys. Res. Commun. 2006, 344, 525–530. [Google Scholar] [CrossRef]
- Hoshino, T.; Nakaya, T.; Araki, W.; Suzuki, K.; Suzuki, T.; Mizushima, T. Endoplasmic reticulum chaperones inhibit the production of amyloid-β peptides. Biochem. J. 2007, 402, 581–589. [Google Scholar] [CrossRef] [Green Version]
- Dixit, S.; Fessel, J.P.; Harrison, F.E. Mitochondrial dysfunction in the APP/PSEN1 mouse model of Alzheimer’s disease and a novel protective role for ascorbate. Free Radic. Biol. Med. 2017, 112, 515–523. [Google Scholar] [CrossRef] [PubMed]
- Pérez, M.J.; Ponce, D.P.; Aranguiz, A.; Behrens, M.I.; Quintanilla, R.A. Mitochondrial permeability transition pore contributes to mitochondrial dysfunction in fibroblasts of patients with sporadic Alzheimer’s disease. Redox Biol. 2018, 19, 290–300. [Google Scholar] [CrossRef] [PubMed]
- Parekh, A.B. Mitochondrial regulation of intracellular Ca2+ signaling: More than just simple Ca2+ buffers. Physiology 2003, 18, 252–256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silva, D.F.; Selfridge, J.E.; Lu, J.; E, L.; Roy, N.; Hutfles, L.; Burns, J.M.; Michaelis, E.K.; Yan, S.; Cardoso, S.M.; et al. Bioenergetic flux, mitochondrial mass and mitochondrial morphology dynamics in AD and MCI cybrid cell lines. Hum. Mol. Genet. 2013, 22, 3931–3946. [Google Scholar] [CrossRef] [PubMed]
- Schrepfer, E.; Scorrano, L. Mitofusins, from mitochondria to metabolism. Mol. Cell 2016, 61, 683–694. [Google Scholar] [CrossRef] [Green Version]
- García-Escudero, V.; Martín-Maestro, P.; Perry, G.; Avila, J. Deconstructing mitochondrial dysfunction in Alzheimer disease. Oxid. Med. Cell Longev. 2013, 2013, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Wang, W.; Li, L.; Perry, G.; Lee, H.; Zhu, X. Oxidative stress and mitochondrial dysfunction in Alzheimer’s disease. Biochim. Biophys. Acta Mol. Basis Dis. 2014, 1842, 1240–1247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Su, B.; Siedlak, S.L.; Moreira, P.I.; Fujioka, H.; Wang, Y.; Casadesus, G.; Zhu, X. Amyloid-β overproduction causes abnormal mitochondrial dynamics via differential modulation of mitochondrial fission/fusion proteins. Proc. Natl. Acad. Sci. USA 2008, 105, 19318–19323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eysert, F.; Kinoshita, P.F.; Mary, A.; Vaillant-Beuchot, L.; Checler, F.; Chami, M. Molecular dysfunctions of mitochondria-associated membranes (MAMs) in Alzheimer’s disease. Int. J. Mol. Sci. 2020, 21, 9521. [Google Scholar] [CrossRef] [PubMed]
- Muñoz, J.P.; Ivanova, S.; Sánchez-Wandelmer, J.; Martínez-Cristóbal, P.; Noguera, E.; Sancho, A.; Díaz-Ramos, A.; Hernández-Alvarez, M.I.; Sebastián, D.; Mauvezin, C.; et al. Mfn2 modulates the UPR and mitochondrial function via repression of PERK. EMBO J. 2013, 32, 2348–2361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verfaillie, T.; Rubio, N.; Garg, A.D.; Bultynck, G.; Rizzuto, R.; Decuypere, J.-P.; Piette, J.; Linehan, C.; Gupta, S.; Samali, A.; et al. PERK is required at the ER-mitochondrial contact sites to convey apoptosis after ROS-based ER stress. Cell Death Differ. 2012, 19, 1880–1891. [Google Scholar] [CrossRef] [Green Version]
- Carreras-Sureda, A.; Jaña, F.; Urra, H.; Durand, S.; Mortenson, D.E.; Sagredo, A.; Bustos, G.; Hazari, Y.; Ramos-Fernández, E.; Sassano, M.L.; et al. Non-canonical function of IRE1α determines mitochondria-associated endoplasmic reticulum composition to control calcium transfer and bioenergetics. Nat. Cell Biol. 2019, 21, 755–767. [Google Scholar] [CrossRef]
- Pająk, B.; Kania, E.; Orzechowski, A. Killing me softly: Connotations to unfolded protein response and oxidative stress in Alzheimer’s disease. Oxid. Med. Cell Longev. 2016, 2016, 1–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, G.; Mongillo, M.; Chin, K.-T.; Harding, H.; Ron, D.; Marks, A.R.; Tabas, I. Role of ERO1-α–mediated stimulation of inositol 1,4,5-triphosphate receptor activity in endoplasmic reticulum stress–induced apoptosis. J. Cell Biol. 2009, 186, 783–792. [Google Scholar] [CrossRef] [Green Version]
- Olaisen, C.; Müller, R.; Nedal, A.; Otterlei, M. PCNA-interacting peptides reduce Akt phosphorylation and TLR-mediated cytokine secretion suggesting a role of PCNA in cellular signaling. Cell Signal. 2015, 27, 1478–1487. [Google Scholar] [CrossRef] [Green Version]
- Frade, J.M.; López-Sánchez, N. A novel hypothesis for Alzheimer disease based on neuronal tetraploidy induced by p75 NTR. Cell Cycle 2010, 9, 1934–1941. [Google Scholar] [CrossRef]
- Jensen, H.L.B.; Lillenes, M.S.; Rabano, A.; Günther, C.-C.; Riaz, T.; Kalayou, S.T.; Ulstein, I.D.; Bøhmer, T.; Tønjum, T. Expression of nucleotide excision repair in Alzheimer’s disease is higher in brain tissue than in blood. Neurosci. Lett. 2018, 672, 53–58. [Google Scholar] [CrossRef]
- Anandatheerthavarada, H.K.; Biswas, G.; Robin, M.-A.; Avadhani, N.G. Mitochondrial targeting and a novel transmembrane arrest of Alzheimer’s amyloid precursor protein impairs mitochondrial function in neuronal cells. J. Cell Biol. 2003, 161, 41–54. [Google Scholar] [CrossRef]
- Hansson Petersen, C.A.; Alikhani, N.; Behbahani, H.; Wiehager, B.; Pavlov, P.F.; Alafuzoff, I.; Leinonen, V.; Ito, A.; Winblad, B.; Glaser, E.; et al. The amyloid-peptide is imported into mitochondria via the TOM import machinery and localized to mitochondrial cristae. Proc. Natl. Acad. Sci. USA 2008, 105, 13145–13150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Devi, L.; Prabhu, B.M.; Galati, D.F.; Avadhani, N.G.; Anandatheerthavarada, H.K. Accumulation of amyloid precursor protein in the mitochondrial import channels of human Alzheimer’s disease brain is associated with mitochondrial dysfunction. J. Neurosci. 2006, 26, 9057–9068. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lustbader, J.W.; Cirilli, M.; Lin, C.; Xu, H.W.; Takuma, K.; Wang, N.; Caspersen, C.; Chen, X.; Pollak, S.; Chaney, M.; et al. ABAD directly links Abeta to mitochondrial toxicity in Alzheimer’s disease. Science 2004, 304, 448–452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manczak, M.; Anekonda, T.S.; Henson, E.; Park, B.S.; Quinn, J.; Reddy, P.H. Mitochondria are a direct site of A beta accumulation in Alzheimer’s disease neurons: Implications for free radical generation and oxidative damage in disease progression. Hum. Mol. Genet. 2006, 15, 1437–1449. [Google Scholar] [CrossRef] [PubMed]
- Walls, K.C.; Coskun, P.; Gallegos-Perez, J.L.; Zadourian, N.; Freude, K.; Rasool, S.; Blurton-Jones, M.; Green, K.N.; LaFerla, F.M. Swedish Alzheimer mutation induces mitochondrial dysfunction mediated by HSP60 mislocalization of amyloid precursor protein (APP) and beta-amyloid. J. Biol. Chem. 2012, 287, 30317–30327. [Google Scholar] [CrossRef] [Green Version]
- Pavlov, P.F.; Petersen, C.H.; Glaser, E.; Ankarcrona, M. Mitochondrial accumulation of APP and Aβ: Significance for Alzheimer disease pathogenesis. J. Cell Mol. Med. 2009, 13, 4137–4145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simonovitch, S.; Schmukler, E.; Masliah, E.; Pinkas-Kramarski, R.; Michaelson, D.M. The effects of APOE4 on mitochondrial dynamics and proteins in vivo. J. Alzheimer’s Dis. 2019, 70, 861–875. [Google Scholar] [CrossRef] [PubMed]
- Beck, J.S.; Mufson, E.J.; Counts, S.E. Evidence for mitochondrial UPR gene activation in familial and sporadic Alzheimer’s disease. Curr. Alzheimer Res. 2016, 13, 610–614. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primary Antibody | Dilution | Species | Company | Catalog Number | Location |
|---|---|---|---|---|---|
| APP | 1:4000 | Rabbit | Sigma-Aldrich | A8717 | St. Louis, MO, USA |
| β-Actin | 1:10,000 | Mouse | Sigma-Aldrich | A5316 | St. Louis, MO, USA |
| BiP/GRP78 | 1:1000 | Mouse | BD Transduction | 610978 | San Jose, CA, USA |
| DRP1 | 1:1000 | Rabbit | Cell Signaling | 8570 | Danvers, MA, USA |
| ERO1α | 1:1000 | Mouse | Santa Cruz Biotechnology | sc-100805 | Santa Cruz, CA, USA |
| FIS-1 | 1:500 | Rabbit | Novus Biologicals | NB100-56646 | Littleton, CO, USA |
| Hsp60 | 1:1000 | Mouse | BD Transduction | 611563 | San Jose, CA, USA |
| IRE1α | 1:500 | Rabbit | Cell Signaling | 3294 | Danvers, MA, USA |
| MCU | 1:1000 | Rabbit | Cell Signaling | 14997 | Danvers, MA, USA |
| Mfn1 | 1:1000 | Rabbit | Santa Cruz Biotechnology | sc-50330 | Santa Cruz, CA, USA |
| Mfn2 | 1:1000 | Mouse | Santa Cruz Biotechnology | sc-100560 | Santa Cruz, CA, USA |
| mtTFA | 1:1000 | Goat | Santa Cruz Biotechnology | sc-23588 | Santa Cruz, CA, USA |
| ND1 | 1:1000 | Goat | Santa Cruz Biotechnology | sc-20493 | Santa Cruz, CA, USA |
| NRF-1 | 1:1000 | Rabbit | Santa Cruz Biotechnology | sc-33771 | Santa Cruz, CA, USA |
| PCNA | 1:1000 | Mouse | Santa Cruz Biotechnology | sc-25280 | Santa Cruz, CA, USA |
| PERK | 1:500 | Rabbit | Cell Signaling | 3192 | Danvers, MA, USA |
| p-DRP1 | 1:500 | Rabbit | Cell Signaling | 3455 | Danvers, MA, USA |
| Sigma1R | 1:2000 | Goat | Santa Cruz Biotechnology | sc-22948 | Santa Cruz, CA, USA |
| Tom40 | 1:1000 | Mouse | Santa Cruz Biotechnology | sc-365467 | Santa Cruz, CA, USA |
| VDAC1 | 1:1000 | Mouse | Santa Cruz Biotechnology | sc-390996 | Santa Cruz, CA, USA |
| Secondary Antibody | Dilution | Species | Company | Catalog Number | Location |
|---|---|---|---|---|---|
| IgG (anti-goat) | 1:10,000 | Rabbit | Santa Cruz Biotechnology | sc-2771 | Santa Cruz, CA, USA |
| IgG (anti-mouse) | 1:10,000 | Goat | Thermo Fisher | 31320 | Waltham, MA, USA |
| IgG (anti-rabbit) | 1:20,000 | Goat | GE Healthcare | NIF1317 | Chicago, IL, USA |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fernandes, T.; Resende, R.; Silva, D.F.; Marques, A.P.; Santos, A.E.; Cardoso, S.M.; Domingues, M.R.; Moreira, P.I.; Pereira, C.F. Structural and Functional Alterations in Mitochondria-Associated Membranes (MAMs) and in Mitochondria Activate Stress Response Mechanisms in an In Vitro Model of Alzheimer’s Disease. Biomedicines 2021, 9, 881. https://doi.org/10.3390/biomedicines9080881
Fernandes T, Resende R, Silva DF, Marques AP, Santos AE, Cardoso SM, Domingues MR, Moreira PI, Pereira CF. Structural and Functional Alterations in Mitochondria-Associated Membranes (MAMs) and in Mitochondria Activate Stress Response Mechanisms in an In Vitro Model of Alzheimer’s Disease. Biomedicines. 2021; 9(8):881. https://doi.org/10.3390/biomedicines9080881
Chicago/Turabian StyleFernandes, Tânia, Rosa Resende, Diana F. Silva, Ana P. Marques, Armanda E. Santos, Sandra M. Cardoso, M. Rosário Domingues, Paula I. Moreira, and Cláudia F. Pereira. 2021. "Structural and Functional Alterations in Mitochondria-Associated Membranes (MAMs) and in Mitochondria Activate Stress Response Mechanisms in an In Vitro Model of Alzheimer’s Disease" Biomedicines 9, no. 8: 881. https://doi.org/10.3390/biomedicines9080881
APA StyleFernandes, T., Resende, R., Silva, D. F., Marques, A. P., Santos, A. E., Cardoso, S. M., Domingues, M. R., Moreira, P. I., & Pereira, C. F. (2021). Structural and Functional Alterations in Mitochondria-Associated Membranes (MAMs) and in Mitochondria Activate Stress Response Mechanisms in an In Vitro Model of Alzheimer’s Disease. Biomedicines, 9(8), 881. https://doi.org/10.3390/biomedicines9080881