
Au2phen and Auoxo6, Two Dinuclear Oxo-Bridged Gold(III) Compounds, Induce Apoptotic Signaling in Human Ovarian A2780 Cancer Cells

 ,
,  , , ,
, , ,  ,
,  ,
,  and
and
Abstract
:1. Introduction
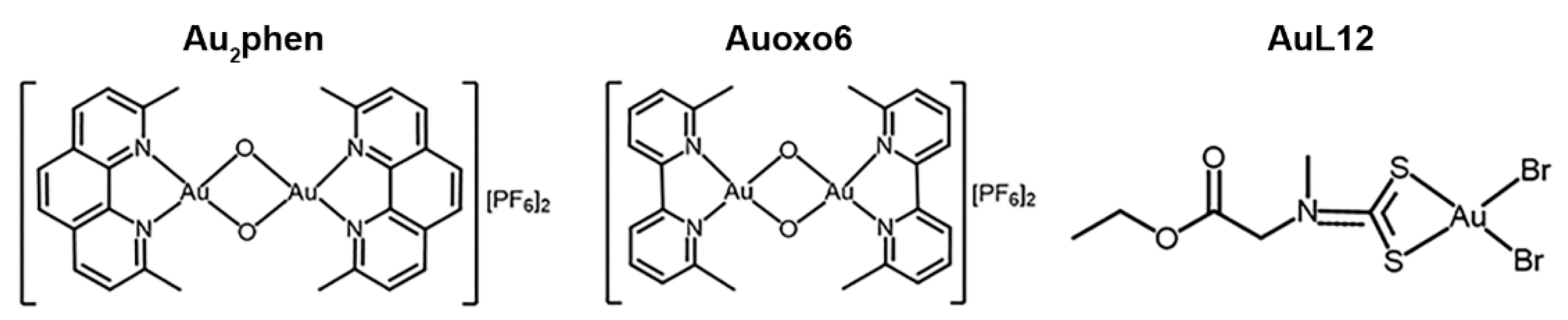
2. Materials and Methods
2.1. Materials
2.2. Chemical Synthesis and Drug Treatment
2.3. Cell Line and Culture Conditions
2.4. Study of the Cytotoxic Effects on A2780 Cell Line
2.5. Assessment of Apoptosis by Flow Cytometry
2.6. Thioredoxin Reductase Activity Assay
2.7. Intracellular ROS Evaluation
2.8. Measurement of Mitochondrial Membrane Potential
2.9. Mitochondrial Respiration Analysis
2.10. Lactate Production Assay
2.11. Western Blot Analysis
2.12. Statistical Analysis
3. Results
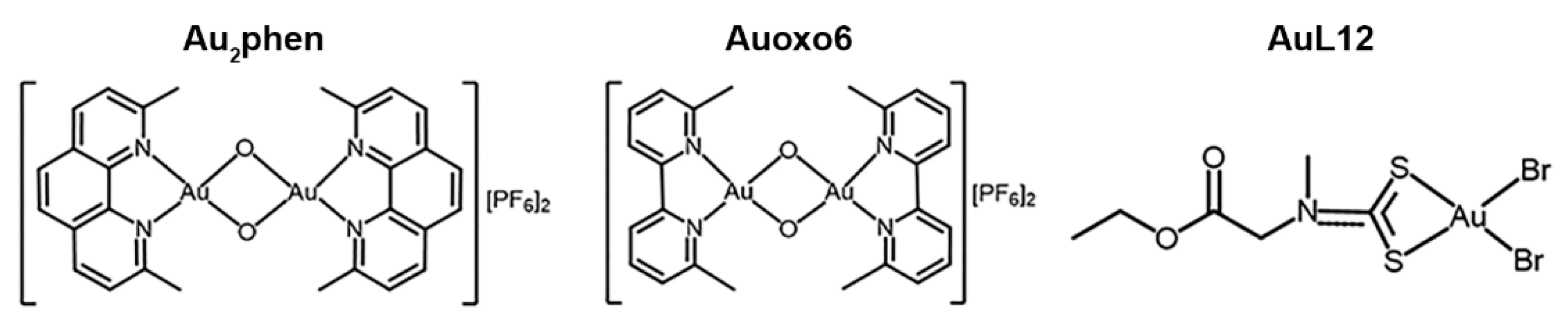
3.1. Antiproliferative Activity of Au2phen, Auoxo6, AuL12
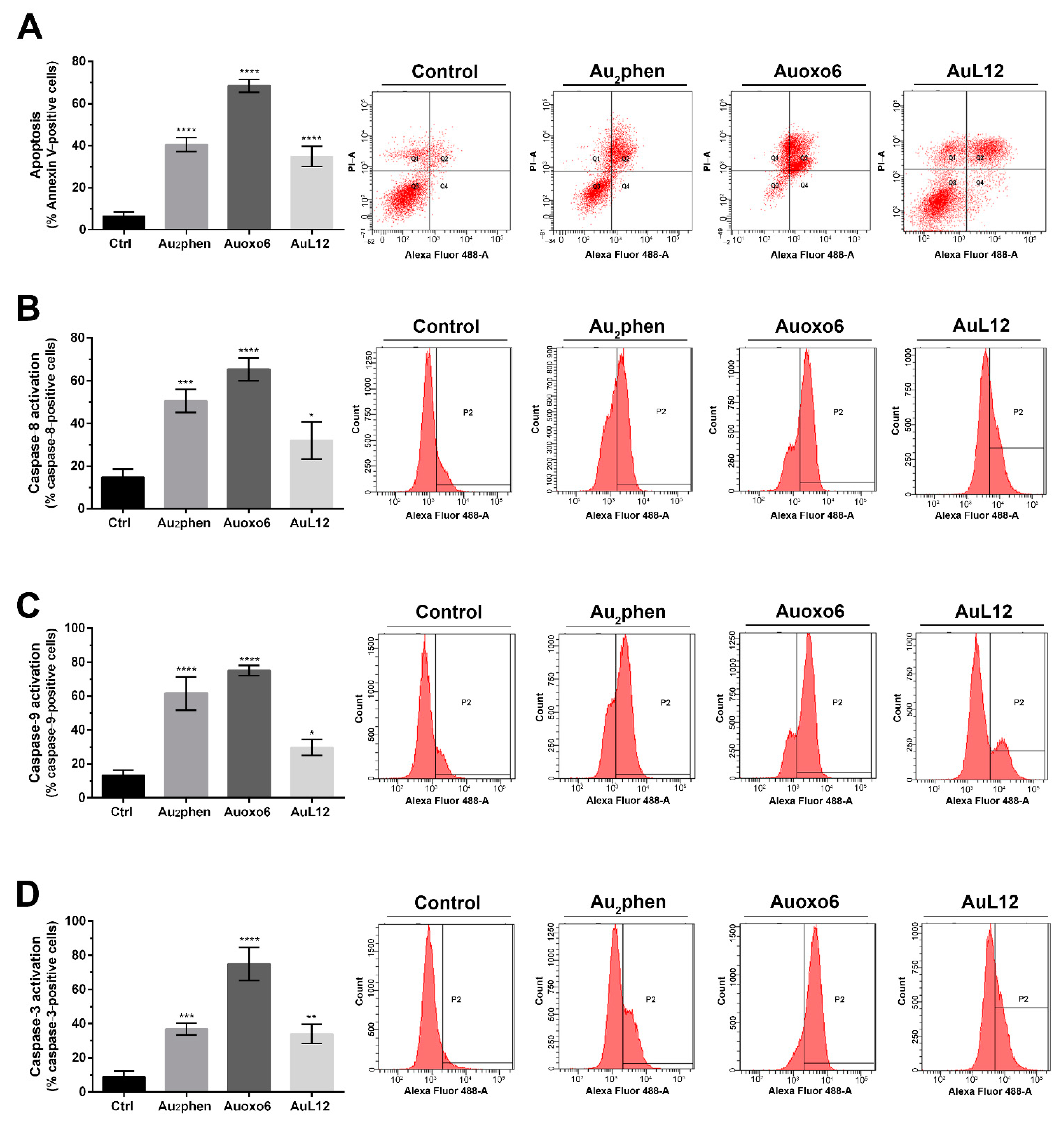
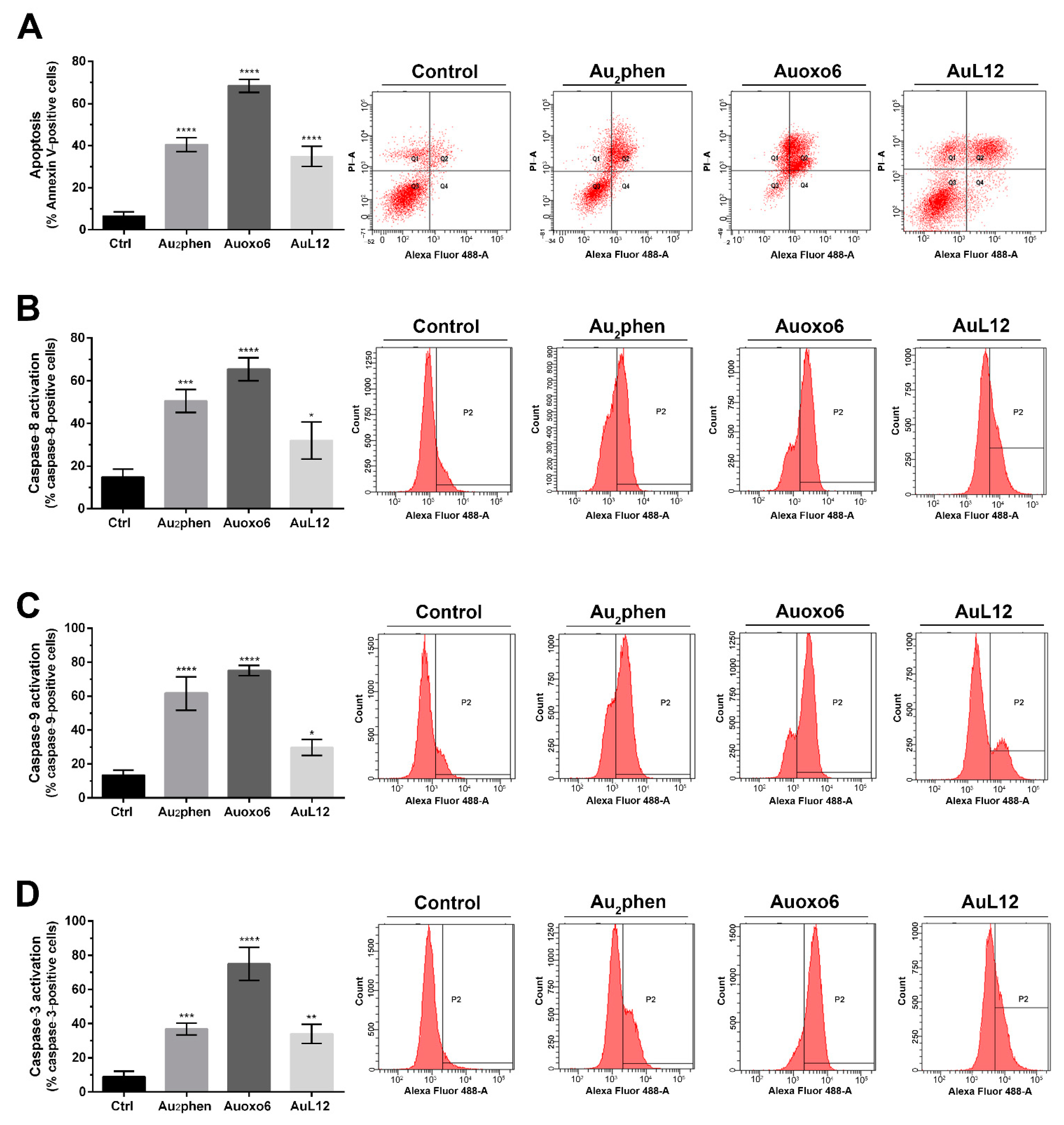
3.2. Apoptotic Profile Analysis and Caspase Activation
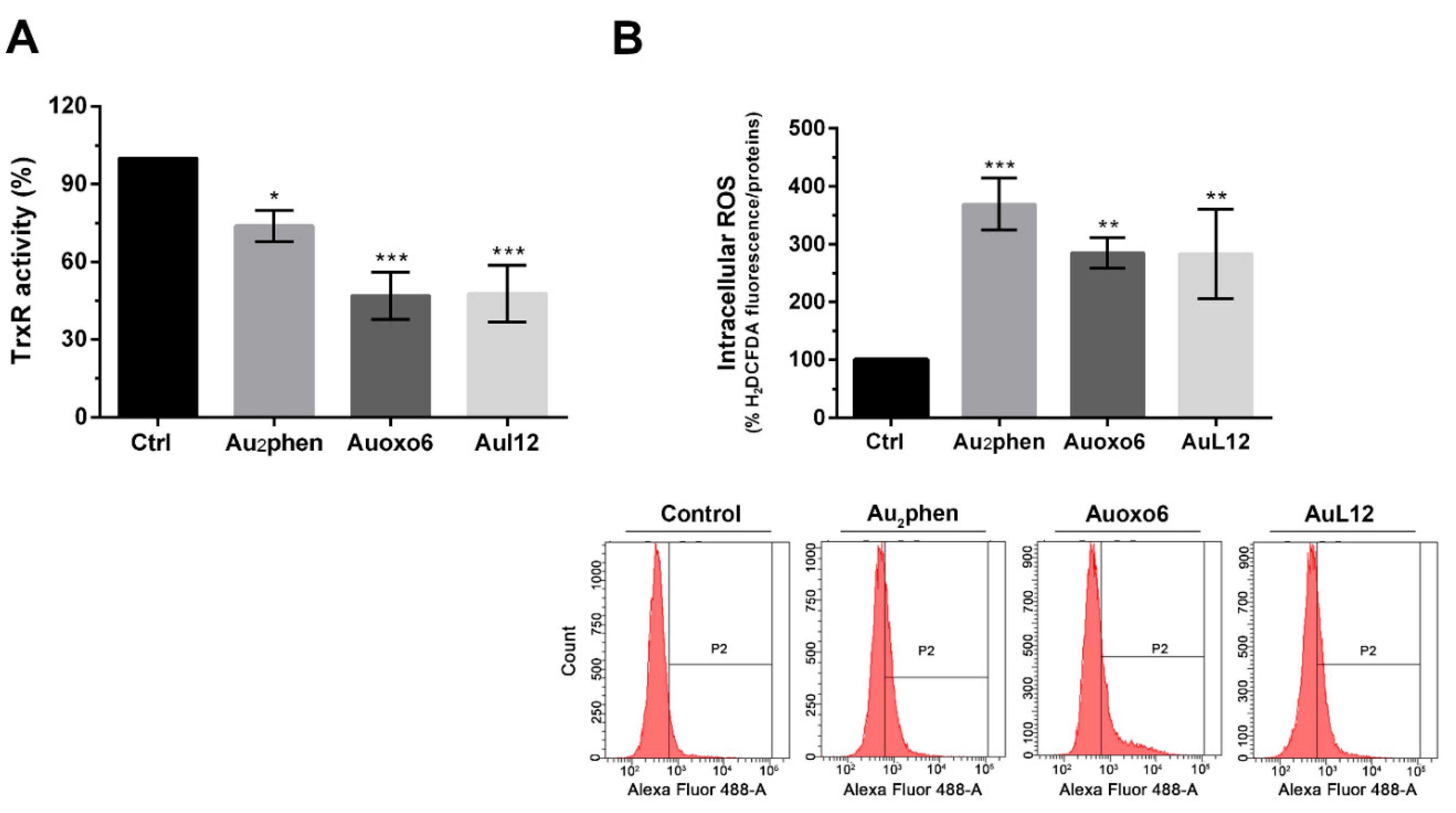
3.3. Effects on Thioredoxin Reductase Activity and Cell Redox Balance
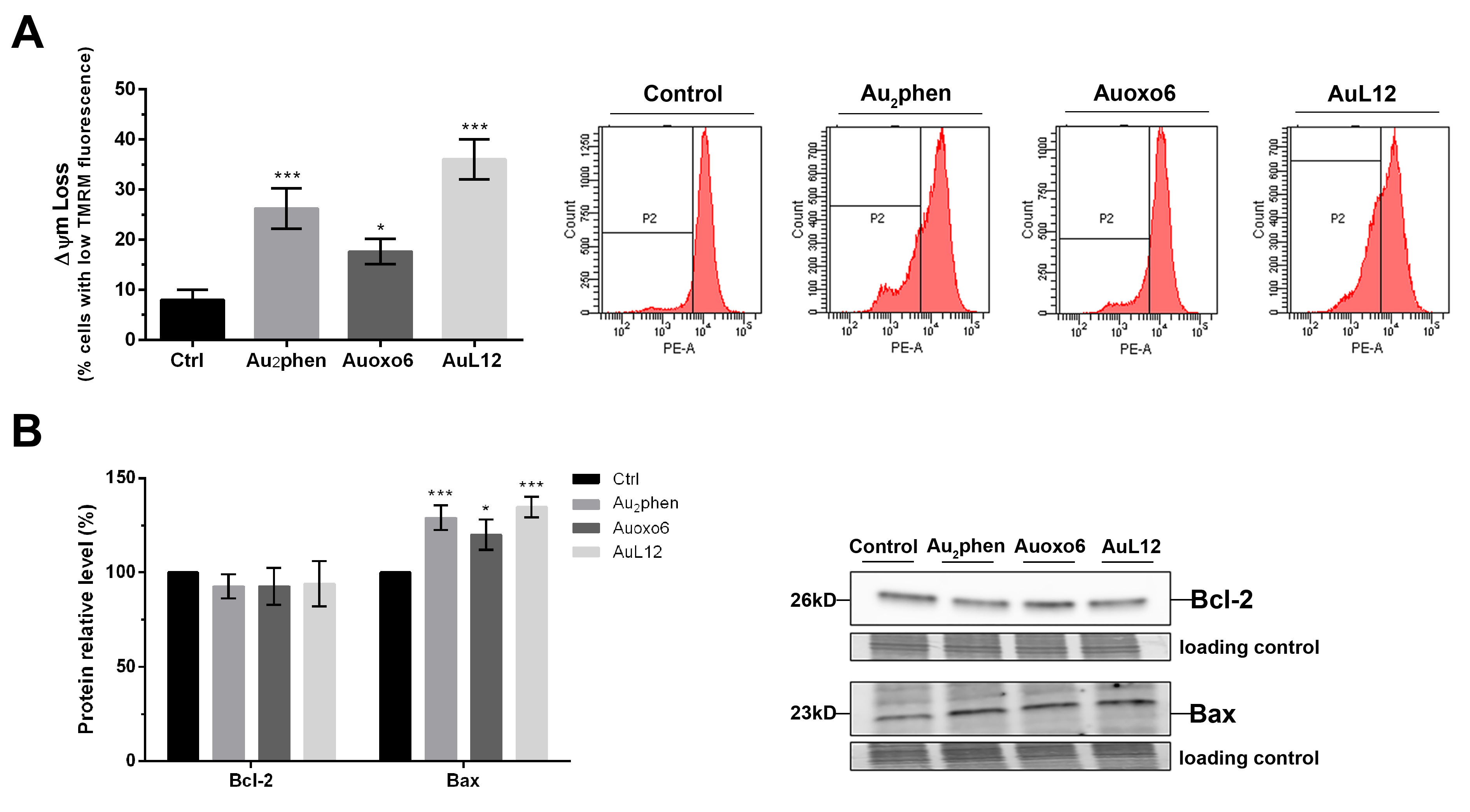
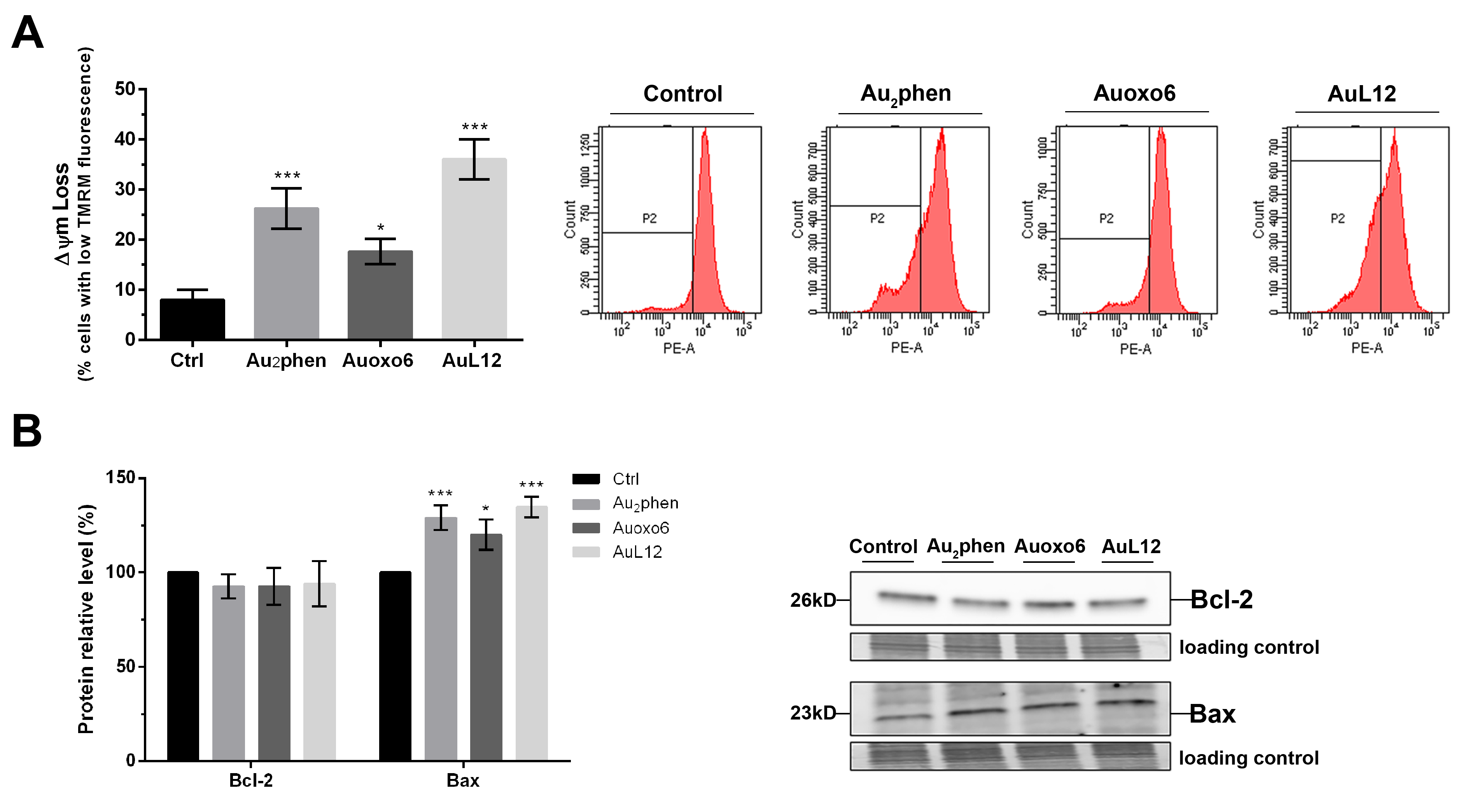
3.4. Involvement of Mitochondrial Membrane Potential Impairment in Gold(III)-Induced Apoptosis
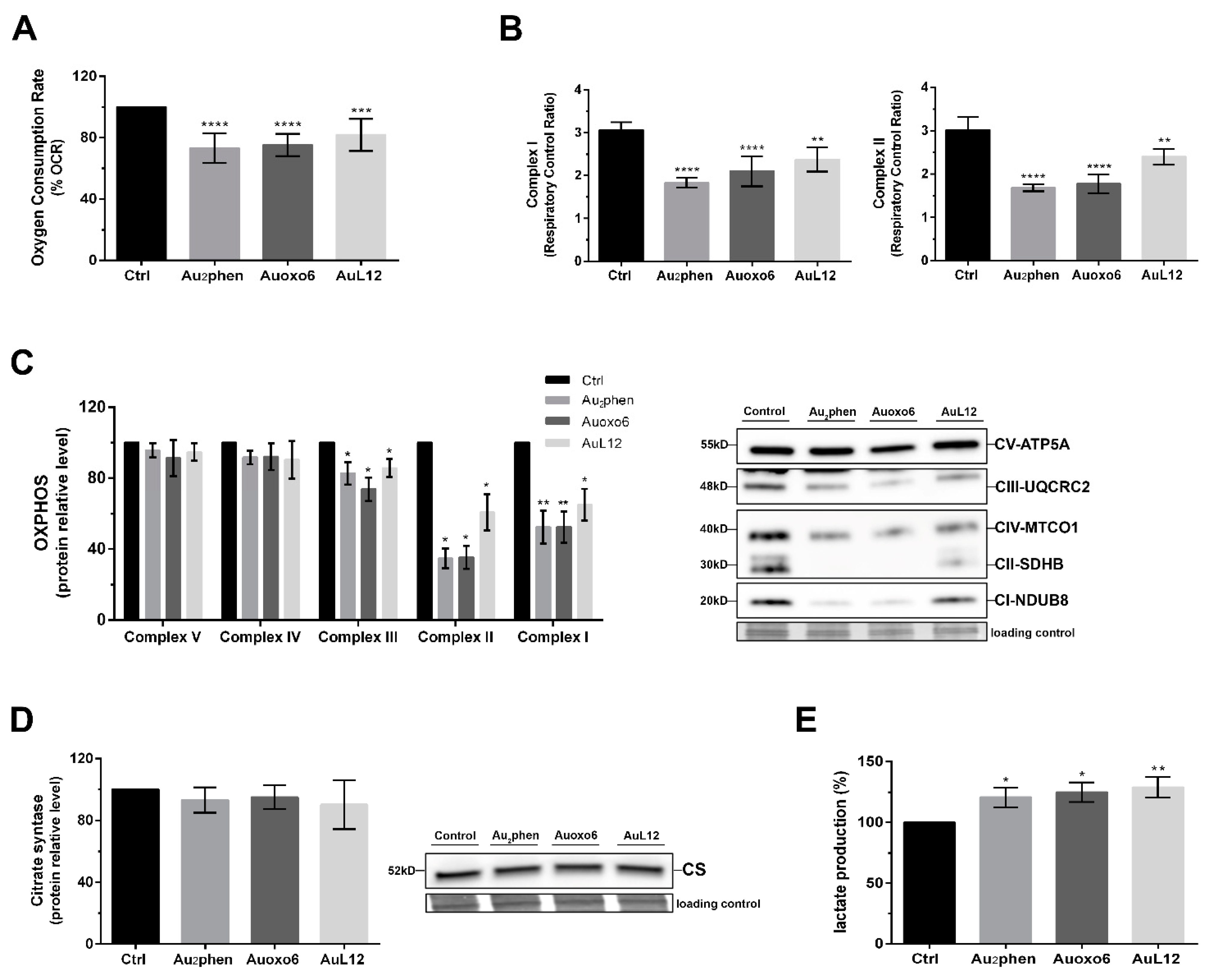
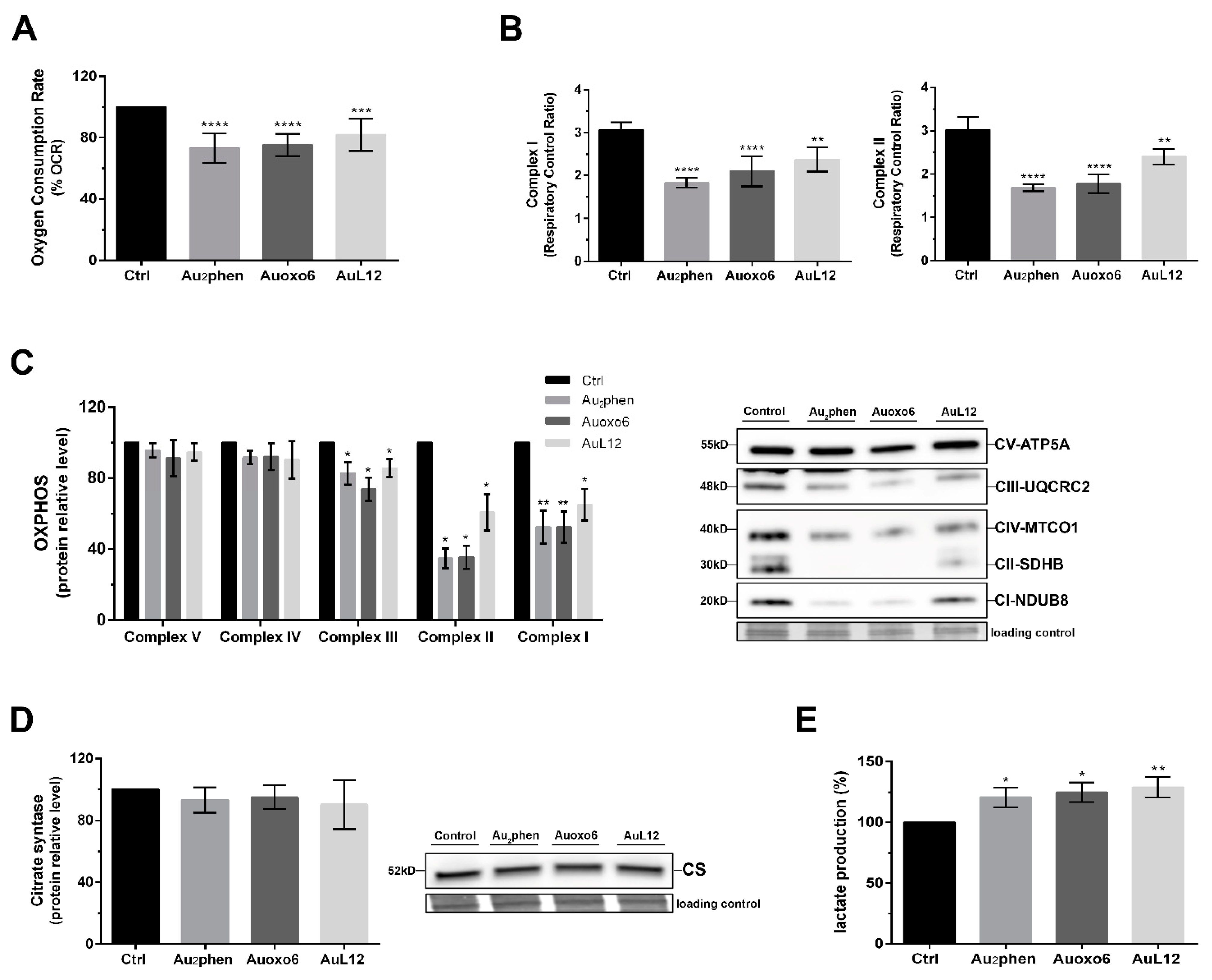
3.5. Metabolic Changes Elicited by Gold(III) Compounds
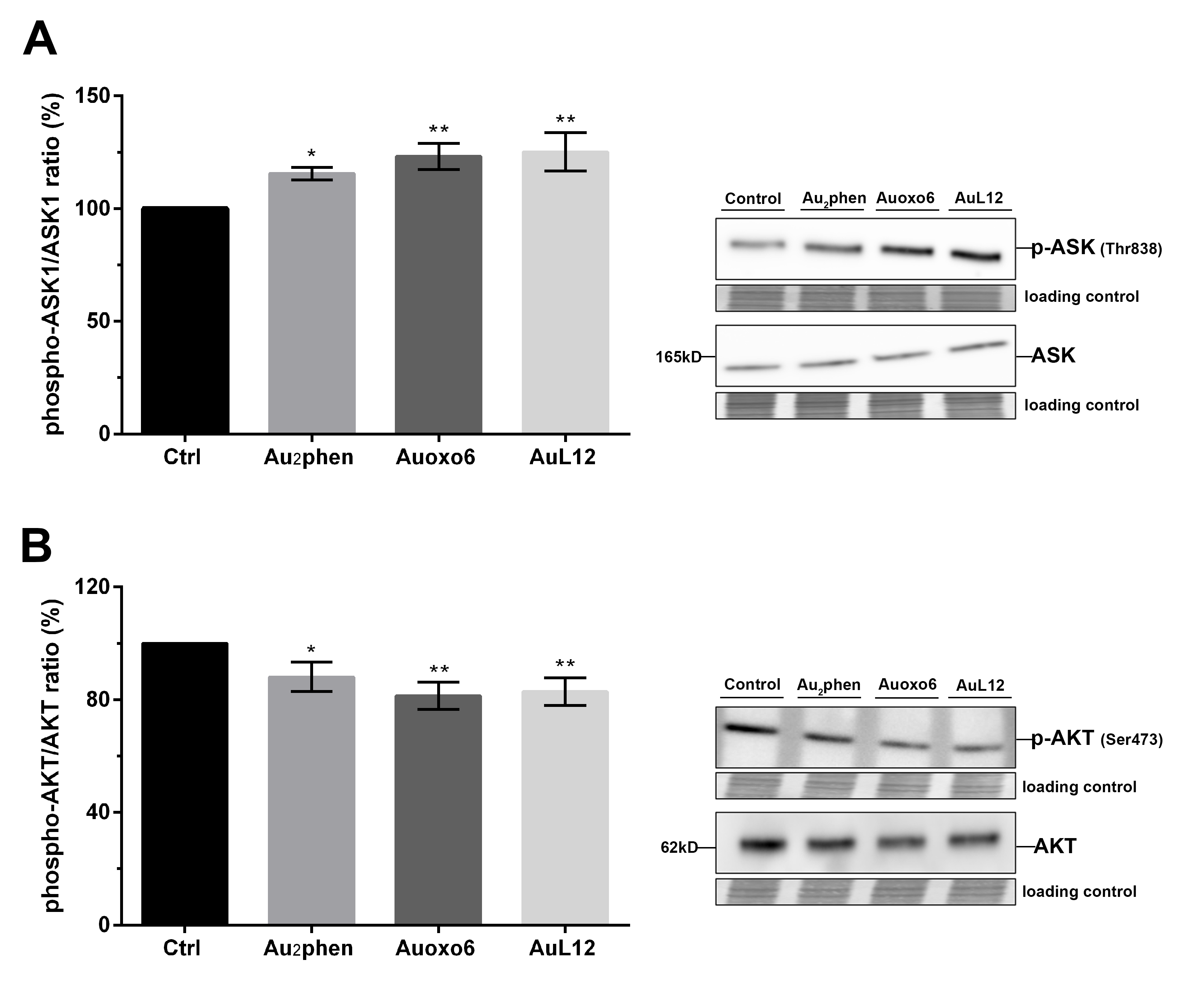
3.6. Contribution of ASK1 and AKT in Gold(III)-Induced Apoptotic Signaling
4. Discussion
4.1. Antiproliferative and Proapoptotic Effects
4.2. TrxR Inhibition and ROS Unbalance
4.3. Impairment of Mitochondrial Functions
4.4. Involvement of ASK1-p38MAPK and PI3K/AKT Signal Pathways
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Nobili, S.; Mini, E.; Landini, I.; Gabbiani, C.; Casini, A.; Messori, L. Gold Compounds as Anticancer Agents: Chemistry, Cellular Pharmacology, and Preclinical Studies. Med. Res. Rev. 2010, 30, 550–580. [Google Scholar] [CrossRef]
- Zou, T.; Lum, C.T.; Lok, C.-N.; Zhang, J.-J.; Che, C.-M. Chemical Biology of Anticancer Gold(III) and Gold(I) Complexes. Chem. Soc. Rev. 2015, 44, 8786–8801. [Google Scholar] [CrossRef]
- Casini, A.; Kelter, G.; Gabbiani, C.; Cinellu, M.A.; Minghetti, G.; Fregona, D.; Fiebig, H.-H.; Messori, L. Chemistry, Antiproliferative Properties, Tumor Selectivity, and Molecular Mechanisms of Novel Gold(III) Compounds for Cancer Treatment: A Systematic Study. JBIC J. Biol. Inorg. Chem. 2009, 14, 1139–1149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aldinucci, D.; Ronconi, L.; Fregona, D. Groundbreaking Gold(III) Anticancer Agents. Drug Discov. Today 2009, 14, 1075–1076. [Google Scholar] [CrossRef]
- Bertrand, B.; Williams, M.R.M.; Bochmann, M. Gold(III) Complexes for Antitumor Applications: An Overview. Chem. Eur. J. 2018, 24, 11840–11851. [Google Scholar] [CrossRef] [PubMed]
- Fiskus, W.; Saba, N.; Shen, M.; Ghias, M.; Liu, J.; Gupta, S.D.; Chauhan, L.; Rao, R.; Gunewardena, S.; Schorno, K.; et al. Auranofin Induces Lethal Oxidative and Endoplasmic Reticulum Stress and Exerts Potent Preclinical Activity against Chronic Lymphocytic Leukemia. Cancer Res. 2014, 74, 2520–2532. [Google Scholar] [CrossRef] [Green Version]
- Gamberi, T.; Pratesi, A.; Messori, L.; Massai, L. Proteomics as a Tool to Disclose the Cellular and Molecular Mechanisms of Selected Anticancer Gold Compounds. Coord. Chem. Rev. 2021, 438, 213905. [Google Scholar] [CrossRef]
- Casini, A.; Hartinger, C.; Gabbiani, C.; Mini, E.; Dyson, P.J.; Keppler, B.K.; Messori, L. Gold(III) Compounds as Anticancer Agents: Relevance of Gold-Protein Interactions for Their Mechanism of Action. J. Inorg. Biochem. 2008, 102, 564–575. [Google Scholar] [CrossRef] [PubMed]
- Gabbiani, C.; Massai, L.; Scaletti, F.; Michelucci, E.; Maiore, L.; Cinellu, M.A.; Messori, L. Protein Metalation by Metal-Based Drugs: Reactions of Cytotoxic Gold Compounds with Cytochrome c and Lysozyme. J. Biol. Inorg. Chem. 2012, 17, 1293–1302. [Google Scholar] [CrossRef]
- Cinellu, M.A.; Minghetti, G.; Pinna, M.V.; Stoccoro, S.; Zucca, A.; Manassero, M.; Sansoni, M. Μ-Oxo and Alkoxo Complexes of Gold(III) with 6-Alkyl-2,2′-Bipyridines. Synthesis, Characterization and X-Ray Structures. J. Chem. Soc. Dalton Trans. 1998, 1735–1742. [Google Scholar] [CrossRef]
- Casini, A.; Cinellu, M.A.; Minghetti, G.; Gabbiani, C.; Coronnello, M.; Mini, E.; Messori, L. Structural and Solution Chemistry, Antiproliferative Effects, and DNA and Protein Binding Properties of a Series of Dinuclear Gold(III) Compounds with Bipyridyl Ligands. J. Med. Chem. 2006, 49, 5524–5531. [Google Scholar] [CrossRef] [PubMed]
- Gabbiani, C.; Casini, A.; Messori, L.; Guerri, A.; Cinellu, M.A.; Minghetti, G.; Corsini, M.; Rosani, C.; Zanello, P.; Arca, M. Structural Characterization, Solution Studies, and DFT Calculations on a Series of Binuclear Gold(III) Oxo Complexes: Relationships to Biological Properties. Inorg. Chem. 2008, 47, 2368–2379. [Google Scholar] [CrossRef]
- Cinellu, M.A.; Maiore, L.; Manassero, M.; Casini, A.; Arca, M.; Fiebig, H.-H.; Kelter, G.; Michelucci, E.; Pieraccini, G.; Gabbiani, C.; et al. [Au2(Phen2Me)2(μ-O)2](PF6)2, a Novel Dinuclear Gold(III) Complex Showing Excellent Antiproliferative Properties. ACS Med. Chem. Lett. 2010, 1, 336–339. [Google Scholar] [CrossRef] [Green Version]
- Ronconi, L.; Giovagnini, L.; Marzano, C.; Bettìo, F.; Graziani, R.; Pilloni, G.; Fregona, D. Gold Dithiocarbamate Derivatives as Potential Antineoplastic Agents: Design, Spectroscopic Properties, and In Vitro Antitumor Activity. Inorg. Chem. 2005, 44, 1867–1881. [Google Scholar] [CrossRef] [PubMed]
- Nardon, C.; Boscutti, G.; Gabbiani, C.; Massai, L.; Pettenuzzo, N.; Fassina, A.; Messori, L.; Fregona, D. Cell and Cell-Free Mechanistic Studies on Two Gold(III) Complexes with Proven Antitumor Properties. Eur. J. Inorg. Chem. 2017, 2017, 1737–1744. [Google Scholar] [CrossRef] [Green Version]
- Chiara, F.; Gambalunga, A.; Sciacovelli, M.; Nicolli, A.; Ronconi, L.; Fregona, D.; Bernardi, P.; Rasola, A.; Trevisan, A. Chemotherapeutic Induction of Mitochondrial Oxidative Stress Activates GSK-3 α/β and Bax, Leading to Permeability Transition Pore Opening and Tumor Cell Death. Cell Death Dis. 2012, 3, e444. [Google Scholar] [CrossRef]
- Marzo, T.; Massai, L.; Pratesi, A.; Stefanini, M.; Cirri, D.; Magherini, F.; Becatti, M.; Landini, I.; Nobili, S.; Mini, E.; et al. Replacement of the Thiosugar of Auranofin with Iodide Enhances the Anticancer Potency in a Mouse Model of Ovarian Cancer. ACS Med. Chem. Lett. 2019, 10. [Google Scholar] [CrossRef]
- Becatti, M.; Barygina, V.; Mannucci, A.; Emmi, G.; Prisco, D.; Lotti, T.; Fiorillo, C.; Taddei, N. Sirt1 Protects against Oxidative Stress-Induced Apoptosis in Fibroblasts from Psoriatic Patients: A New Insight into the Pathogenetic Mechanisms of Psoriasis. Int. J. Mol. Sci. 2018, 19, 1572. [Google Scholar] [CrossRef] [Green Version]
- Pini, A.; Boccalini, G.; Baccari, M.C.; Becatti, M.; Garella, R.; Fiorillo, C.; Calosi, L.; Bani, D.; Nistri, S. Protection from Cigarette Smoke-Induced Vascular Injury by Recombinant Human Relaxin-2 (Serelaxin). J. Cell. Mol. Med. 2016, 20, 891–902. [Google Scholar] [CrossRef]
- Becatti, M.; Fiorillo, C.; Barygina, V.; Cecchi, C.; Lotti, T.; Prignano, F.; Silvestro, A.; Nassi, P.; Taddei, N. SIRT1 Regulates MAPK Pathways in Vitiligo Skin: Insight into the Molecular Pathways of Cell Survival. J. Cell. Mol. Med. 2014, 18, 514–529. [Google Scholar] [CrossRef]
- Magherini, F.; Fiaschi, T.; Valocchia, E.; Becatti, M.; Pratesi, A.; Marzo, T.; Massai, L.; Gabbiani, C.; Landini, I.; Nobili, S.; et al. Antiproliferative Effects of Two Gold(I)-N-Heterocyclic Carbene Complexes in A2780 Human Ovarian Cancer Cells: A Comparative Proteomic Study. Oncotarget 2018, 9, 28042–28068. [Google Scholar] [CrossRef]
- Baracca, A.; Chiaradonna, F.; Sgarbi, G.; Solaini, G.; Alberghina, L.; Lenaz, G. Mitochondrial Complex I Decrease Is Responsible for Bioenergetic Dysfunction in K-Ras Transformed Cells. Biochim. Biophys. Acta 2010, 1797, 314–323. [Google Scholar] [CrossRef] [Green Version]
- Rueden, C.T.; Schindelin, J.; Hiner, M.C.; DeZonia, B.E.; Walter, A.E.; Arena, E.T.; Eliceiri, K.W. ImageJ2: ImageJ for the next Generation of Scientific Image Data. BMC Bioinform. 2017, 18, 529. [Google Scholar] [CrossRef]
- Rigobello, M.P.; Messori, L.; Marcon, G.; Agostina Cinellu, M.; Bragadin, M.; Folda, A.; Scutari, G.; Bindoli, A. Gold Complexes Inhibit Mitochondrial Thioredoxin Reductase: Consequences on Mitochondrial Functions. J. Inorg. Biochem. 2004, 98, 1634–1641. [Google Scholar] [CrossRef]
- Marzano, C.; Gandin, V.; Folda, A.; Scutari, G.; Bindoli, A.; Rigobello, M.P. Inhibition of Thioredoxin Reductase by Auranofin Induces Apoptosis in Cisplatin-Resistant Human Ovarian Cancer Cells. Free Radic. Biol. Med. 2007, 42, 872–881. [Google Scholar] [CrossRef]
- Bindoli, A.; Rigobello, M.P.; Scutari, G.; Gabbiani, C.; Casini, A.; Messori, L. Thioredoxin Reductase: A Target for Gold Compounds Acting as Potential Anticancer Drugs. Coord. Chem. Rev. 2009, 253, 1692–1707. [Google Scholar] [CrossRef]
- Berners-Price, S.J.; Filipovska, A. Gold Compounds as Therapeutic Agents for Human Diseases. Metallomics 2011, 3, 863–873. [Google Scholar] [CrossRef]
- Schuh, E.; Pflüger, C.; Citta, A.; Folda, A.; Rigobello, M.P.; Bindoli, A.; Casini, A.; Mohr, F. Gold(I) Carbene Complexes Causing Thioredoxin 1 and Thioredoxin 2 Oxidation as Potential Anticancer Agents. J. Med. Chem. 2012, 55, 5518–5528. [Google Scholar] [CrossRef] [PubMed]
- Fan, C.; Zheng, W.; Fu, X.; Li, X.; Wong, Y.-S.; Chen, T. Enhancement of Auranofin-Induced Lung Cancer Cell Apoptosis by Selenocystine, a Natural Inhibitor of TrxR1 in Vitro and in Vivo. Cell Death Dis. 2014, 5, e1191. [Google Scholar] [CrossRef] [Green Version]
- Gandin, V.; Fernandes, A.P.; Rigobello, M.P.; Dani, B.; Sorrentino, F.; Tisato, F.; Björnstedt, M.; Bindoli, A.; Sturaro, A.; Rella, R.; et al. Cancer Cell Death Induced by Phosphine Gold(I) Compounds Targeting Thioredoxin Reductase. Biochem. Pharm. 2010, 79, 90–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saggioro, D.; Rigobello, M.P.; Paloschi, L.; Folda, A.; Moggach, S.A.; Parsons, S.; Ronconi, L.; Fregona, D.; Bindoli, A. Gold(III)-Dithiocarbamato Complexes Induce Cancer Cell Death Triggered by Thioredoxin Redox System Inhibition and Activation of ERK Pathway. Chem. Biol. 2007, 14, 1128–1139. [Google Scholar] [CrossRef]
- Rigobello, M.P.; Scutari, G.; Boscolo, R.; Bindoli, A. Induction of Mitochondrial Permeability Transition by Auranofin, a Gold(I)-Phosphine Derivative. Br. J. Pharm. 2002, 136, 1162–1168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McKeage, M.J.; Maharaj, L.; Berners-Price, S.J. Mechanisms of Cytotoxicity and Antitumor Activity of Gold(I) Phosphine Complexes: The Possible Role of Mitochondria. Coord. Chem. Rev. 2002, 232, 127–135. [Google Scholar] [CrossRef]
- Wang, Y.; He, Q.-Y.; Sun, R.W.-Y.; Che, C.-M.; Chiu, J.-F. Gold(III) Porphyrin 1a Induced Apoptosis by Mitochondrial Death Pathways Related to Reactive Oxygen Species. Cancer Res. 2005, 65, 11553–11564. [Google Scholar] [CrossRef]
- Barnard, P.J.; Berners-Price, S.J. Targeting the Mitochondrial Cell Death Pathway with Gold Compounds. Coord. Chem. Rev. 2007, 251, 1889–1902. [Google Scholar] [CrossRef]
- Li, Y.; Liu, G.-F.; Tan, C.-P.; Ji, L.-N.; Mao, Z.-W. Antitumor Properties and Mechanisms of Mitochondria-Targeted Ag(I) and Au(I) Complexes Containing N-Heterocyclic Carbenes Derived from Cyclophanes. Metallomics 2014, 6, 1460–1468. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Youle, R.J. The Role of Mitochondria in Apoptosis. Annu Rev. Genet. 2009, 43, 95–118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harris, M.H.; Thompson, C.B. The Role of the Bcl-2 Family in the Regulation of Outer Mitochondrial Membrane Permeability. Cell Death Differ. 2000, 7, 1182–1191. [Google Scholar] [CrossRef]
- Arnér, E.S.J.; Holmgren, A. Physiological Functions of Thioredoxin and Thioredoxin Reductase. Eur. J. Biochem. 2000, 267, 6102–6109. [Google Scholar] [CrossRef]
- Zhang, J.; Li, X.; Han, X.; Liu, R.; Fang, J. Targeting the Thioredoxin System for Cancer Therapy. Trends Pharmacol. Sci. 2017, 38, 794–808. [Google Scholar] [CrossRef]
- Zhang, R.; Al-Lamki, R.; Bai, L.; Streb, J.W.; Miano, J.M.; Bradley, J.; Min, W. Thioredoxin-2 Inhibits Mitochondria-Located ASK1-Mediated Apoptosis in a JNK-Independent Manner. Circ. Res. 2004, 94, 1483–1491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Psenakova, K.; Hexnerova, R.; Srb, P.; Obsilova, V.; Veverka, V.; Obsil, T. The Redox-Active Site of Thioredoxin Is Directly Involved in Apoptosis Signal-Regulating Kinase 1 Binding That Is Modulated by Oxidative Stress. FEBS J. 2020, 287, 1626–1644. [Google Scholar] [CrossRef] [PubMed]
- Shu, N.; Hägglund, P.; Cai, H.; Hawkins, C.L.; Davies, M.J. Modification of Cys Residues in Human Thioredoxin-1 by p-Benzoquinone Causes Inhibition of Its Catalytic Activity and Activation of the ASK1/P38-MAPK Signalling Pathway. Redox Biol. 2020, 29, 101400. [Google Scholar] [CrossRef] [PubMed]
- Saitoh, M.; Nishitoh, H.; Fujii, M.; Takeda, K.; Tobiume, K.; Sawada, Y.; Kawabata, M.; Miyazono, K.; Ichijo, H. Mammalian Thioredoxin Is a Direct Inhibitor of Apoptosis Signal-Regulating Kinase (ASK) 1. EMBO J. 1998, 17, 2596–2606. [Google Scholar] [CrossRef] [Green Version]
- Marzano, C.; Ronconi, L.; Chiara, F.; Giron, M.C.; Faustinelli, I.; Cristofori, P.; Trevisan, A.; Fregona, D. Gold(III)-Dithiocarbamato Anticancer Agents: Activity, Toxicology and Histopathological Studies in Rodents. Int. J. Cancer 2011, 129, 487–496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marcon, G.; Carotti, S.; Coronnello, M.; Messori, L.; Mini, E.; Orioli, P.; Mazzei, T.; Cinellu, M.A.; Minghetti, G. Gold(III) Complexes with Bipyridyl Ligands: Solution Chemistry, Cytotoxicity, and DNA Binding Properties. J. Med. Chem. 2002, 45, 1672–1677. [Google Scholar] [CrossRef] [PubMed]
- Park, S.-J.; Kim, I.-S. The Role of P38 MAPK Activation in Auranofin-Induced Apoptosis of Human Promyelocytic Leukaemia HL-60 Cells. Br. J. Pharmacol. 2005, 146, 506–513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tonissen, K.F.; Di Trapani, G. Thioredoxin System Inhibitors as Mediators of Apoptosis for Cancer Therapy. Mol. Nutr. Food Res. 2009, 53, 87–103. [Google Scholar] [CrossRef]
- Lu, J.; Holmgren, A. Thioredoxin System in Cell Death Progression. Antioxid. Redox Signal. 2012, 17, 1738–1747. [Google Scholar] [CrossRef]
- Cheng, X.; Holenya, P.; Can, S.; Alborzinia, H.; Rubbiani, R.; Ott, I.; Wölfl, S. A TrxR Inhibiting Gold(I) NHC Complex Induces Apoptosis through ASK1-P38-MAPK Signaling in Pancreatic Cancer Cells. Mol. Cancer 2014, 13, 221. [Google Scholar] [CrossRef] [Green Version]
- Mendoza, M.C.; Er, E.E.; Blenis, J. The Ras-ERK and PI3K-MTOR Pathways: Cross-Talk and Compensation. Trends Biochem. Sci. 2011, 36, 320–328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Hu, J.; Wu, S.; Wang, L.; Cao, X.; Zhang, X.; Dai, B.; Cao, M.; Shao, R.; Zhang, R.; et al. Auranofin-Mediated Inhibition of PI3K/AKT/MTOR Axis and Anticancer Activity in Non-Small Cell Lung Cancer Cells. Oncotarget 2016, 7, 3548–3558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, C.; Liu, Z.; Li, M.; Li, X.; Wong, Y.-S.; Ngai, S.-M.; Zheng, W.; Zhang, Y.; Chen, T. Enhancement of Auranofin-Induced Apoptosis in MCF-7 Human Breast Cells by Selenocystine, a Synergistic Inhibitor of Thioredoxin Reductase. PLoS ONE 2013, 8, e53945. [Google Scholar] [CrossRef] [Green Version]
- Hyter, S.; Hirst, J.; Pathak, H.; Pessetto, Z.Y.; Koestler, D.C.; Raghavan, R.; Pei, D.; Godwin, A.K. Developing a Genetic Signature to Predict Drug Response in Ovarian Cancer. Oncotarget 2018, 9, 14828. [Google Scholar] [CrossRef] [PubMed]
- Pessetto, Z.Y.; Weir, S.J.; Sethi, G.; Broward, M.A.; Godwin, A.K. Drug Repurposing for Gastrointestinal Stromal Tumor. Mol. Cancer 2013, 12, 1299–1309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Menconi, A.; Marzo, T.; Massai, L.; Pratesi, A.; Severi, M.; Petroni, G.; Antonuzzo, L.; Messori, L.; Pillozzi, S.; Cirri, D. Anticancer Effects against Colorectal Cancer Models of Chloro(Triethylphosphine)Gold(I) Encapsulated in PLGA–PEG Nanoparticles. Biometals 2021. [Google Scholar] [CrossRef] [PubMed]
- Holenya, P.; Can, S.; Rubbiani, R.; Alborzinia, H.; Jünger, A.; Cheng, X.; Ott, I.; Wölfl, S.; Solary, E.; Garrido, C.; et al. Detailed Analysis of Pro-Apoptotic Signaling and Metabolic Adaptation Triggered by a N-Heterocyclic Carbene–Gold(i) Complex. Metallomics 2014, 6, 1591–1601. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound (µM) ± SD 1 | Cell Lines | ||
|---|---|---|---|
| A2780 | PNT1 | HEK293 | |
| Au2phen | 0.80 ± 0.02 µM | 11.80 ± 0.25 µM | 4.18 ± 0.05 µM |
| Auoxo6 | 1.14 ± 0.18 µM | 18.40 ± 0.30 µM | 6.16 ± 0.04 µM |
| AuL12 | 4.00 ± 0.03 µM | 12.30 ± 0.17 µM | 10.70 ± 0.36 µM |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gorini, G.; Magherini, F.; Fiaschi, T.; Massai, L.; Becatti, M.; Modesti, A.; Messori, L.; Gamberi, T. Au2phen and Auoxo6, Two Dinuclear Oxo-Bridged Gold(III) Compounds, Induce Apoptotic Signaling in Human Ovarian A2780 Cancer Cells. Biomedicines 2021, 9, 871. https://doi.org/10.3390/biomedicines9080871
Gorini G, Magherini F, Fiaschi T, Massai L, Becatti M, Modesti A, Messori L, Gamberi T. Au2phen and Auoxo6, Two Dinuclear Oxo-Bridged Gold(III) Compounds, Induce Apoptotic Signaling in Human Ovarian A2780 Cancer Cells. Biomedicines. 2021; 9(8):871. https://doi.org/10.3390/biomedicines9080871
Chicago/Turabian StyleGorini, Giulia, Francesca Magherini, Tania Fiaschi, Lara Massai, Matteo Becatti, Alessandra Modesti, Luigi Messori, and Tania Gamberi. 2021. "Au2phen and Auoxo6, Two Dinuclear Oxo-Bridged Gold(III) Compounds, Induce Apoptotic Signaling in Human Ovarian A2780 Cancer Cells" Biomedicines 9, no. 8: 871. https://doi.org/10.3390/biomedicines9080871
APA StyleGorini, G., Magherini, F., Fiaschi, T., Massai, L., Becatti, M., Modesti, A., Messori, L., & Gamberi, T. (2021). Au2phen and Auoxo6, Two Dinuclear Oxo-Bridged Gold(III) Compounds, Induce Apoptotic Signaling in Human Ovarian A2780 Cancer Cells. Biomedicines, 9(8), 871. https://doi.org/10.3390/biomedicines9080871