
Efficient Selection of Recombinant Fluorescent Vaccinia Virus Strains and Rapid Virus Titer Determination by Using a Multi-Well Plate Imaging System



Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Lines
2.2. Vaccinia Virus Strains
2.3. Plasmid
2.4. Recombinant Vaccinia Virus Generation by Homologous Recombination
2.5. Fluorescence-Based Viral Plaque Assay
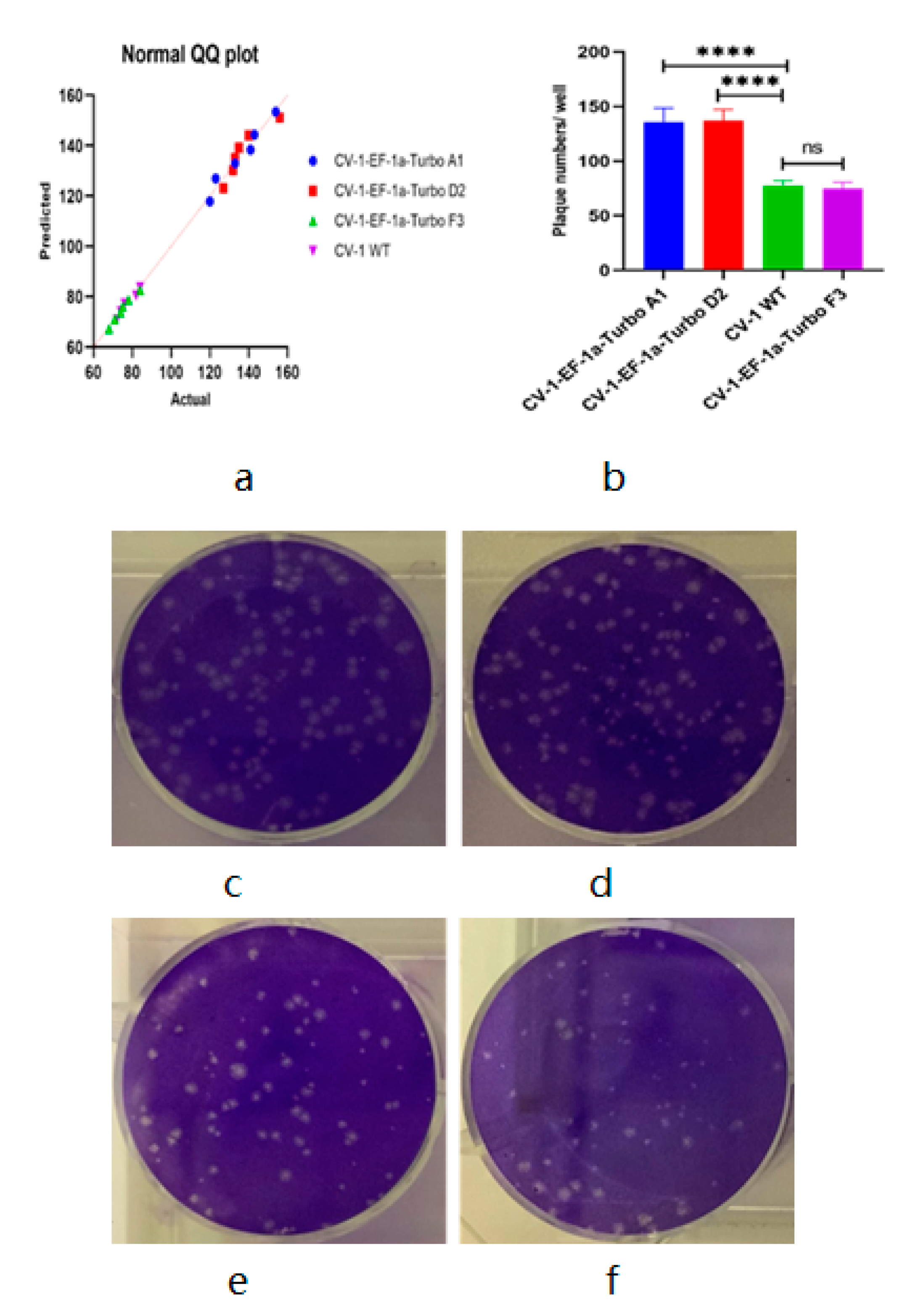
2.6. Viral Plaque Assay for Statistical Analysis
2.7. Statistical Analysis
2.8. Software
3. Results
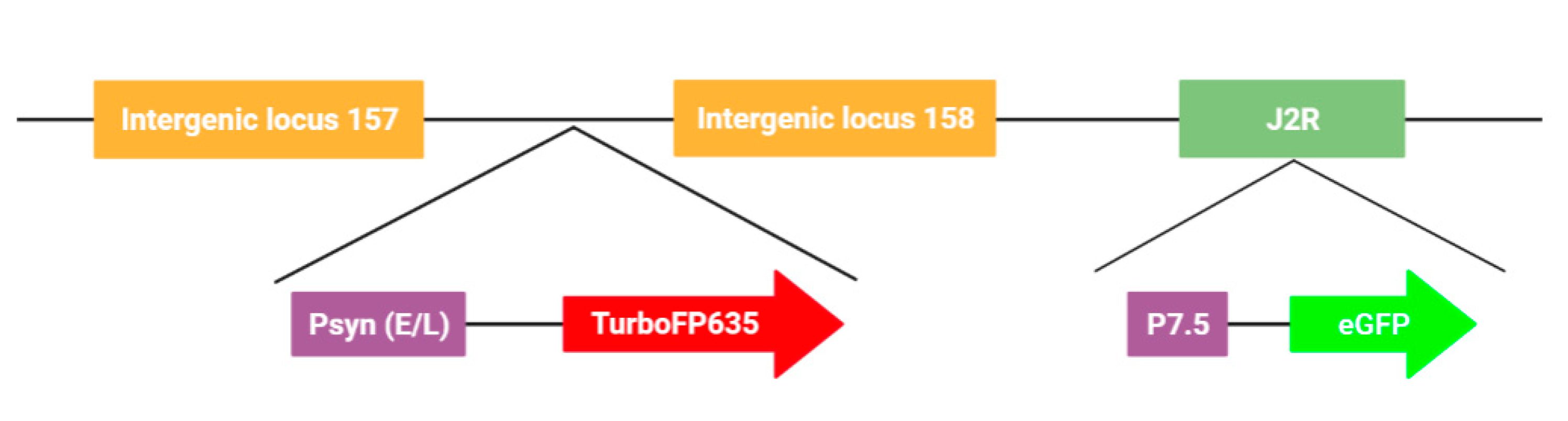
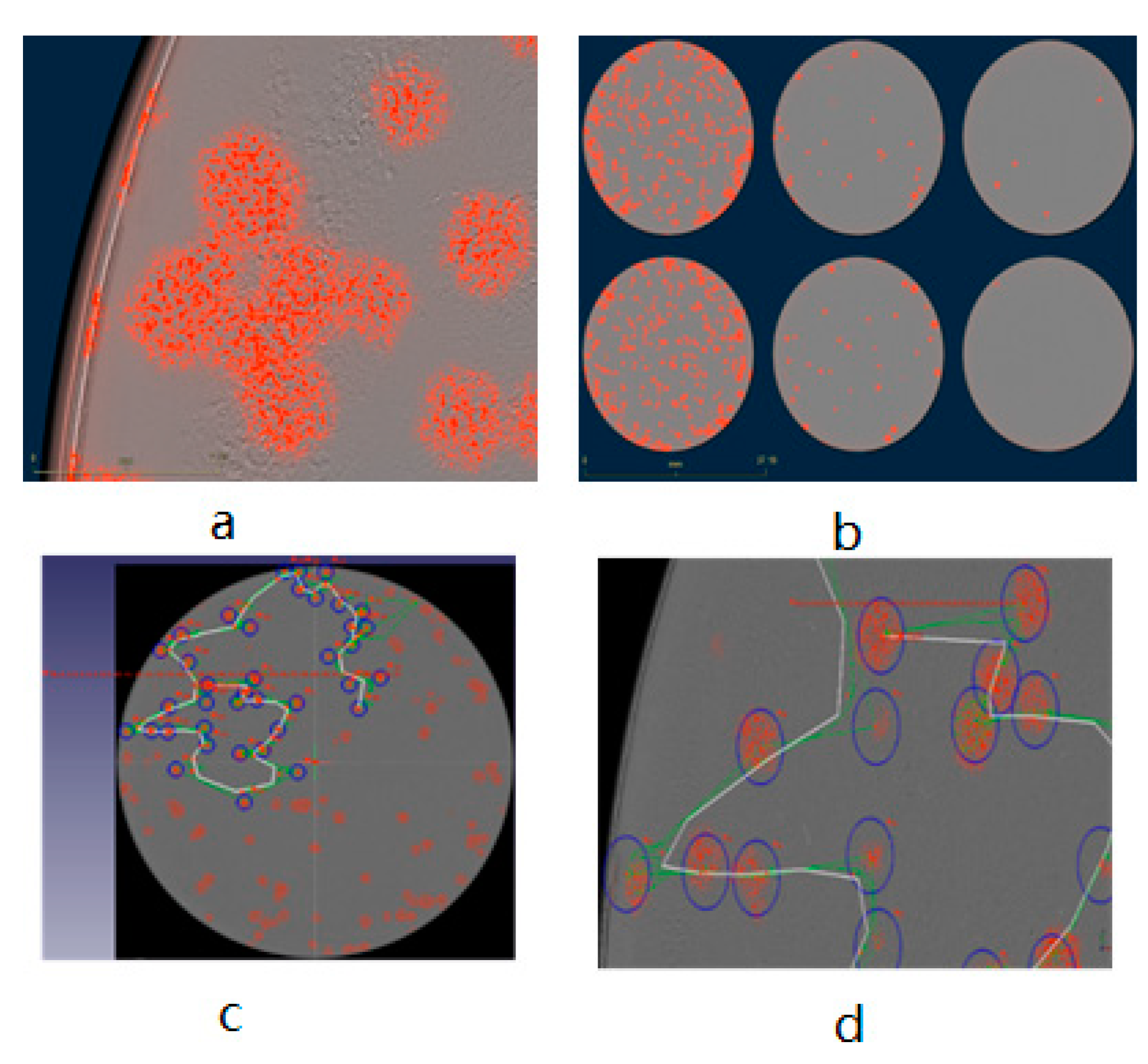
3.1. Generation, Selection, and Purification of the New L1c-Ig-Turbo-TK-eGFP Strain Expressing eGFP and Turbofp635 Proteins
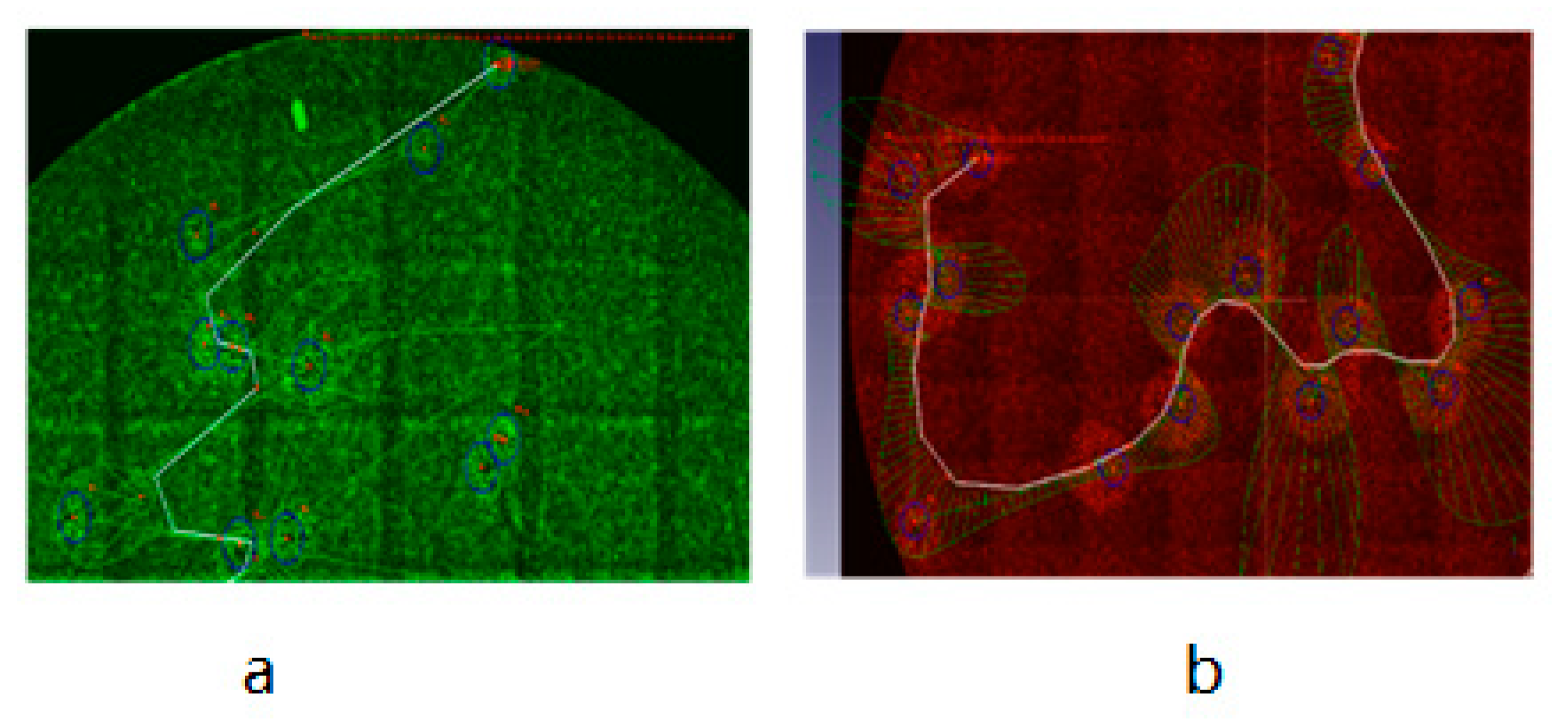
3.2. Vaccinia Virus Titer Determination Based on Fluorescence-Dependent Plaque Assay
3.3. Development of a Plaque Assay for Non-Fluorescent rVACV on the Basis of Cell Lines Expressing Fluorescent Proteins
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A

References
- Walsh, S.R.; Dolin, R. Vaccinia viruses: Vaccines against smallpox and vectors against infectious diseases and tumors. Expert Rev Vaccines 2011, 10, 1221–1240. [Google Scholar] [CrossRef]
- Guo, Z.S.; Lu, B.; Guo, Z.; Giehl, E.; Feist, M.; Dai, E.; Liu, W.; Storkus, W.J.; He, Y.; Liu, Z.; et al. Vaccinia virus-mediated cancer immunotherapy: Cancer vaccines and oncolytics. J. Immunother. Cancer. 2019, 7, 6. [Google Scholar] [CrossRef]
- Jenne, L.; Schuler, G.; Steinkasserer, A. Viral vectors for dendritic cell-based immunotherapy. Trends Immunol. 2001, 22, 102–107. [Google Scholar] [CrossRef]
- Aghi, M.K.; Martuza, R.L. Oncolytic viral therapies—The clinical experience. Oncogene 2005, 24, 7802–7816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thorne, S.H.; Kirn, D.H. Future directions for the field of oncolytic virotherapy: A perspective on the use of vaccinia virus. Expert Opin. Biol. Ther. 2004, 4, 1307–1321. [Google Scholar] [CrossRef]
- Jhawar, S.R.; Thandoni, A.; Bommareddy, P.K.; Hassan, S.; Kohlhapp, F.J.; Goyal, S.; Schenkel, J.; Silk, A.W.; Zloza, A. Oncolytic Viruses—Natural and Genetically Engineered Cancer Immunotherapies. Front. Oncol. 2017, 7, 202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, Z.S.; Bartlett, D.L. Vaccinia as a vector for gene delivery. Expert Opin. Biol. Ther. 2004, 4, 901–917. [Google Scholar] [CrossRef]
- Smith, G.L.; Benfield, C.; de Motes, C.M.; Mazzon, M.; Ember, S.W.J.; Ferguson, B.; Sumner, R.P. Vaccinia virus immune evasion: Mechanisms, virulence and immunogenicity. J. Gen. Virol. 2013, 94, 2367–2392. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Sampedro, L.; Gomez, C.E.; Mejías-Pérez, E.; Pérez-Jiménez, E.; Oliveros, J.C.; Esteban, M. Attenuated and Replication-Competent Vaccinia Virus Strains M65 and M101 with Distinct Biology and Immunogenicity as Potential Vaccine Candidates against Pathogens. J. Virol. 2013, 87, 6955–6974. [Google Scholar] [CrossRef] [Green Version]
- Wyatt, L.S.; Earl, P.L.; Moss, B. Generation of Recombinant Vaccinia Viruses. Curr. Protoc. Protein Sci. 2017, 89, 5.13.1–5.13.18. [Google Scholar] [CrossRef]
- Nakano, E.; Panicali, D.; Paoletti, E. Molecular genetics of vaccinia virus: Demonstration of marker rescue. Proc. Natl. Acad. Sci. USA 1982, 79, 1593–1596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, M.; Zhang, W.; Wang, J.; Al Yaghchi, C.; Ahmed, J.; Chard, L.; Lemoine, N.; Wang, Y. Efficiently Editing the Vaccinia Virus Genome by Using the CRISPR-Cas9 System. J. Virol. 2015, 89, 5176–5179. [Google Scholar] [CrossRef] [Green Version]
- Yuan, M.; Gao, X.; Chard, L.; Ali, Z.; Ahmed, J.; Li, Y.; Liu, P.; Lemoine, N.; Wang, Y. A marker-free system for highly efficient construction of vaccinia virus vectors using CRISPR Cas9. Mol. Ther. Methods Clin. Dev. 2015, 2, 15035. [Google Scholar] [CrossRef] [PubMed]
- Kosicki, M.; Tomberg, K.; Bradley, A. Repair of double-strand breaks induced by CRISPR–Cas9 leads to large deletions and complex rearrangements. Nat. Biotechnol. 2018, 36, 765–771. [Google Scholar] [CrossRef] [PubMed]
- Jasperse, B.; O’Connell, C.M.; Wang, Y.; Verardi, P.H. EPPIC (Efficient Purification by Parental Inducer Constraint) Platform for Rapid Generation of Recombinant Vaccinia Viruses. Mol. Ther. Methods Clin. Dev. 2020, 17, 731–738. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.S.; Liu, Z.; Sathaiah, M.; Wang, J.; Ravindranathan, R.; Kim, E.; Huang, S.; Kenniston, T.W.; Bell, J.C.; Zeh, H.J.; et al. Rapid Generation of Multiple Loci-Engineered Marker-free Poxvirus and Characterization of a Clinical-Grade Oncolytic Vaccinia Virus. Mol. Ther. Methods Clin. Dev. 2017, 7, 112–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jonsson, N.; Gullberg, M.; Lindberg, A.M. Real-time polymerase chain reaction as a rapid and efficient alternative to estimation of picornavirus titers by tissue culture infectious dose 50% or plaque forming units. Microbiol. Immunol. 2009, 53, 149–154. [Google Scholar] [CrossRef] [PubMed]
- Fox, J.C.; Emery, V.C. Quantification of viruses in clinical samples. Rev. Med Virol. 1992, 2, 195–203. [Google Scholar] [CrossRef]
- Cooper, P. A method for producing plaques in agar suspensions of animal cells. Virol. 1955, 1, 397–401. [Google Scholar] [CrossRef]
- Dulbecco, R. Production of Plaques in Monolayer Tissue Cultures by Single Particles of an Animal Virus. Proc. Natl. Acad. Sci. USA 1952, 38, 747–752. [Google Scholar] [CrossRef] [Green Version]
- Khatib, R.; Chason, J.L.; Silberberg, B.K.; Lerner, A.M. Age-Dependent Pathogenicity of Group B Coxsackieviruses in Swiss-Webster Mice: Infectivity for Myocardium and Pancreas. J. Infect. Dis. 1980, 141, 394–403. [Google Scholar] [CrossRef]
- Dulbecco, R.; Vogt, M. Plaque formation and isolation of pure lines with poliomyelitis viruses. J. Exp. Med. 1954, 99, 167–182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- WHO. WHO Global Influenza Surveillance Network: Manual for the Laboratory Diagnosis and Virological Surveillance of In-Fluenza; World Health Organization: Geneva, Switzerland, 2011. [Google Scholar]
- Marintcheva, B. Introduction to Viral Structure, Diversity and Biology; Elsevier: Amsterdam, The Netherlands, 2018; pp. 1–26. [Google Scholar] [CrossRef]
- Smither, S.J.; Lear-Rooney, C.; Biggins, J.; Pettitt, J.; Lever, M.S.; Olinger, G. Comparison of the plaque assay and 50% tissue culture infectious dose assay as methods for measuring filovirus infectivity. J. Virol. Methods 2013, 193, 565–571. [Google Scholar] [CrossRef] [PubMed]
- Baer, A.; Kehn-Hall, K. Viral Concentration Determination Through Plaque Assays: Using Traditional and Novel Overlay Systems. J. Vis. Exp. 2014, 93, e52065. [Google Scholar] [CrossRef] [PubMed]
- Cacciabue, M.; Currá, A.; Gismondi, M.I. ViralPlaque: A Fiji macro for automated assessment of viral plaque statistics. PeerJ 2019, 7, e7729. [Google Scholar] [CrossRef] [PubMed]
- Yin, J.; McCaskill, J. Replication of viruses in a growing plaque: A reaction-diffusion model. Biophys. J. 1992, 61, 1540–1549. [Google Scholar] [CrossRef]
- Smee, D.F.; Hurst, B.L.; Evans, W.J.; Clyde, N.; Wright, S.; Peterson, C.; Jung, K.-H.; Day, C.W. Evaluation of cell viability dyes in antiviral assays with RNA viruses that exhibit different cytopathogenic properties. J. Virol. Methods 2017, 246, 51–57. [Google Scholar] [CrossRef]
- Ye, M.; Wilhelm, M.; Gentschev, I.; Szalay, A. A Modified Limiting Dilution Method for Monoclonal Stable Cell Line Selection Using a Real-Time Fluorescence Imaging System: A Practical Workflow and Advanced Applications. Methods Protoc. 2021, 4, 16. [Google Scholar] [CrossRef]
- Gentschev, I.; Adelfinger, M.; Josupeit, R.; Rudolph, S.; Ehrig, K.; Donat, U.; Weibel, S.; Chen, N.G.; Yu, Y.A.; Zhang, Q.; et al. Preclinical Evaluation of Oncolytic Vaccinia Virus for Therapy of Canine Soft Tissue Sarcoma. PLoS ONE 2012, 7, e37239. [Google Scholar] [CrossRef]
- Frentzen, A.; Yu, Y.A.; Chen, N.; Zhang, Q.; Weibel, S.; Raab, V.; Szalay, A.A. Anti-VEGF single-chain antibody GLAF-1 encoded by oncolytic vaccinia virus significantly enhances antitumor therapy. Proc. Natl. Acad. Sci. USA 2009, 106, 12915–12920. [Google Scholar] [CrossRef] [Green Version]
- Kelly, K.J.; Woo, Y.; Brader, P.; Yu, Z.; Riedl, C.; Lin, S.-F.; Chen, N.; Yu, Y.A.; Rusch, V.; Szalay, A.A.; et al. Novel Oncolytic Agent GLV-1h68 Is Effective Against Malignant Pleural Mesothelioma. Hum. Gene Ther. 2008, 19, 774–782. [Google Scholar] [CrossRef] [PubMed]
- Matrosovich, M.; Matrosovich, T.; Garten, W.; Klenk, H.-D. New low-viscosity overlay medium for viral plaque assays. Virol. J. 2006, 3, 63. [Google Scholar] [CrossRef] [Green Version]
- Masci, A.L.; Menesale, E.B.; Chen, W.-C.; Co, C.; Lu, X.; Bergelson, S. Integration of Fluorescence Detection and Image-Based Automated Counting Increases Speed, Sensitivity, and Robustness of Plaque Assays. Mol. Ther. Methods Clin. Dev. 2019, 14, 270–274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arias-Arias, J.; Corrales-Aguilar, E.; Mora-Rodríguez, R. A Fluorescent Real-Time Plaque Assay Enables Single-Cell Analysis of Virus-Induced Cytopathic Effect by Live-Cell Imaging. Viruses 2021, 13, 1193. [Google Scholar] [CrossRef]
- Lorenzo, M.M.; Galindo, I.; Blasco, R. Construction and Isolation of Recombinant Vaccinia Virus Using Genetic Markers. Vaccinia Virus Poxvirol. 2004, 269, 15–30. [Google Scholar] [CrossRef]
- ClinicalTrials.gov Identifier: NCT02759588. Available online: https://clinicaltrials.gov/ct2/show/study/NCT02759588 (accessed on 21 July 2021).




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Plate No. | Total Number of Viral Plaques | Number of eGFP-Positive Plaques | Recombination Rate |
|---|---|---|---|
| 1 | 522 | 3 | 0.57% |
| 2 | 550 | 5 | 0.91% |
| 3 | 567 | 3 | 0.53% |
| 4 | 493 | 4 | 0.81% |
| 5 | 487 | 6 | 1.23% |
| 6 | 475 | 3 | 0.63% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ye, M.; Keicher, M.; Gentschev, I.; Szalay, A.A. Efficient Selection of Recombinant Fluorescent Vaccinia Virus Strains and Rapid Virus Titer Determination by Using a Multi-Well Plate Imaging System. Biomedicines 2021, 9, 1032. https://doi.org/10.3390/biomedicines9081032
Ye M, Keicher M, Gentschev I, Szalay AA. Efficient Selection of Recombinant Fluorescent Vaccinia Virus Strains and Rapid Virus Titer Determination by Using a Multi-Well Plate Imaging System. Biomedicines. 2021; 9(8):1032. https://doi.org/10.3390/biomedicines9081032
Chicago/Turabian StyleYe, Mingyu, Markus Keicher, Ivaylo Gentschev, and Aladar A. Szalay. 2021. "Efficient Selection of Recombinant Fluorescent Vaccinia Virus Strains and Rapid Virus Titer Determination by Using a Multi-Well Plate Imaging System" Biomedicines 9, no. 8: 1032. https://doi.org/10.3390/biomedicines9081032
APA StyleYe, M., Keicher, M., Gentschev, I., & Szalay, A. A. (2021). Efficient Selection of Recombinant Fluorescent Vaccinia Virus Strains and Rapid Virus Titer Determination by Using a Multi-Well Plate Imaging System. Biomedicines, 9(8), 1032. https://doi.org/10.3390/biomedicines9081032