
Glucose-Dependent Insulinotropic Polypeptide Suppresses Foam Cell Formation of Macrophages through Inhibition of the Cyclin-Dependent Kinase 5-CD36 Pathway

 ,
,  , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials and Reagents
2.2. Animal Experiments
2.3. Characteristics and Biochemical Parameters in Mice
2.4. Cholesterol Esterification Assay of Macrophages Extracted from Mice
2.5. Experiments of U937 Macrophages
2.6. Dil-ox-LDL Uptake into Macrophages
2.7. Levels of Gene Expression
2.8. Statistical Analysis
3. Results
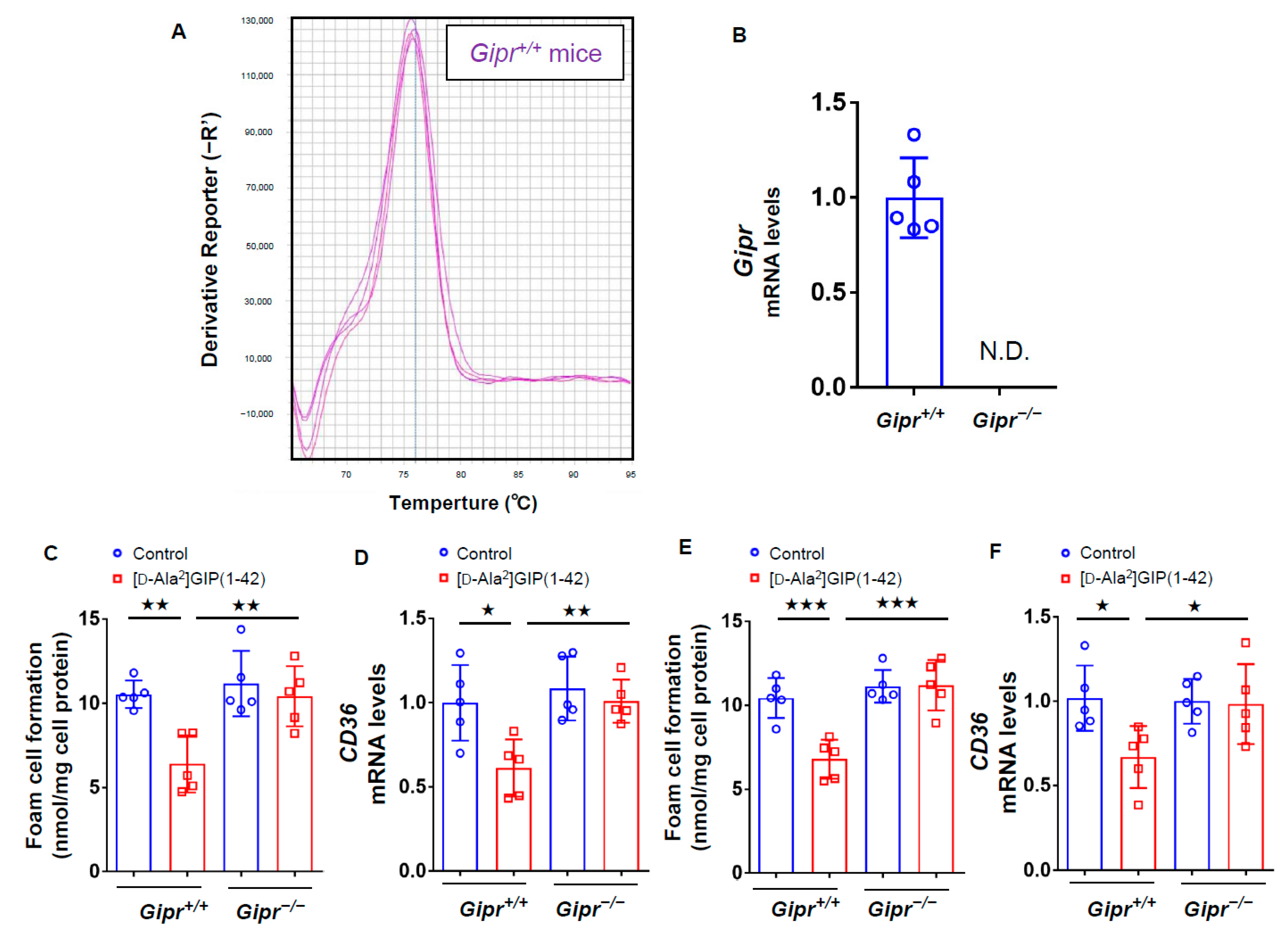
3.1. Characteristics and Biochemical Data of Gipr−/− Mice and Gipr+/+ Mice
3.2. Effects of [D-Ala2]GIP(1–42) on Foam Cell Formation of, and CD36 Expression in, Macrophages Isolated from Gipr−/− Mice and Gipr+/+ Mice
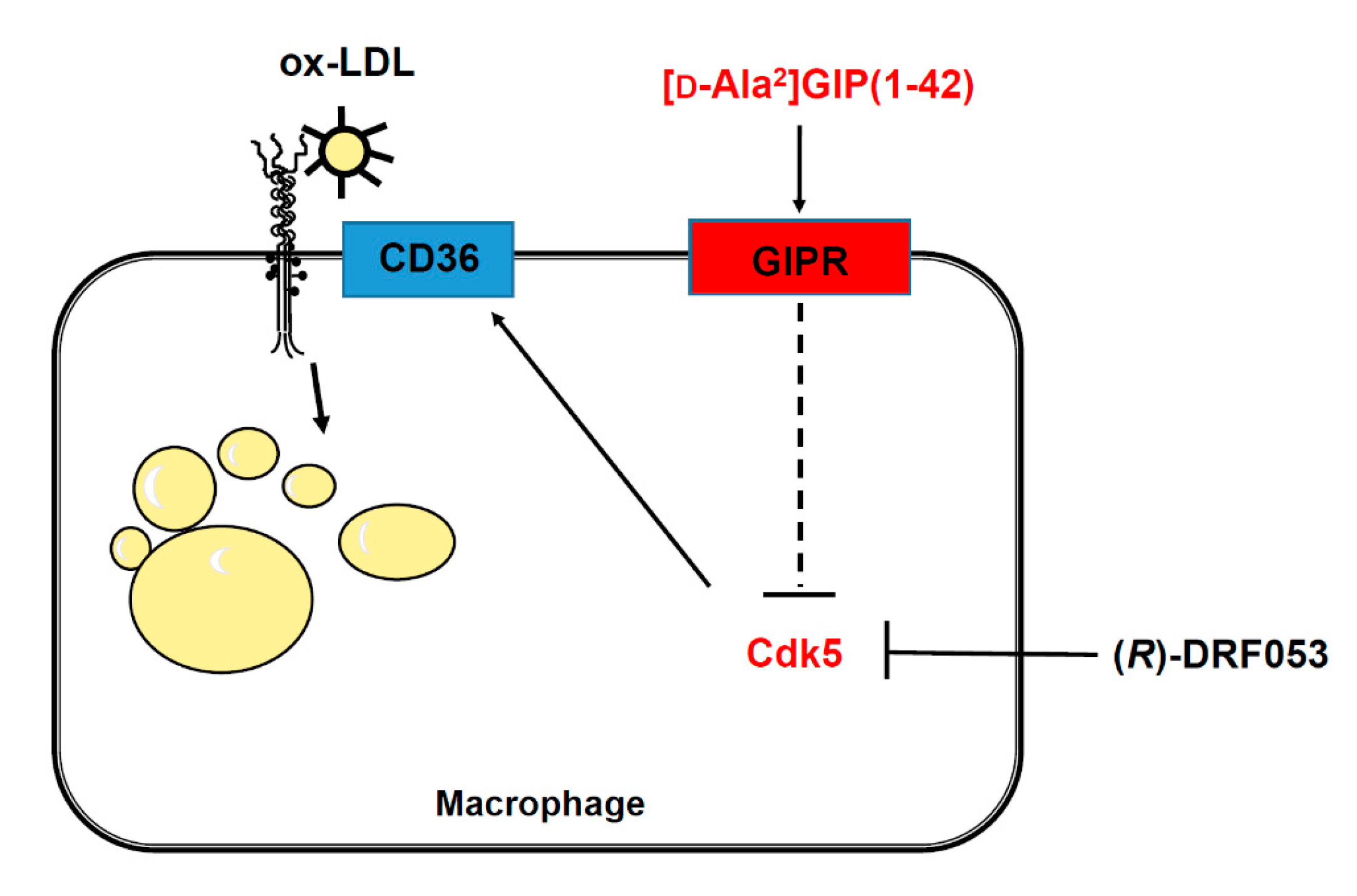
3.3. Effects of [D-Ala2]GIP(1–42) and (R)-DRF053 on U937 Macrophages
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Disclosure
References
- Rao Kondapally Seshasai, S.; Kaptoge, S.; Thompson, A.; Di Angelantonio, E.; Gao, P.; Sarwar, N.; Whincup, P.H.; Mukamal, K.J.; Gillum, R.F.; Holme, I.; et al. Diabetes mellitus, fasting glucose, and risk of cause-specific death. N. Engl. J. Med. 2011, 364, 829–841. [Google Scholar] [PubMed] [Green Version]
- Low Wang, C.C.; Hess, C.N.; Hiatt, W.R.; Goldfine, A.B. Clinical update: Cardiovascular disease in diabetes mellitus: Atherosclerotic cardiovascular disease and heart failure in type 2 diabetes mellitus—Mechanisms, management, and clinical considerations. Circulation 2016, 133, 2459–2502. [Google Scholar] [CrossRef]
- Glass, C.K.; Witztum, J.L. Atherosclerosis: The road ahead. Cell 2001, 104, 503–516. [Google Scholar] [CrossRef] [Green Version]
- Lusis, A.J. Atherosclerosis. Nature 2000, 407, 233–241. [Google Scholar] [CrossRef] [PubMed]
- Allahverdian, S.; Pannu, P.S.; Francis, G.A. Contribution of monocyte-derived macrophages and smooth muscle cells to arterial foam cell formation. Cardiovasc. Res. 2012, 95, 165–172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drucker, D.J. The biology of incretin hormones. Cell Metab. 2006, 3, 153–165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baggio, L.L.; Drucker, D.J. Biology of incretins: GLP-1 and GIP. Gastroenterology 2007, 132, 2131–2157. [Google Scholar] [CrossRef]
- Seino, Y.; Fukushima, M.; Yabe, D. GIP and GLP-1, the two incretin hormones: Similarities and differences. J. Diabetes Investig. 2010, 1, 8–23. [Google Scholar] [CrossRef] [Green Version]
- Fukami, A.; Seino, Y.; Ozaki, N.; Yamamoto, M.; Sugiyama, C.; Sakamoto-Miura, E.; Himeno, T.; Takagishi, Y.; Tsunekawa, S.; Ali, S.; et al. Ectopic expression of GIP in pancreatic β-cells maintains enhanced insulin secretion in mice with complete absence of proglucagon-derived peptides. Diabetes 2013, 62, 510–518. [Google Scholar] [CrossRef] [Green Version]
- Mori, Y.; Matsui, T.; Hirano, T.; Yamagishi, S.I. GIP as a potential therapeutic target for atherosclerotic cardiovascular disease—A systematic review. Int. J. Mol. Sci. 2020, 21, 1509. [Google Scholar] [CrossRef] [Green Version]
- Mori, Y.; Kushima, H.; Koshibu, M.; Saito, T.; Hiromura, M.; Kohashi, K.; Terasaki, M.; Seino, Y.; Yamada, Y.; Hirano, T. Glucose-dependent insulinotropic polypeptide suppresses peripheral arterial remodeling in male mice. Endocrinology 2018, 159, 2717–2732. [Google Scholar] [CrossRef] [Green Version]
- Kahles, F.; Liberman, A.; Halim, C.; Rau, M.; Möllmann, J.; Mertens, R.W.; Rückbeil, M.; Diepolder, I.; Walla, B.; Diebold, S.; et al. The incretin hormone GIP is upregulated in patients with atherosclerosis and stabilizes plaques in ApoE-/-mice by blocking monocyte/macrophage activation. Mol. Metab. 2018, 14, 150–157. [Google Scholar] [CrossRef] [PubMed]
- Ojima, A.; Matsui, T.; Maeda, S.; Takeuchi, M.; Yamagishi, S. Glucose-dependent insulinotropic polypeptide (GIP) inhibits signaling pathways of advanced glycation end products (AGEs) in endothelial cells via its antioxidative properties. Horm. Metab. Res. 2012, 44, 501–505. [Google Scholar] [CrossRef]
- Nagashima, M.; Watanabe, T.; Terasaki, M.; Tomoyasu, M.; Nohtomi, K.; Kim-Kaneyama, J.; Miyazaki, A.; Hirano, T. Native incretins prevent the development of atherosclerotic lesions in apolipoprotein E knockout mice. Diabetologia 2011, 54, 2649–2659. [Google Scholar] [CrossRef] [Green Version]
- Nogi, Y.; Nagashima, M.; Terasaki, M.; Nohtomi, K.; Watanabe, T.; Hirano, T. Glucose-dependent insulinotropic polypeptide prevents the progression of macrophage-driven atherosclerosis in diabetic apolipoprotein E-null mice. PLoS ONE 2012, 7, e35683. [Google Scholar] [CrossRef]
- Ingham, M.; Schwartz, G.K. Cell-cycle therapeutics come of age. J. Clin. Oncol. 2017, 35, 2949–2959. [Google Scholar] [CrossRef] [PubMed]
- Cheng, W.; Yang, Z.; Wang, S.; Li, Y.; Wei, H.; Tian, X.; Kan, Q. Recent development of CDK inhibitors: An overview of CDK/inhibitor co-crystal structures. Eur. J. Med. Chem. 2019, 164, 615–639. [Google Scholar] [CrossRef] [PubMed]
- Bai, B.; Liang, Y.; Xu, C.; Lee, M.Y.; Xu, A.; Wu, D.; Vanhoutte, P.M.; Wang, Y. Cuclin-dependent kinase 5-mediated hyperphosphorylation of sirtuin-1 contributes to the development of endothelial senescence and atherosclerosis. Circulation 2012, 126, 729–740. [Google Scholar] [CrossRef] [Green Version]
- Na, Y.R.; Jung, D.; Gu, G.J.; Jang, A.R.; Suh, Y.H.; Seok, S.H. The early synthesis of p35 and activation of CDK5 in LPS-stimulated macrophages suppresses interleukin-10 production. Sci. Signal. 2015, 8, ra121. [Google Scholar] [CrossRef]
- Ahmed, D.; Sharma, M. Cyclin-dependent kinase 5/p35/p39: A novel and imminent therapeutic target for diabetes mellitus. Int. J. Endocrinol. 2011, 2011, 530274. [Google Scholar] [CrossRef] [Green Version]
- Roufayel, R.; Murshid, N. CDK5: Key regulation of atherosclerosis and cell survival. Biomedicines 2019, 7, 88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hellmich, M.R.; Pant, H.C.; Wada, E.; Battey, J.F. Neuronal cdc2-like kinase: A cdc2-related protein kinase with predominantly neuronal expression. Proc. Natl. Acad. Sci. USA 1992, 89, 10867–10871. [Google Scholar] [CrossRef] [Green Version]
- Dhavan, R.; Tsai, L.H. A decade of CDK5. Nat. Rev. Mol. Cell Biol. 2001, 2, 749–759. [Google Scholar] [CrossRef]
- Merk, H.; Zhang, S.; Lehr, T.; Muller, C.; Ulrich, M.; Bibb, J.A.; Adams, R.H.; Bracher, F.; Zahler, S.; Vollmar, A.M.; et al. Inhibition of endothelial Cdk5 reduces tumor growth by promoting non-productive angiogenesis. Oncotarget 2016, 7, 6088–6104. [Google Scholar] [CrossRef]
- Yashima, H.; Terasaki, M.; Sotokawauchi, A.; Matsui, T.; Mori, Y.; Saito, T.; Osaka, N.; Kushima, H.; Hiromura, M.; Ohara, M.; et al. AGE-RAGE axis stimulates oxidized LDL uptake into macrophages through cyclin-dependent kinase 5-CD36 pathway via oxidative stress generation. Int. J. Mol. Sci. 2020, 21, 9263. [Google Scholar] [CrossRef]
- Kume, S.; Takeya, M.; Mori, T.; Araki, N.; Suzuki, H.; Horiuchi, S.; Kodama, T.; Miyauchi, Y.; Takahashi, K. Immunohistochemical and ultrastructural detection of advanced glycation end products in atherosclerotic lesions of human aorta with a novel specific monoclonal antibody. Am. J. Pathol. 1995, 147, 654–667. [Google Scholar]
- Wang, Z.Q.; Jing, L.L.; Yan, J.C.; Sun, Z.; Bao, Z.Y.; Shao, C.; Pang, Q.W.; Geng, Y.; Zhang, L.L.; Li, L.H. Role of AGEs in the progression and regression of atherosclerotic plaques. Glycoconj. J. 2018, 35, 443–450. [Google Scholar] [CrossRef] [PubMed]
- Hassen, N.M.; Wouters, K.; Hujiberts, M.S.; Gijbels, M.J.; Sluimer, J.C.; Scheijen, J.L.; Heeneman, S.; Biessen, E.A.; Daemen, M.J.; Brownlee, M.; et al. Higher levels of advanced glycation endproducts in human carotid atherosclerotic plaques are associated with a repture-prone phenotype. Eur. Heart J. 2014, 35, 1137–1146. [Google Scholar] [CrossRef] [Green Version]
- Han, X.; Ma, W.; Zhu, Y.; Sun, X.; Liu, N. Advanced glycation end products enhance macrophage polarization to the Ma phenotype via the HIF-1α/PDK4 pathway. Mol. Cell Endocrinol. 2020, 514, 110878. [Google Scholar] [CrossRef]
- Bijnen, M.; Beelen, N.; Wetzels, S.; Gaar, J.V.; Vroomen, M.; Wijnands, E.; Scheijen, J.L.; van de Waarenburg, M.P.H.; Gijbels, M.J.; Cleutjens, J.P.; et al. RAGE deficiency dose not affect non-alcholic steatohepatitis and atherosclerosis in Western type diet-fed Ldlr(−/−) mice. Sci. Rep. 2018, 8, 15256. [Google Scholar] [CrossRef]
- Miyawaki, K.; Yamada, Y.; Yano, H.; Niwa, H.; Ban, N.; Ihara, Y.; Kubota, A.; Fujimoto, S.; Kajikawa, M.; Kuroe, A.; et al. Glucose intolerance caused by a defect in the entero-insular axis: A study in gastric inhibitory polypeptide receptor knockout mice. Proc. Natl. Acad. Sci. USA 1999, 96, 14843–14847. [Google Scholar] [CrossRef] [Green Version]
- Terasaki, M.; Yashima, H.; Mori, Y.; Saito, T.; Matsui, T.; Hiromura, M.; Kushima, H.; Osaka, N.; Ohara, M.; Fukui, T.; et al. A dipeptidyl peptidase-4 inhibitor inhibits foam cell formation of macrophages in type 1 diabetes via suppression of CD36 expression. Int. J. Mol. Sci. 2020, 21, 4811. [Google Scholar] [CrossRef]
- Terasaki, M.; Hiromura, M.; Mori, Y.; Kohashi, K.; Nagashima, M.; Kushima, H.; Watanabe, T.; Hirano, T. Amelioration of hyperglycemia with a sodium-glucose cotransporter 2 inhibitor prevents macrophage-driven atherosclerosis through macrophage foam cell formation suppression in type 1 and type 2 diabetic mice. PLoS ONE 2015, 10, e0143396. [Google Scholar] [CrossRef]
- Terasaki, M.; Nagashima, M.; Nohtomi, K.; Kohashi, K.; Tomoyasu, M.; Sinmura, K.; Nogi, Y.; Katayama, Y.; Sato, K.; Itoh, F.; et al. Preventive effect of dipeptidyl peptidase-4 inhibitor on atherosclerosis is mainly attributable to incretin’s actions in nondiabetic and diabetic apolipoprotein E-null mice. PLoS ONE 2013, 8, e70933. [Google Scholar] [CrossRef]
- Terasaki, M.; Nagashima, M.; Watanabe, T.; Nohtomi, K.; Mori, Y.; Miyazaki, A.; Hirano, T. Effects of PKF275-055, a dipeptidyl peptidase-4 inhibitor, on the development of atherosclerotic lesions in apolipoprotein E-null mice. Metabolism 2012, 61, 974–977. [Google Scholar] [CrossRef]
- Terasaki, M.; Hiromura, M.; Mori, Y.; Kohashi, K.; Kushima, H.; Koshibu, M.; Saito, T.; Yashima, H.; Watanabe, T.; Hirano, T. A dipeptidyl peptidase-4 inhibitor suppresses macrophage foam cell formation in diabetic db/db mice and type 2 diabetes patients. Int. J. Endocrinol. 2018, 2018. [Google Scholar] [CrossRef] [Green Version]
- Song, M.G.; Ryoo, I.G.; Choi, H.Y.; Choi, B.H.; Kim, S.T.; Heo, T.H.; Lee, J.Y.; Park, P.H.; Kwak, M.K. NRF2 signaling negatively regulates phorbol-12-Myristate-13-acetate (PMA)-induced differentiation of human monocytic U937 cells into pro-inflammatory macrophages. PLoS ONE 2015, 10, e0134235. [Google Scholar] [CrossRef]
- Tusiimire, J.; Wallace, J.; Woods, N.; Dufton, M.J.; Parkinson, J.A.; Abbott, G.; Clements, C.J.; Young, L.; Park, J.K.; Jeon, J.W.; et al. Effect of bee venom and its fractions of pro-inflammatory cytokines in PMA-differentiated U937 cells co-stimulated with LPS. Vaccines 2016, 4, 11. [Google Scholar] [CrossRef] [Green Version]
- Hida, A.; Kawakami, A.; Nakashima, T.; Yamasaki, S.; Sakai, H.; Urayama, S.; Ida, H.; Nakamura, H.; Migita, K.; Kawabe, Y.; et al. Nuclear factor-kappaB and caspases co-operatively regulate the activation and apoptosis of human macrophages. Immunology 2000, 99, 553–560. [Google Scholar] [CrossRef]
- Whyte, J.; Roberts, A.D.; Morley, K.A.; Sharp, R.J.; Marsh, P.D. Phagocytosis of mycobacteria by U937 cells: A rapid method for monitoring uptake and separating phagocytosed and free bacteria by magnetic beads. Lett. Appl. Microbiol. 2000, 30, 90–94. [Google Scholar] [CrossRef]
- Ewart, M.A.; Kennedy, S. AMPK and vasculoprotection. Pharmacol. Ther. 2011, 131, 242–253. [Google Scholar] [CrossRef]
- Carling, D.; Sanders, M.J.; Woods, A. The regulation of AMP-activated protein kinase by upstream kinases. Int. J. Obes. 2008, 32 (Suppl. S4), S55–S59. [Google Scholar] [CrossRef] [Green Version]
- Gao, F.; Chen, J.; Zhu, H. A potential strategy for treating atherosclerosis: Improving endothelial function via AMP-activated protein kinase. Sci. China Life Sci. 2018, 61, 1024–1029. [Google Scholar] [CrossRef]
- Sakamaki, J.; Fu, A.; Reeks, C.; Baird, S.; Depatie, C.; Al Azzabi, M.; Bardeesy, N.; Gingras, A.C.; Yee, S.P.; Screaton, R.A. Role of the SIK-2-p35-PJA2 complex in pancreatic β-cell functional compensation. Nat. Cell Biol. 2014, 16, 234–244. [Google Scholar] [CrossRef] [Green Version]
- Fang, K.; Gu, M. Crocin improves insulin sensitivity and ameliorates adiposity by regulating AMPK-CDK5-PPARγ signaling. Biomed Res. Int. 2020, 2020, 9136282. [Google Scholar] [CrossRef] [PubMed]
- Musson, M.C.; Jepeal, L.I.; Mabray, P.D.; Zhdanova, I.V.; Cardoso, W.V.; Wolfe, M.N. Expression of glucose-dependent insulinotropic polypeptide in the zebrafish. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2009, 297, R1803–R1812. [Google Scholar] [CrossRef] [Green Version]
- Kanungo, J.; Zheng, Y.L.; Amin, N.D.; Kaur, S.; Ramchandran, R.; Pant, H.C. Specific inhibition of cyclin-dependent kinase 5 activity induces motor neuron development in vivo. Biocham. Biophys. Res. Commun. 2009, 386, 263–267. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
{kind=link}
| Gipr+/+ Mice | Gipr+/+ Mice +[D-Ala2]GIP(1–42) | Gipr−/− Mice | Gipr−/− Mice +[D-Ala2]GIP(1–42) | |
|---|---|---|---|---|
| Number | 5 | 5 | 5 | 5 |
| Final body weight (g) | 24 ± 0.8 | 24.1 ± 1.7 | 22.3 ± 1.4 | 22.5 ± 0.7 |
| Food Intake (g/day) | 4.0 ± 0.7 | 4.1 ± 1.0 | 4.3 ± 0.5 | 4.3 ± 0.6 |
| SBP (mmHg) | 102 ± 11 | 100 ± 8 | 102 ± 8 | 103 ± 11 |
| DBP (mmHg) | 63 ± 9 | 58 ± 4 | 65 ± 9 | 62 ± 10 |
| Heart rate (bpm) | 538 ± 50 | 561 ± 55 | 591 ± 48 | 594 ± 51 |
| Total-C (mg/dL) | 104 ± 10 | 106 ± 4 | 112 ± 10 | 117 ± 23 |
| HDL-C (mg/dL) | 36 ± 16 | 41 ± 7 | 53 ± 12 | 49 ± 12 |
| Triglycerides (mg/dL) | 99 ± 8 | 103 ± 7 | 52 ± 43 | 58 ± 44 |
| Insulin (ng/mL) | 0.4 ± 0.25 | 0.41 ± 0.13 | 0.43 ± 0.2 | 0.46 ± 0.12 |
| Total-GIP (pmol/L) | 30 ± 8 | 46 ± 18 | 34 ± 5 | 43 ± 7 |
| FBG (mg/dL) | 99 ± 8 | 98 ± 9 | 100 ± 21 | 101 ± 9 |
| HbA1c (%) | 4.7 ± 0.2 | 4.8 ± 0.2 | 4.9 ± 0.1 | 4.9 ± 0.2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Terasaki, M.; Yashima, H.; Mori, Y.; Saito, T.; Shiraga, Y.; Kawakami, R.; Ohara, M.; Fukui, T.; Hirano, T.; Yamada, Y.; et al. Glucose-Dependent Insulinotropic Polypeptide Suppresses Foam Cell Formation of Macrophages through Inhibition of the Cyclin-Dependent Kinase 5-CD36 Pathway. Biomedicines 2021, 9, 832. https://doi.org/10.3390/biomedicines9070832
Terasaki M, Yashima H, Mori Y, Saito T, Shiraga Y, Kawakami R, Ohara M, Fukui T, Hirano T, Yamada Y, et al. Glucose-Dependent Insulinotropic Polypeptide Suppresses Foam Cell Formation of Macrophages through Inhibition of the Cyclin-Dependent Kinase 5-CD36 Pathway. Biomedicines. 2021; 9(7):832. https://doi.org/10.3390/biomedicines9070832
Chicago/Turabian StyleTerasaki, Michishige, Hironori Yashima, Yusaku Mori, Tomomi Saito, Yoshie Shiraga, Raichi Kawakami, Makoto Ohara, Tomoyasu Fukui, Tsutomu Hirano, Yuichiro Yamada, and et al. 2021. "Glucose-Dependent Insulinotropic Polypeptide Suppresses Foam Cell Formation of Macrophages through Inhibition of the Cyclin-Dependent Kinase 5-CD36 Pathway" Biomedicines 9, no. 7: 832. https://doi.org/10.3390/biomedicines9070832
APA StyleTerasaki, M., Yashima, H., Mori, Y., Saito, T., Shiraga, Y., Kawakami, R., Ohara, M., Fukui, T., Hirano, T., Yamada, Y., Seino, Y., & Yamagishi, S.-i. (2021). Glucose-Dependent Insulinotropic Polypeptide Suppresses Foam Cell Formation of Macrophages through Inhibition of the Cyclin-Dependent Kinase 5-CD36 Pathway. Biomedicines, 9(7), 832. https://doi.org/10.3390/biomedicines9070832