
Chronic Kidney Disease Induced by Cisplatin, Folic Acid and Renal Ischemia Reperfusion Induces Anemia and Promotes GATA-2 Activation in Mice

 , ,
, ,  and
and
Abstract
:1. Introduction
2. Methods
2.1. Animals
2.2. Experimental Protocol
2.3. Ischemia Reperfusion
2.4. Folic Acid Nephropathy
2.5. Cisplatin Treatment
2.6. Blood Sampling and Tissue Collection
2.7. Renal Function
2.8. Hemoglobin Analysis
2.9. Blood Count Test
2.10. Real-Time PCR
2.11. Enzyme-Linked Immunosorbent Assay
2.12. Renal Fibrosis Analysis
2.13. Kidney Extraction and Sectioning
2.14. Immunofluorescence
2.15. Statistical Analysis
3. Results
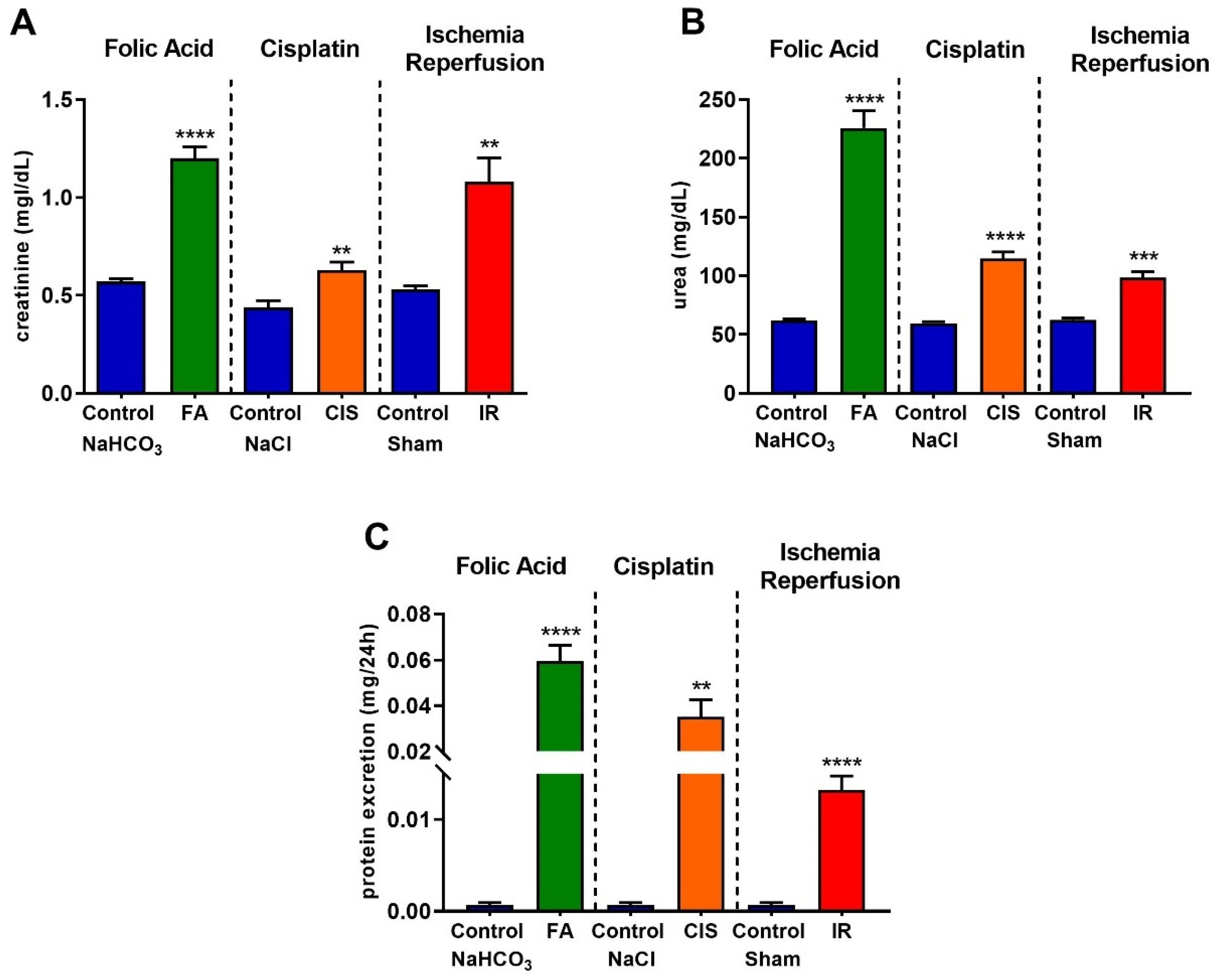
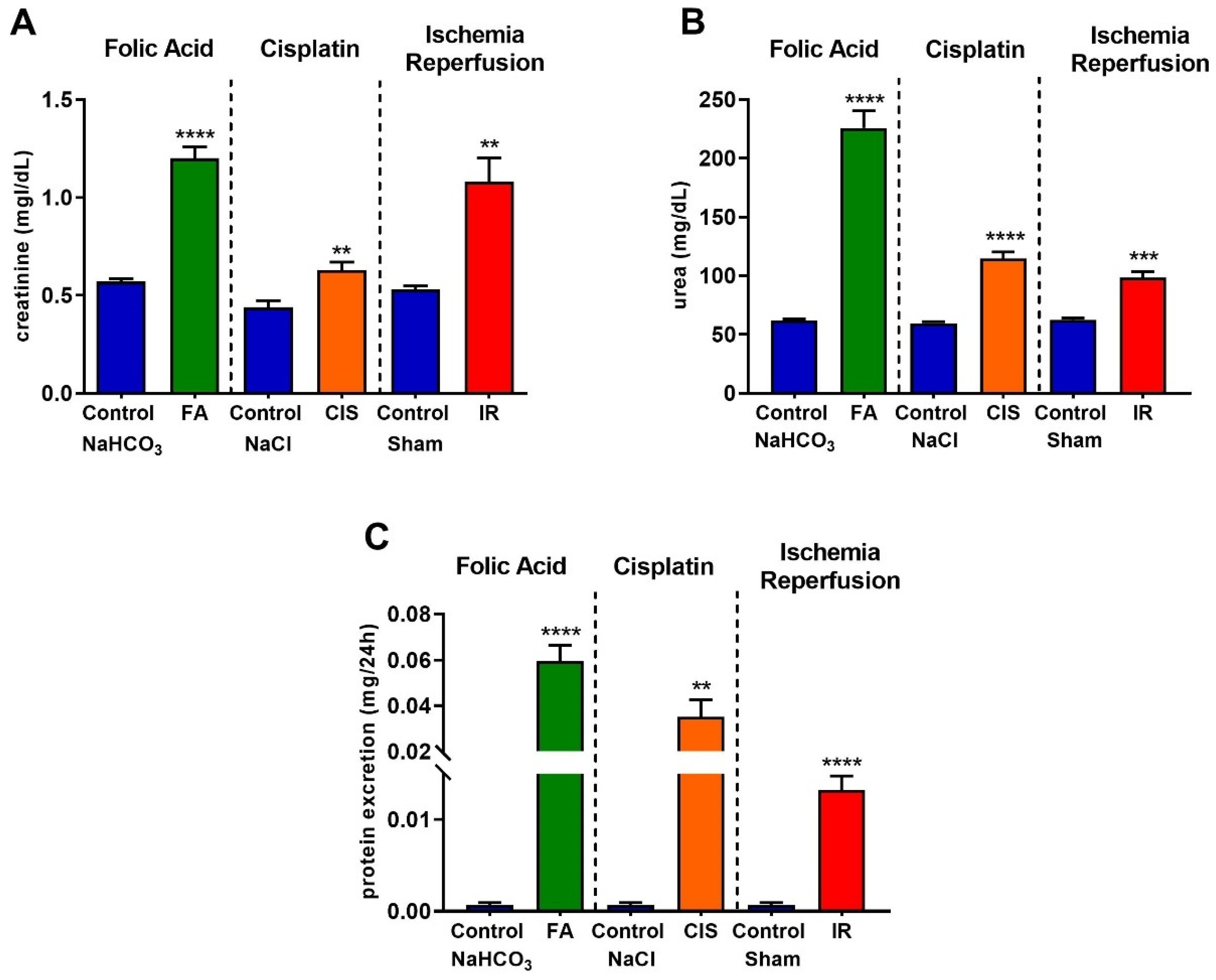
3.1. Folic Acid, Cisplatin and Ischemia-Reperfusion Lead to Renal Dysfunction
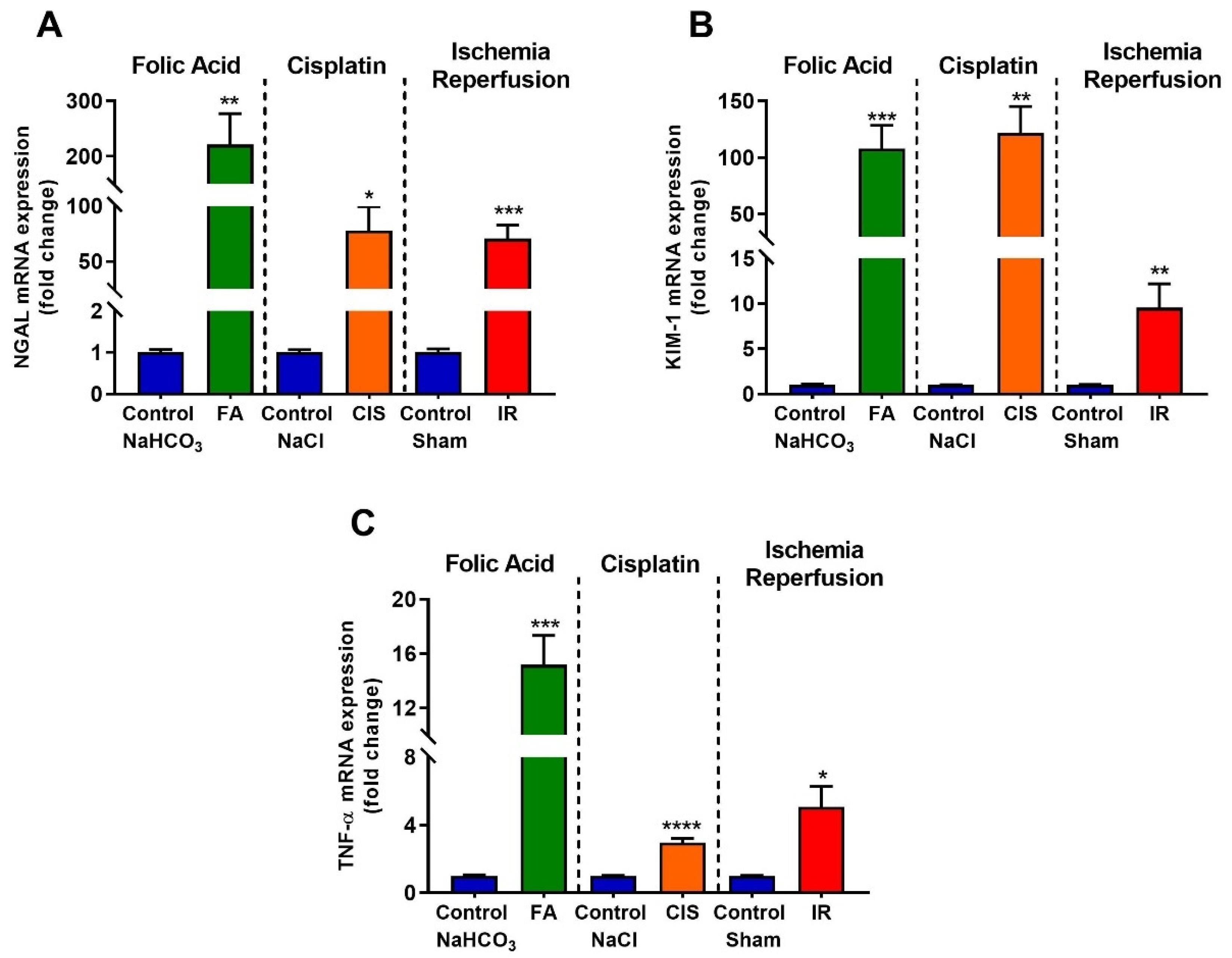
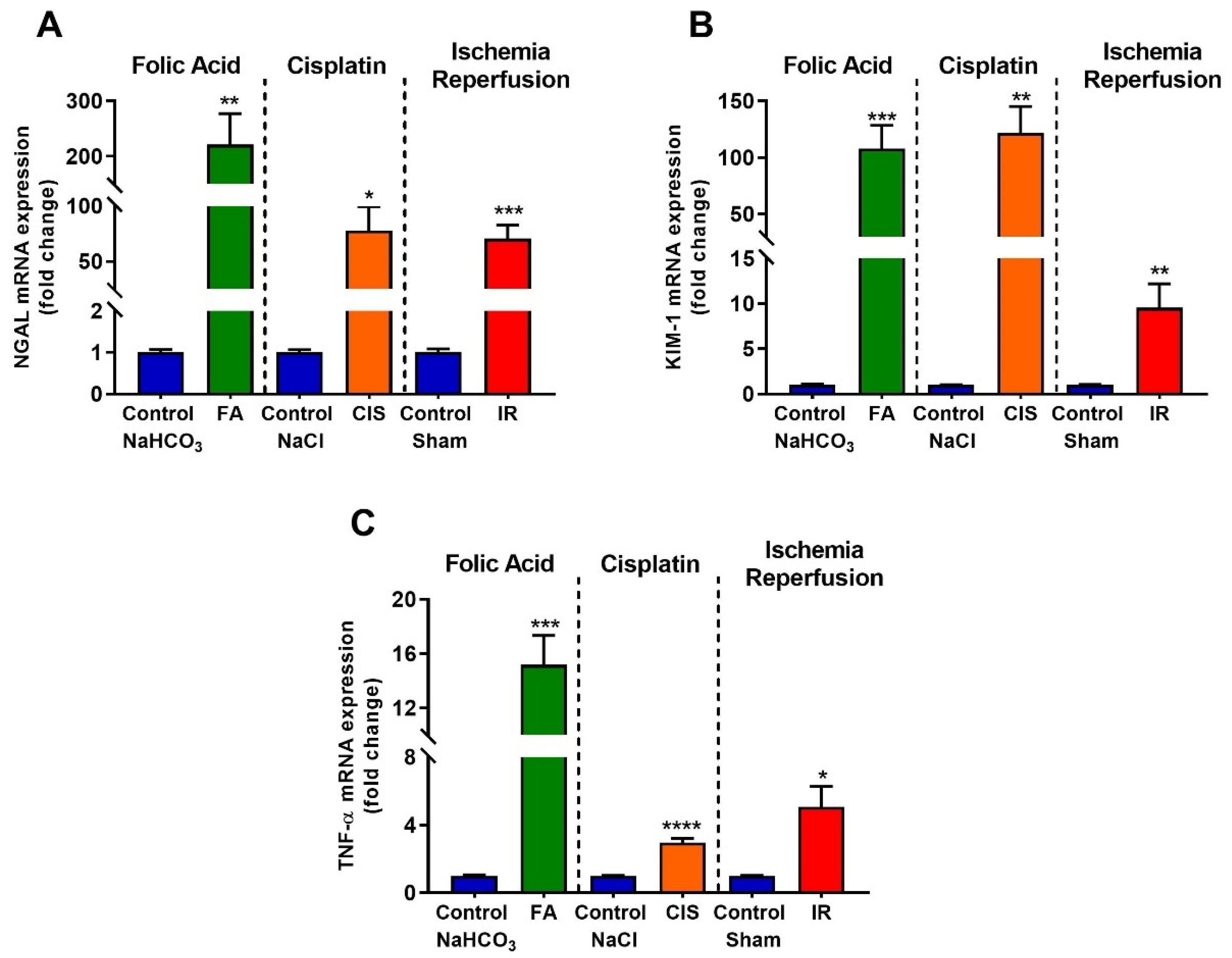
3.2. Folic Acid, Cisplatin and Ischemia-Reperfusion Increase Kidney Injury Markers
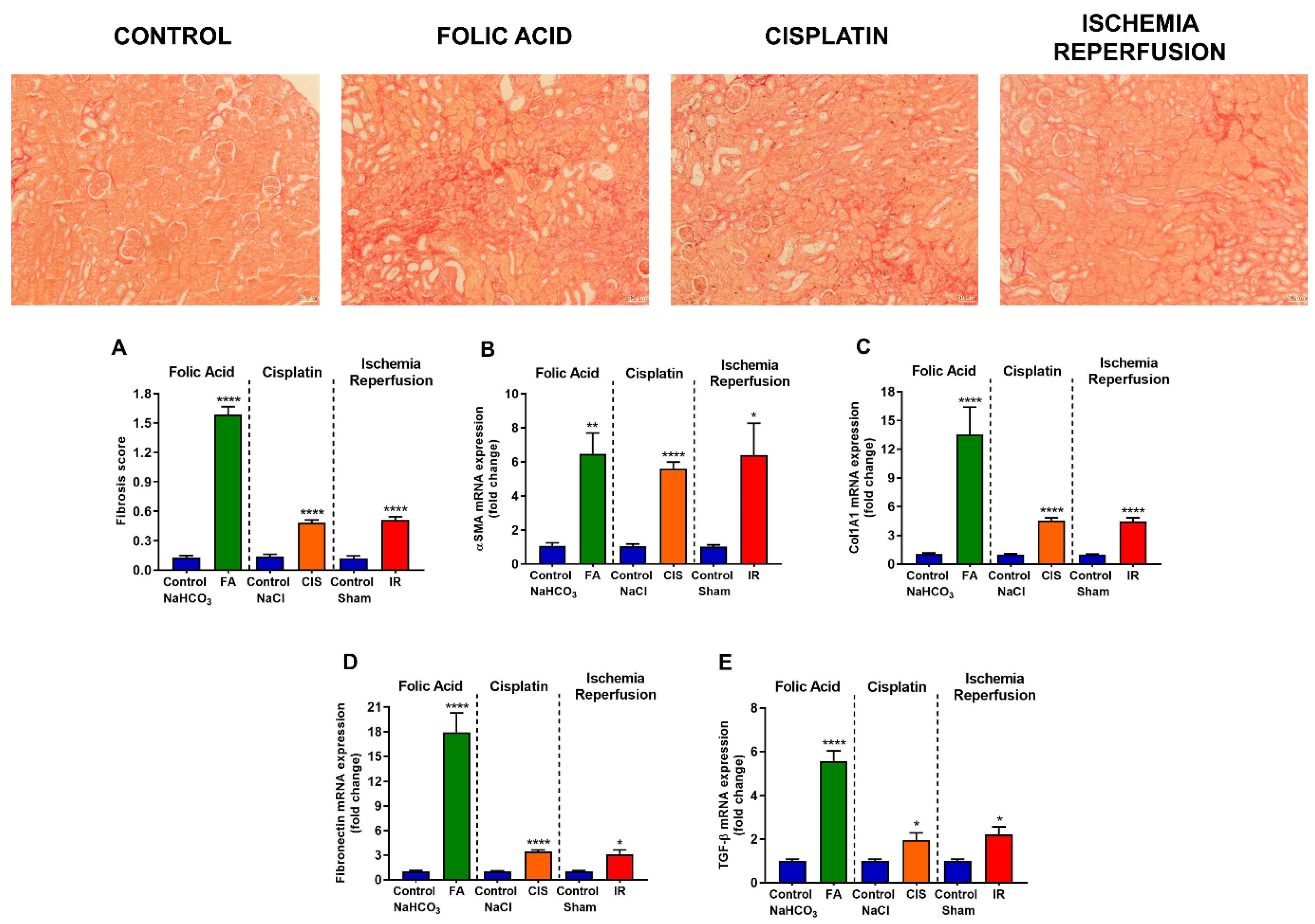
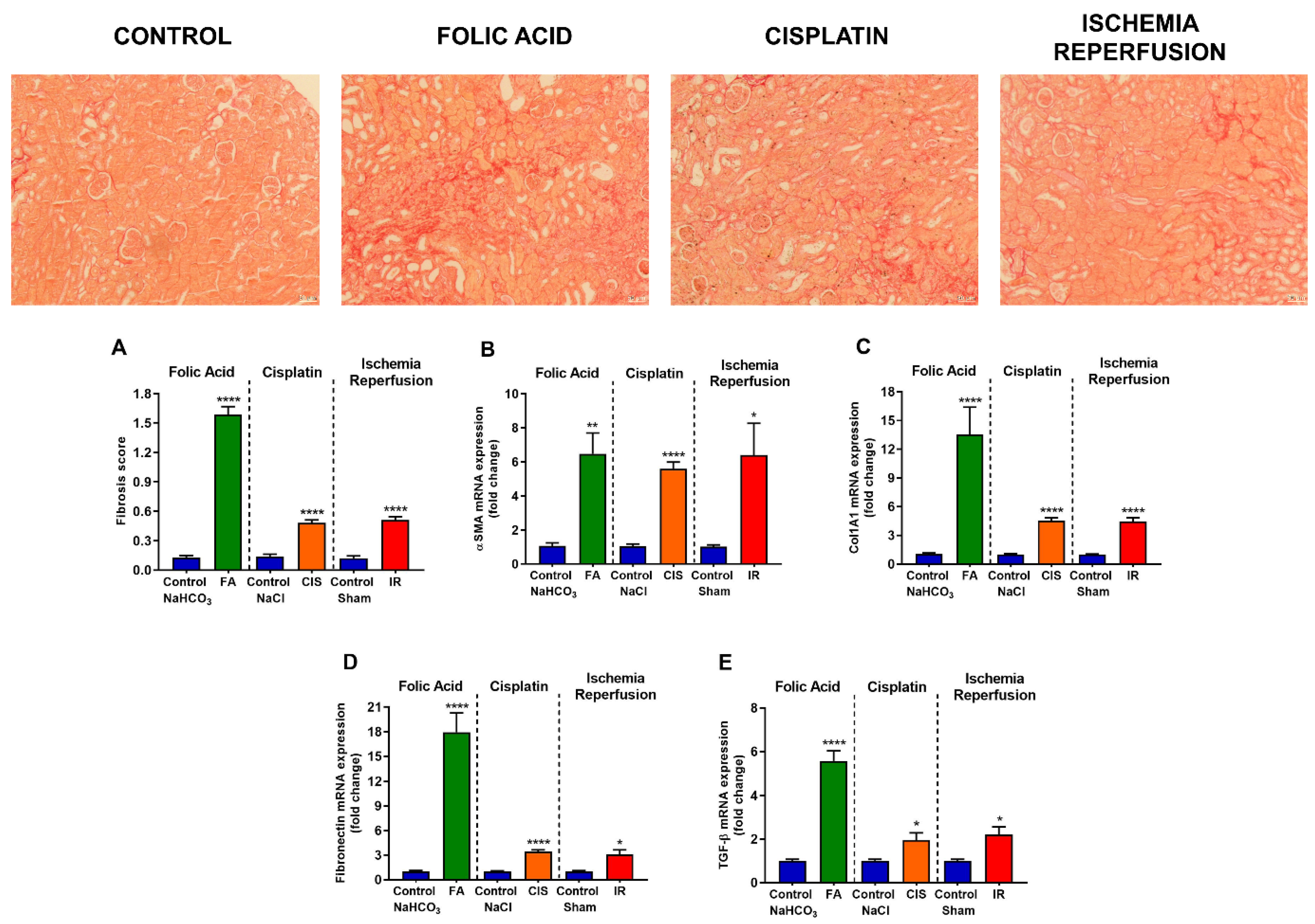
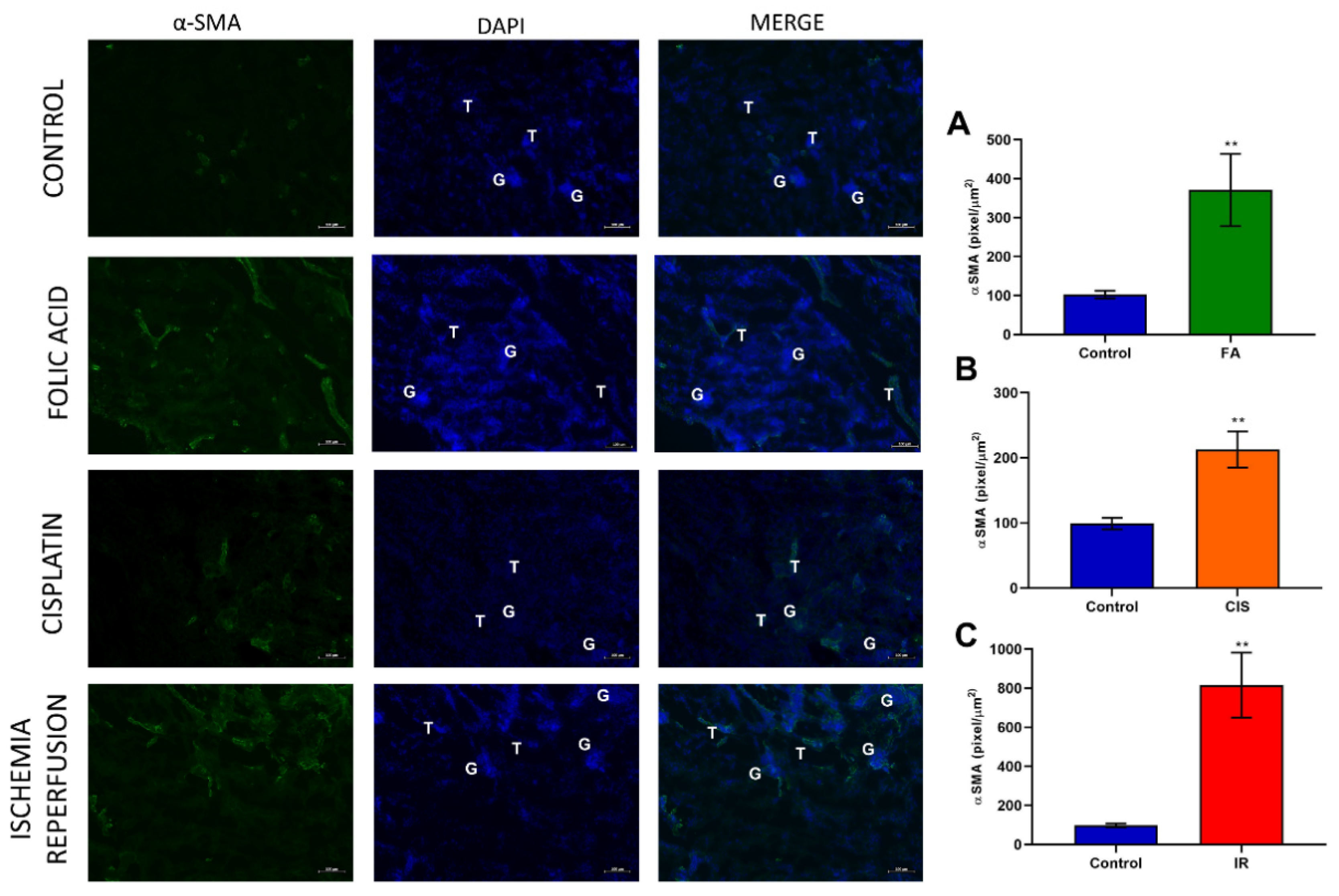
3.3. Folic Acid, Cisplatin and Ischemia-Reperfusion Increase Fibrotic Markers
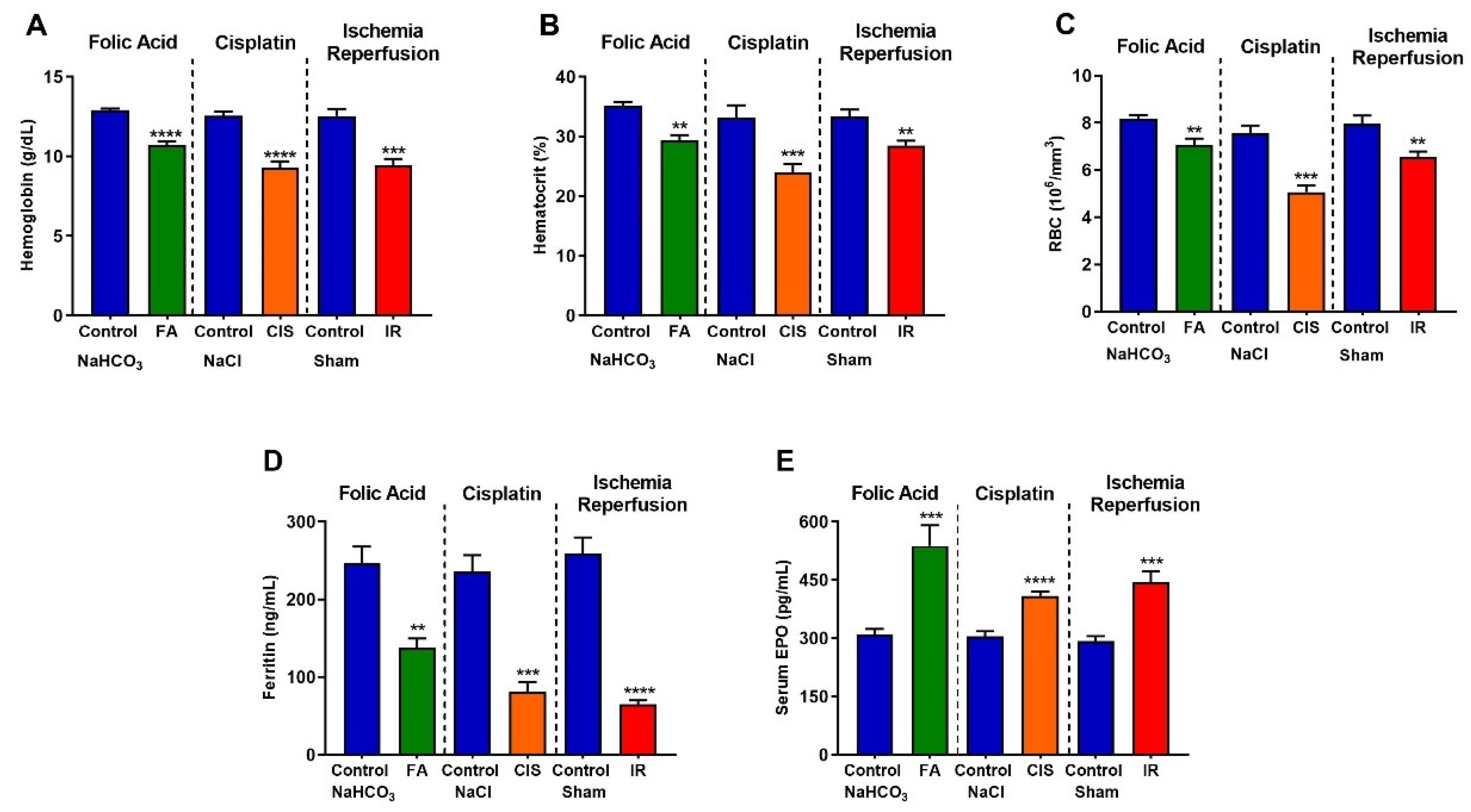
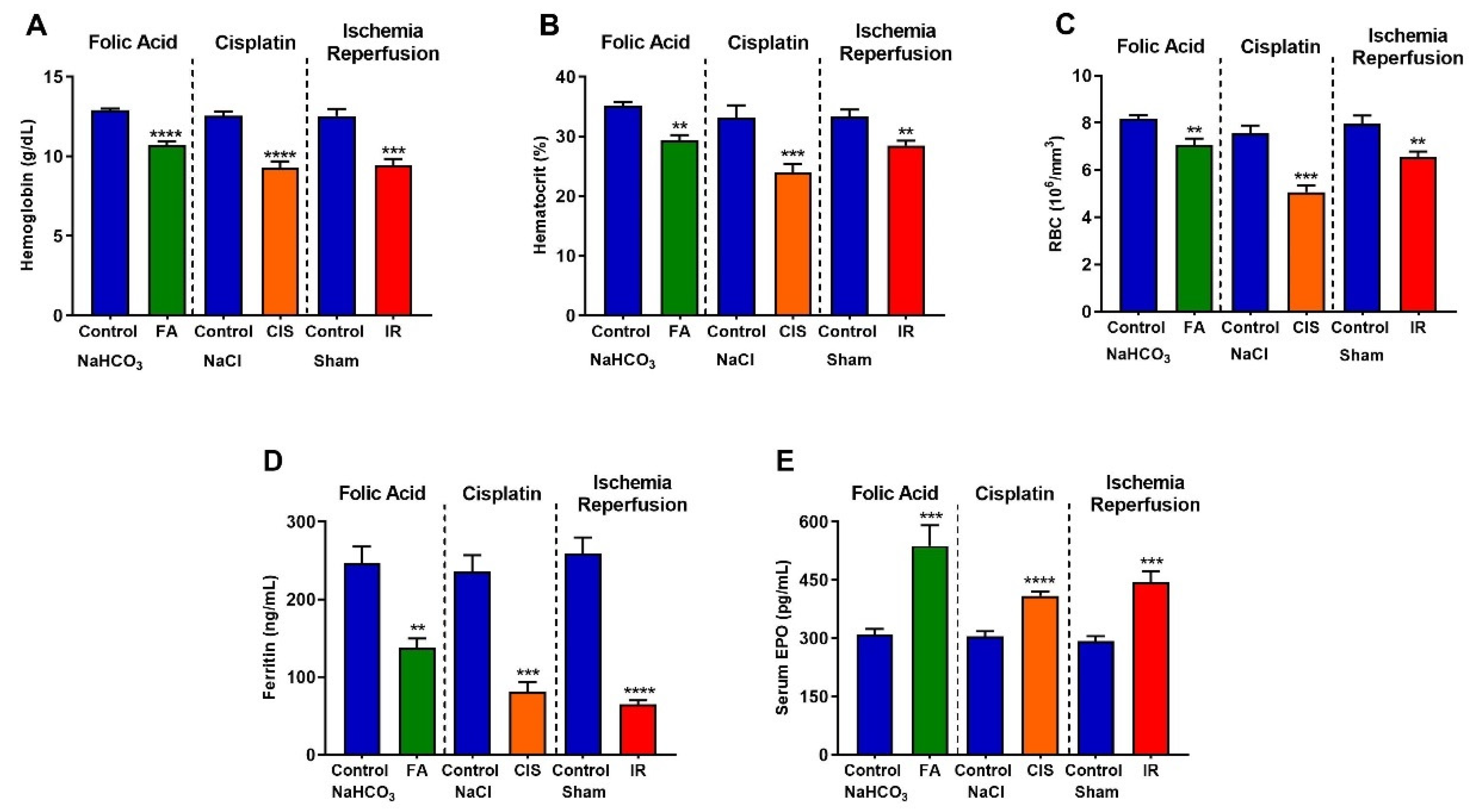
3.4. Folic Acid, Cisplatin and Ischemia-Reperfusion CKD Models Develop Anemia
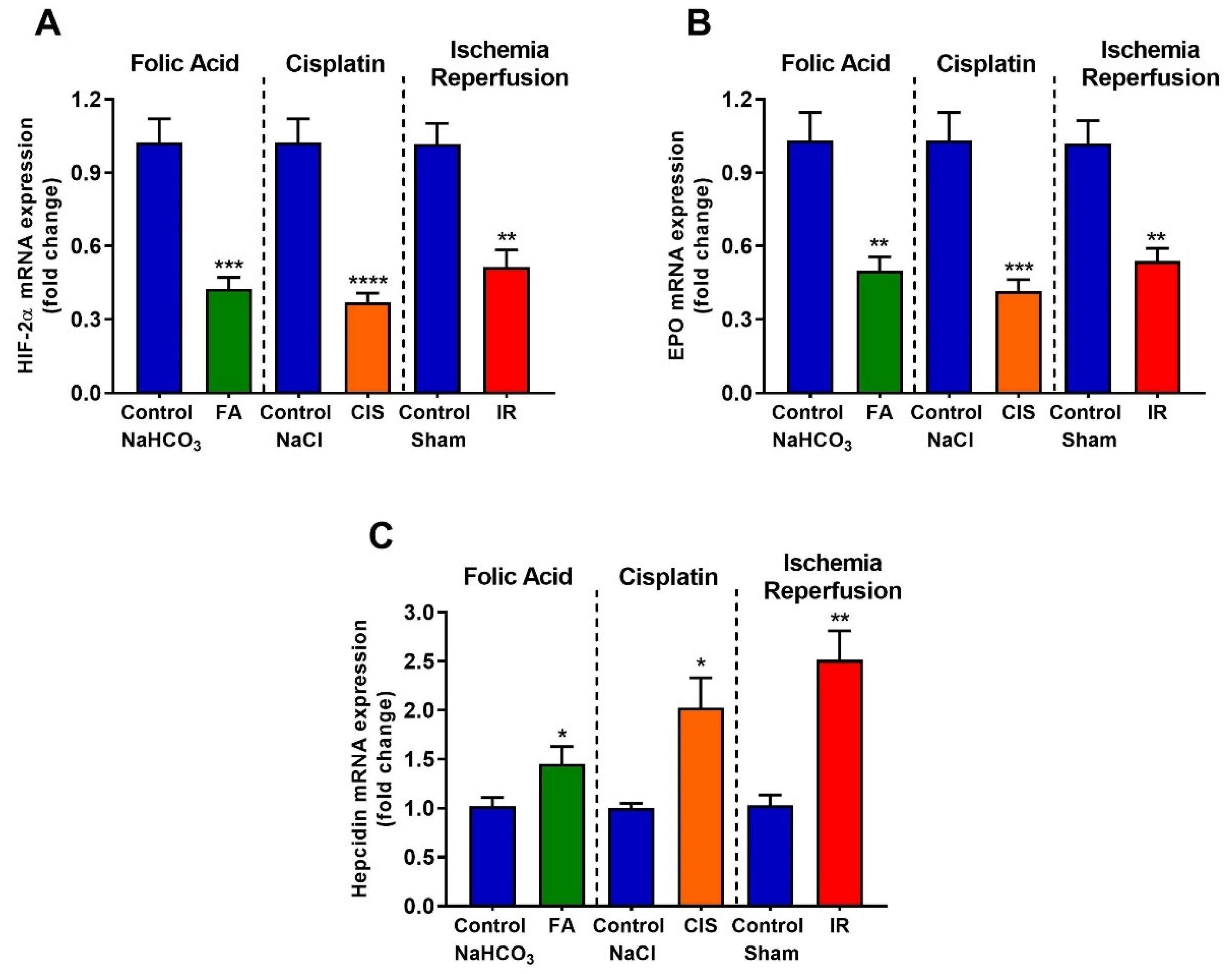
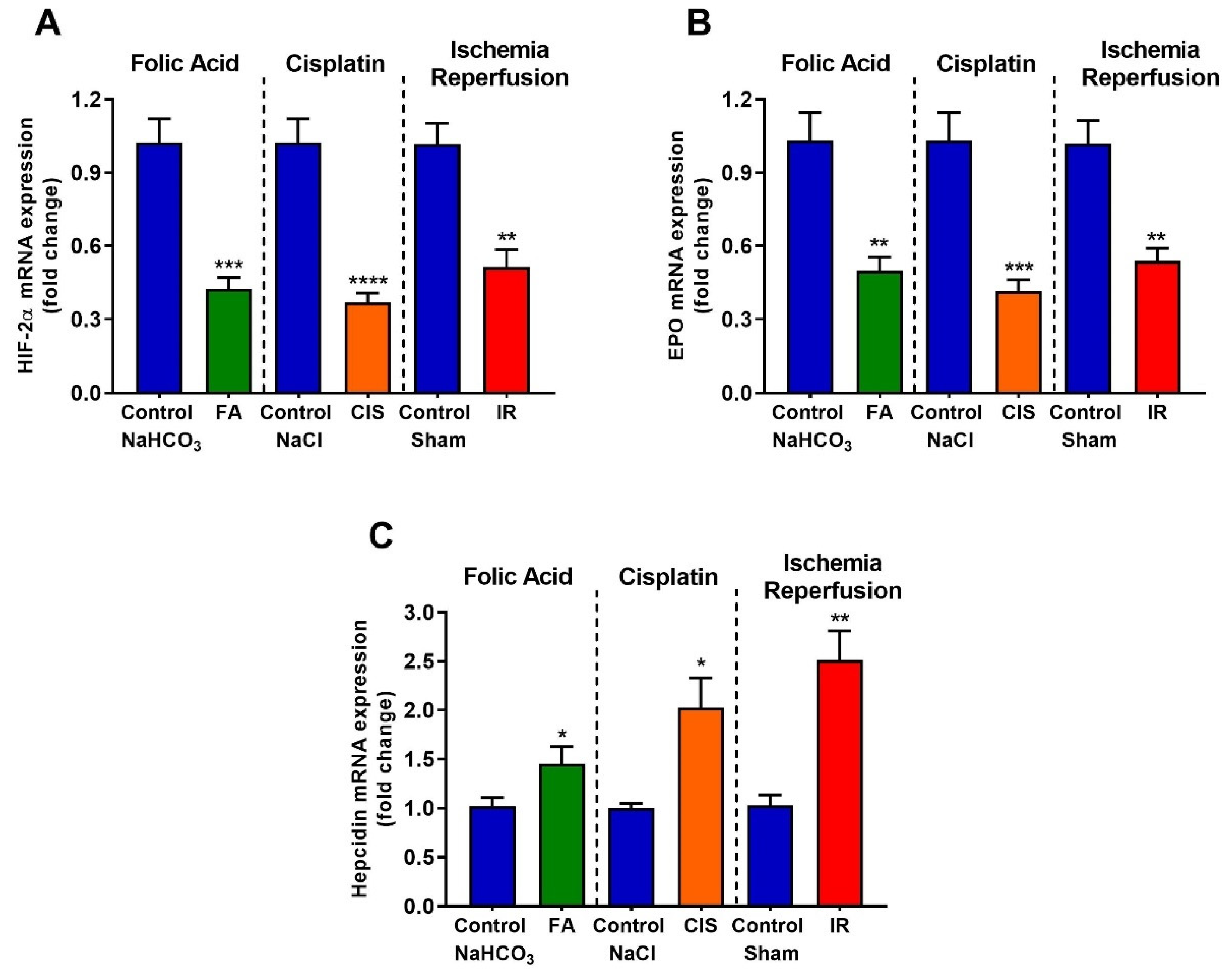
3.5. Folic Acid, Cisplatin and Ischemia-Reperfusion Decrease Renal Expression of Erythropoietin and Hypoxia Inducible Factor-2 Alpha, and Increase Hepcidin Levels
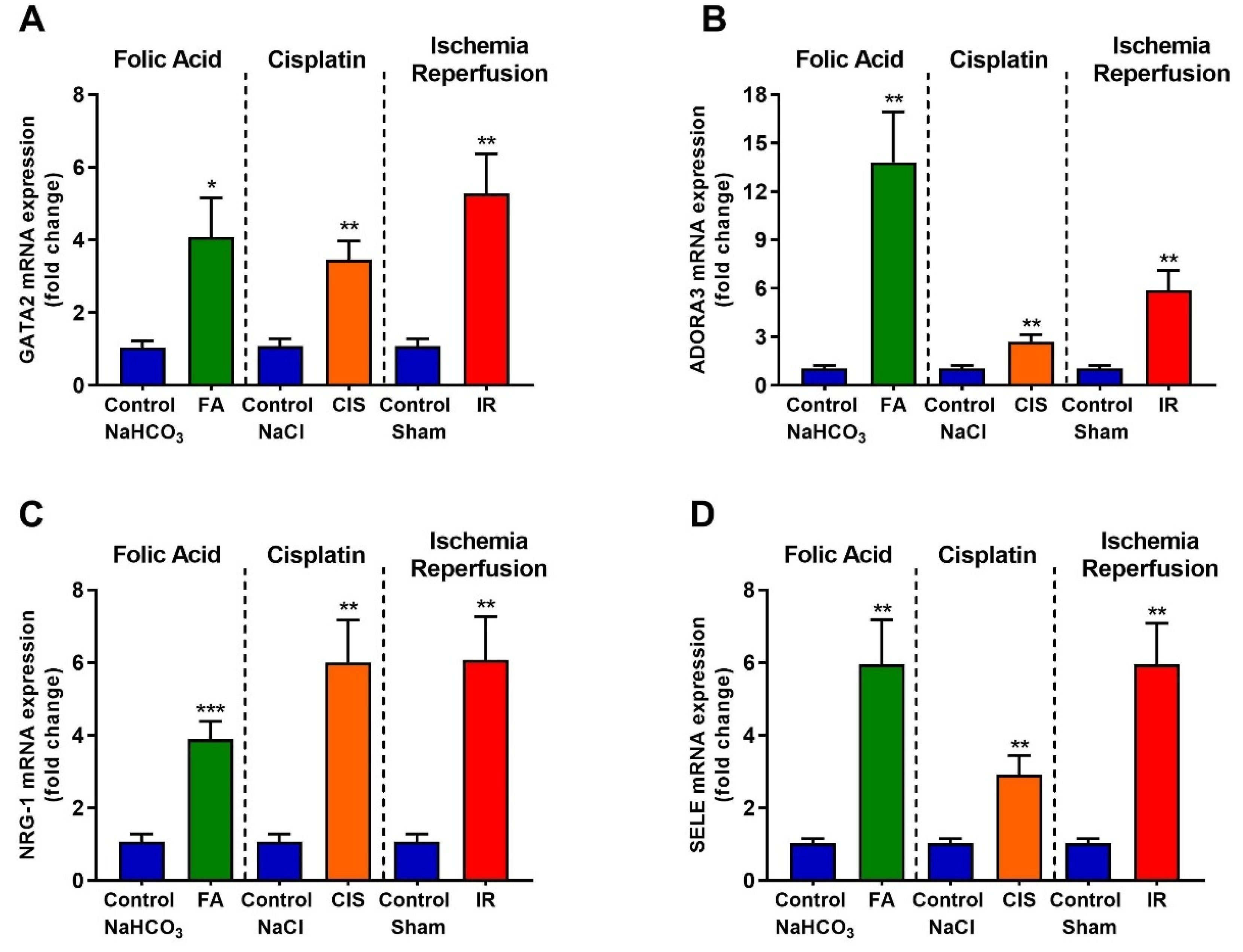
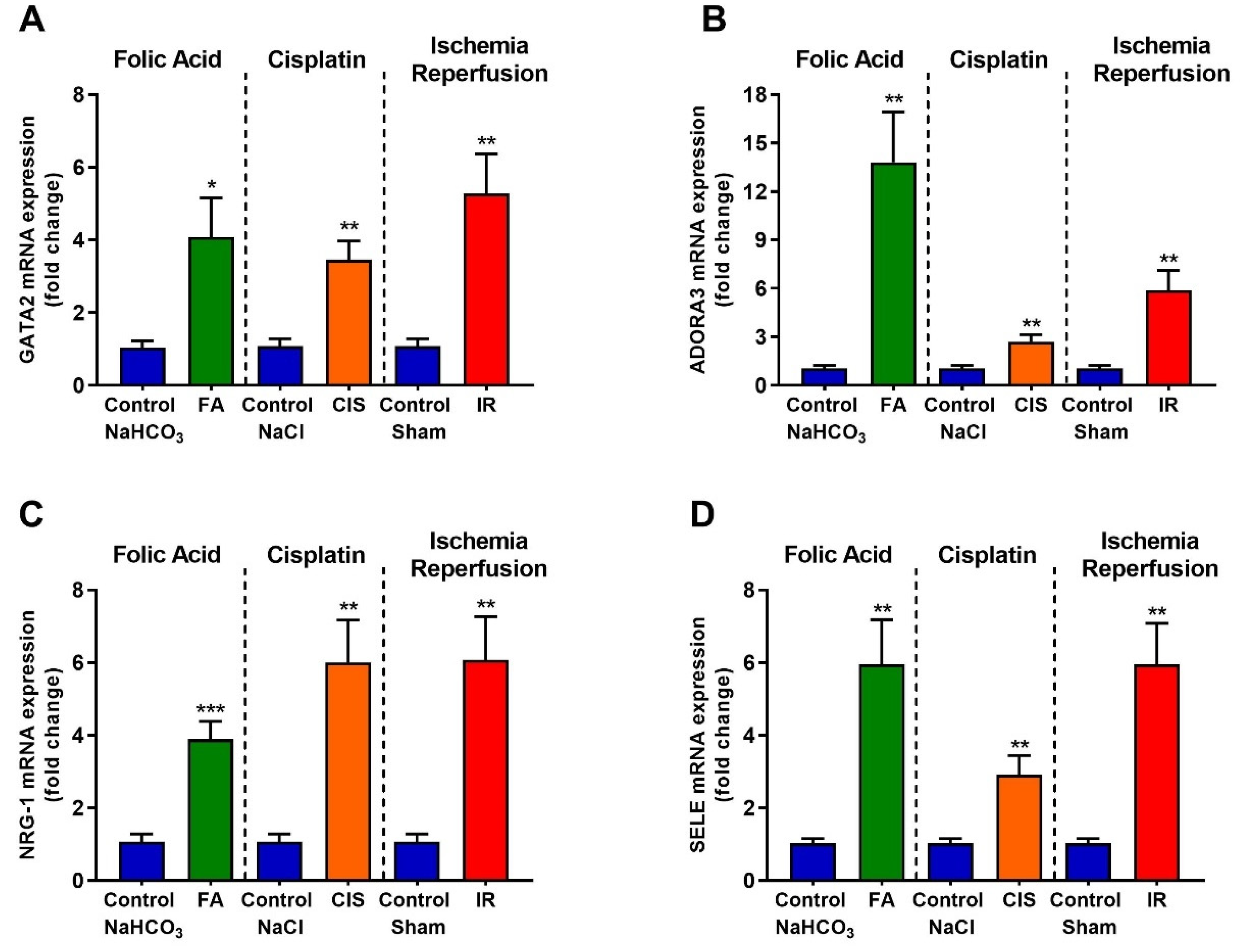
3.6. Folic Acid, Cisplatin and Ischemia-Reperfusion Increase GATA-2 Transcription Factor and the Expression of Its Target Genes
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| αSMA | Alpha smooth muscle actin |
| ADORA3 | Adenosine A3 receptor |
| CKD | Chronic Kidney Disease |
| CIS | Cisplatin |
| COL1A1 | Collagen 1A1 |
| EPO | Erythropoietin |
| FA | Folic acid |
| GATA2 | GATA binding factor 2 or erythroid transcription factor |
| HIF2α | Hypoxia-inducible factor 2 |
| IR | Ischemia reperfusion |
| KIM-1 | Kidney injury molecule-1 |
| NGAL | Neutrophil gelatinase-associated lipocalin |
| NRG-1 | neuregulin 1 |
| SELE | Selectin E |
| TGF-β | Transforming growth factor beta |
| TNF-α | Tumor necrosis factor alpha |
References
- Coresh, J. Update on the Burden of CKD. J. Am. Soc. Nephrol. 2017, 28, 1020–1022. [Google Scholar] [CrossRef] [Green Version]
- He, L.; Wei, Q.; Liu, J.; Yi, M.; Liu, Y.; Liu, H.; Sun, L.; Peng, Y.; Liu, F.; Venkatachalam, M.A.; et al. AKI on CKD: Heightened injury, suppressed repair, and the underlying mechanisms. Kidney Int. 2017, 92, 1071–1083. [Google Scholar] [CrossRef]
- Venkatachalam, M.A.; Weinberg, J.M.; Kriz, W.; Bidani, A.K. Failed Tubule Recovery, AKI-CKD Transition, and Kidney Disease Progression. J. Am. Soc. Nephrol. 2015, 26, 1765–1776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeisberg, M.; Neilson, E.G. Mechanisms of tubulointerstitial fibrosis. J. Am. Soc. Nephrol. 2010, 21, 1819–1834. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hewitson, T.D.; Holt, S.G.; Smith, E.R. Progression of Tubulointerstitial Fibrosis and the Chronic Kidney Disease Phenotype—Role of Risk Factors and Epigenetics. Front. Pharmacol. 2017, 8, 520. [Google Scholar] [CrossRef] [Green Version]
- Hewitson, T.D. Renal tubulointerstitial fibrosis: Common but never simple. Am. J. Physiol. Renal Physiol. 2009, 296, F1239–F1244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sato, Y.; Takahashi, M.; Yanagita, M. Pathophysiology of AKI to CKD progression. Semin. Nephrol. 2020, 40, 206–215. [Google Scholar] [CrossRef]
- Kirita, Y.; Wu, H.; Uchimura, K.; Wilson, P.C.; Humphreys, B.D. Cell profiling of mouse acute kidney injury reveals conserved cellular responses to injury. Proc. Natl. Acad. Sci. USA 2020, 117, 15874–15883. [Google Scholar] [CrossRef] [PubMed]
- Bohle, A.; Christ, H.; Grund, K.E.; Mackensen, S. The role of the interstitium of the renal cortex in renal disease. Contrib. Nephrol. 1979, 16, 109–114. [Google Scholar]
- Eddy, A.A. Molecular insights into renal interstitial fibrosis. J. Am. Soc. Nephrol. 1996, 7, 2495–2508. [Google Scholar] [CrossRef]
- Kuncio, G.S.; Neilson, E.G.; Haverty, T. Mechanisms of tubulointerstitial fibrosis. Kidney Int. 1991, 39, 550–556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Babitt, J.L.; Lin, H.Y. Mechanisms of anemia in CKD. J. Am. Soc. Nephrol. 2012, 23, 1631–1634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rocha, L.A.; Barreto, D.V.; Barreto, F.C.; Dias, C.B.; Moysés, R.; Silva, M.R.; Moura, L.A.; Draibe, S.A.; Jorgetti, V.; Carvalho, A.B.; et al. Serum ferritin level remains a reliable marker of bone marrow iron stores evaluated by histomorphometry in hemodialysis patients. Clin. J. Am. Soc. Nephrol. 2009, 4, 105–109. [Google Scholar] [CrossRef]
- Nissenson, A.R. Achieving target hematocrit in dialysis patients: New concepts in iron management. Am. J. Kidney Dis. 1997, 30, 907–911. [Google Scholar] [CrossRef]
- Nakanishi, T.; Kimura, T.; Kuragano, T. The Hepcidin-Anemia Axis: Pathogenesis of Anemia in Chronic Kidney Disease. Contrib. Nephrol. 2019, 198, 124–134. [Google Scholar] [CrossRef]
- D’Angelo, G. Role of hepcidin in the pathophysiology and diagnosis of anemia. Blood Res. 2013, 48, 10–15. [Google Scholar] [CrossRef]
- Babitt, J.L.; Lin, H.Y. Molecular mechanisms of hepcidin regulation: Implications for the anemia of CKD. Am. J. Kidney Dis. 2010, 55, 726–741. [Google Scholar] [CrossRef] [Green Version]
- Rahman, A.; Yamazaki, D.; Sufiun, A.; Kitada, K.; Hitomi, H.; Nakano, D.; Nishiyama, A. A novel approach to adenine-induced chronic kidney disease associated anemia in rodents. PLoS ONE 2018, 13, e0192531. [Google Scholar] [CrossRef]
- Obara, N.; Suzuki, N.; Kim, K.; Nagasawa, T.; Imagawa, S.; Yamamoto, M. Repression via the GATA box is essential for tissue-specific erythropoietin gene expression. Blood 2008, 111, 5223–5232. [Google Scholar] [CrossRef] [Green Version]
- Imagawa, S.; Nakano, Y.; Obara, N.; Suzuki, N.; Doi, T.; Kodama, T.; Nagasawa, T.; Yamamoto, M. A GATA-specific inhibitor (K-7174) rescues anemia induced by IL-1beta, TNF-alpha, or L-NMMA. FASEB J. 2003, 17, 1742–1744. [Google Scholar] [CrossRef]
- La Ferla, K.; Reimann, C.; Jelkmann, W.; Hellwig-Bürgel, T. Inhibition of erythropoietin gene expression signaling involves the transcription factors GATA-2 and NF-kappaB. FASEB J. 2002, 16, 1811–1813. [Google Scholar] [CrossRef] [PubMed]
- Nasioudis, D.; Witkin, S.S. Neutrophil gelatinase-associated lipocalin and innate immune responses to bacterial infections. Med. Microbiol. Immunol. 2015, 204, 471–479. [Google Scholar] [CrossRef] [PubMed]
- Goetz, D.H.; Holmes, M.A.; Borregaard, N.; Bluhm, M.E.; Raymond, K.N.; Strong, R.K. The neutrophil lipocalin NGAL is a bacteriostatic agent that interferes with siderophore-mediated iron acquisition. Mol. Cell 2002, 10, 1033–1043. [Google Scholar] [CrossRef]
- Lönnberg, M.; Garle, M.; Lönnberg, L.; Birgegård, G. Patients with anaemia can shift from kidney to liver production of erythropoietin as shown by glycoform analysis. J. Pharm. Biomed. Anal. 2013, 81, 187–192. [Google Scholar] [CrossRef] [Green Version]
- Lifshitz, L.; Tabak, G.; Gassmann, M.; Mittelman, M.; Neumann, D. Macrophages as novel target cells for erythropoietin. Haematologica 2010, 95, 1823–1831. [Google Scholar] [CrossRef] [Green Version]
- de Seigneux, S.; Lundby, A.K.; Berchtold, L.; Berg, A.H.; Saudan, P.; Lundby, C. Increased Synthesis of Liver Erythropoietin with CKD. J. Am. Soc. Nephrol. 2016, 27, 2265–2269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shih, H.M.; Wu, C.J.; Lin, S.L. Physiology and pathophysiology of renal erythropoietin-producing cells. J. Formos. Med. Assoc. 2018, 117, 955–963. [Google Scholar] [CrossRef] [PubMed]
- Haase, V.H. Regulation of erythropoiesis by hypoxia-inducible factors. Blood Rev. 2013, 27, 41–53. [Google Scholar] [CrossRef] [Green Version]
- Buck, I.; Morceau, F.; Cristofanon, S.; Heintz, C.; Chateauvieux, S.; Reuter, S.; Dicato, M.; Diederich, M. Tumor necrosis factor alpha inhibits erythroid differentiation in human erythropoietin-dependent cells involving p38 MAPK pathway, GATA-1 and FOG-1 downregulation and GATA-2 upregulation. Biochem. Pharmacol. 2008, 76, 1229–1239. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primers for RT-PCR | ||
|---|---|---|
| Gene | Forward 5’-3’ | Reverse 5’-3’ |
| 18S | CGC CGC TAG AGG TGA AAT TC | TCT TGG CAA ATG CTT TCG C |
| β-actin | CTG GCC TCA CTG TCC ACC TT | CGG ACT CAT CGT ACT CCT GCT T |
| NGAL | ATG TGC AAG TGG CCA CCA CG | CGC ATC CCA GTC AGC CAC AC |
| TNF-α | GCC TCT TCT CAT TCC TGC TTG | CTG ATG AGA GGG AGG CCA TT |
| KIM-1 | TGT CGA GTG GAG ATT CCT GGA TGG T | GGT CTT CCT GTA GCT GTG GGC C |
| TGF-β1 | CAA CAA TTC CTG GCG TTA CCT TGG | GAA AGC CCT GTA TTC CGT CTC CTT |
| Col1A1 | CCC CGG GAC TCC TGG ACT T | GCT CCG ACA CGC CCT CTC TC |
| α-SMA | TTG GAA AAG ATC TGG CAC CAC | GCA GTA GTC ACG AAG GAA TAG |
| Fibronectin | CCT ACG GCC ACT GTG TCA CC | AGT CTG GGT CAC GGC TGT CT |
| HIF-2α | CTG GAC AAA GCC TCC ATC AT | TTG CTG ATG TTT TCC GAC AG |
| EPO | TCC ACT CCG AAC ACT CAC A | CCT CTC CCG TGT ACA GCT T |
| Hepcidin | TCC TGC TTC TCC TCC TTG C | GCT TTC TTC CCC GTG CAA A |
| GATA2 | ATC TCT TCG GCT TCC CAC C | TTG ACG CCA TCC TTG TCC T |
| ADORA3 | GAA CCC CAC TCT GAG GAC C | GCA ATG GCC AAA GGT GTG A |
| NRG-1 | CTA CCC ACC TTG ACC CTG G | TGA GAC CAG AAC AGC GGA G |
| SELE | GAG TTT CAC GTT GCA GGG G | GGC GCA GAT AAG GCT TCA C |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Estrela, G.R.; Freitas-Lima, L.C.; Budu, A.; Arruda, A.C.d.; Perilhão, M.S.; Fock, R.A.; Barrera-Chimal, J.; Araújo, R.C. Chronic Kidney Disease Induced by Cisplatin, Folic Acid and Renal Ischemia Reperfusion Induces Anemia and Promotes GATA-2 Activation in Mice. Biomedicines 2021, 9, 769. https://doi.org/10.3390/biomedicines9070769
Estrela GR, Freitas-Lima LC, Budu A, Arruda ACd, Perilhão MS, Fock RA, Barrera-Chimal J, Araújo RC. Chronic Kidney Disease Induced by Cisplatin, Folic Acid and Renal Ischemia Reperfusion Induces Anemia and Promotes GATA-2 Activation in Mice. Biomedicines. 2021; 9(7):769. https://doi.org/10.3390/biomedicines9070769
Chicago/Turabian StyleEstrela, Gabriel Rufino, Leandro Ceotto Freitas-Lima, Alexandre Budu, Adriano Cleis de Arruda, Mauro Sergio Perilhão, Ricardo Ambrósio Fock, Jonatan Barrera-Chimal, and Ronaldo Carvalho Araújo. 2021. "Chronic Kidney Disease Induced by Cisplatin, Folic Acid and Renal Ischemia Reperfusion Induces Anemia and Promotes GATA-2 Activation in Mice" Biomedicines 9, no. 7: 769. https://doi.org/10.3390/biomedicines9070769
APA StyleEstrela, G. R., Freitas-Lima, L. C., Budu, A., Arruda, A. C. d., Perilhão, M. S., Fock, R. A., Barrera-Chimal, J., & Araújo, R. C. (2021). Chronic Kidney Disease Induced by Cisplatin, Folic Acid and Renal Ischemia Reperfusion Induces Anemia and Promotes GATA-2 Activation in Mice. Biomedicines, 9(7), 769. https://doi.org/10.3390/biomedicines9070769