
Accumulated Knowledge of Activin Receptor-Like Kinase 2 (ALK2)/Activin A Receptor, Type 1 (ACVR1) as a Target for Human Disorders


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Basics of Receptors of the TGF-β Family, Including ALK2/ACVR1
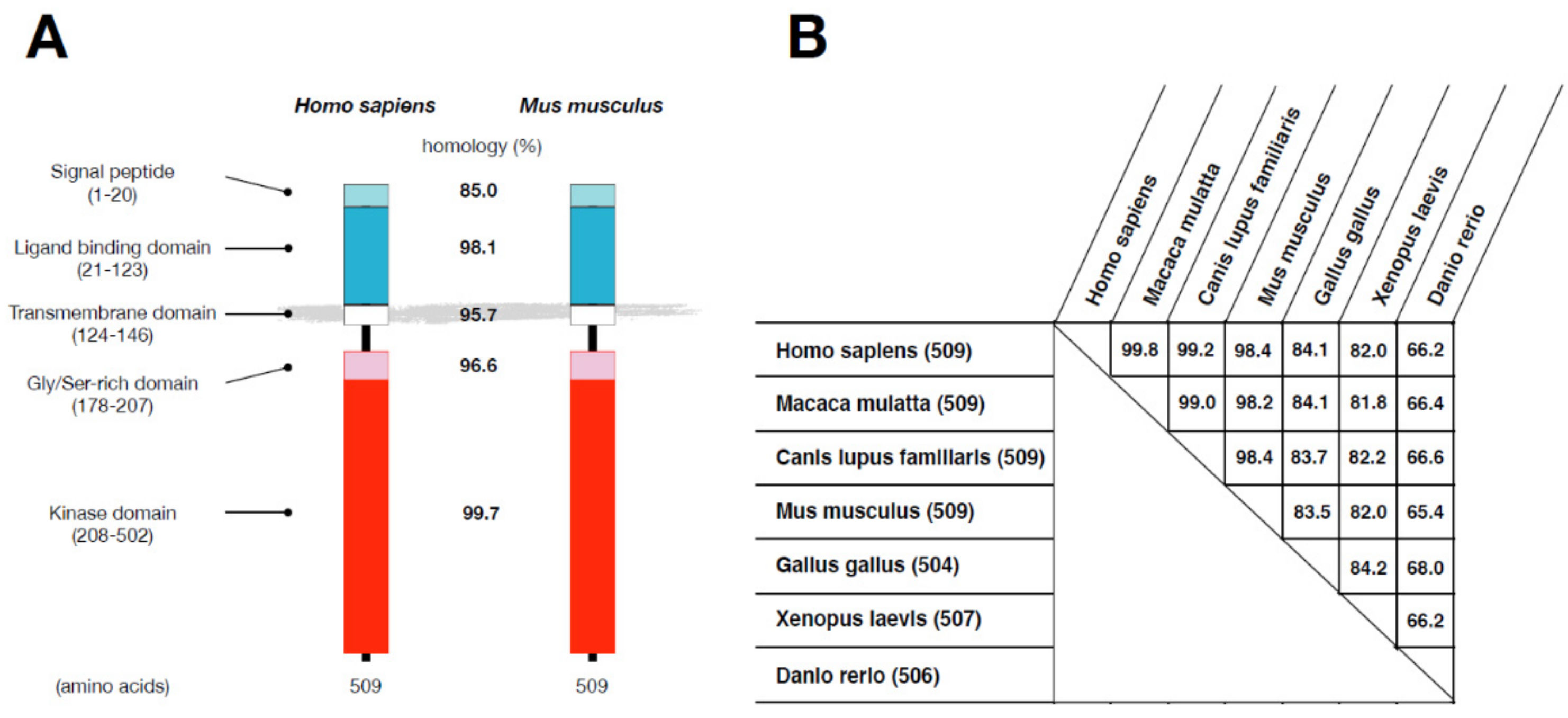
2.1. Protein Structure of ALK2/ACVR1 in Vertebrates
2.2. Type I and Type II Receptors of the TGF-β Family
2.3. The Role of Type I Receptors in Ligand-Induced Signal Transduction
3. Disorders Associated with ALK2/ACVR1
3.1. Fibrodysplasia Ossificans Progressiva (FOP)
3.2. Diffuse Intrinsic Pontine Glioma (DIPG)
3.3. Diffuse Idiopathic Skeletal Hyperostosis (DISH)
3.4. Primary Focal Hyperhidrosis (PFH)
3.5. Congenital Heart Defect (CHD)
4. Model Animals for Studying ALK2-Associated Disorders
4.1. Genomic Structure of Human and Mouse ACVR1 Genes
4.2. Genetically Engineered Animal Models of ALK2/ACVR1-Related Disorder
5. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Shore, E.M.; Xu, M.; Feldman, G.J.; Fenstermacher, D.A.; Cho, T.-J.; Choi, I.H.; Connor, J.M.; Delai, P.; Glaser, D.L.; LeMerrer, M.; et al. A recurrent mutation in the BMP type I receptor ACVR1 causes inherited and sporadic fibrodysplasia ossificans progressiva. Nat. Genet. 2006, 38, 525–527. [Google Scholar] [CrossRef]
- Katagiri, T. Heterotopic bone formation induced by bone morphogenetic protein signaling: Fibrodysplasia ossificans progressiva. J. Oral. Biosci. 2010, 52, 33–41. [Google Scholar] [CrossRef]
- Kaplan, F.S.; Chakkalakal, S.A.; Shore, E.M. Fibrodysplasia ossificans progressiva: Mechanisms and models of skeletal metamorphosis. Dis. Model. Mech. 2012, 5, 756–762. [Google Scholar] [CrossRef]
- Bragdon, B.; Moseychuk, O.; Saldanha, S.; King, D.; Julian, J.; Nohe, A. Bone morphogenetic proteins: A critical review. Cell Signal 2011, 23, 609–620. [Google Scholar] [CrossRef] [PubMed]
- Katagiri, T.; Watabe, T. Bone morphogenetic proteins. Cold Spring Harb. Perspect. Biol. 2016, 8, a021899. [Google Scholar] [CrossRef]
- Wu, G.; Diaz, A.K.; Paugh, B.S.; Rankin, S.L.; Ju, B.; Li, Y.; Zhu, X.; Qu, C.; Chen, X.; Zhang, J.; et al. The genomic landscape of diffuse intrinsic pontine glioma and pediatric non-brainstem high-grade glioma. Nat. Genet. 2014, 46, 444–450. [Google Scholar]
- Buczkowicz, P.; Hoeman, C.; Rakopoulos, P.; Pajovic, S.; Letourneau, L.; Dzamba, M.; Morrison, A.; Lewis, P.; Bouffet, E.; Bartels, U.; et al. Genomic analysis of diffuse intrinsic pontine gliomas identifies three molecular subgroups and recurrent activating ACVR1 mutations. Nat. Genet. 2014, 46, 451–456. [Google Scholar] [CrossRef]
- Taylor, K.R.; Mackay, A.; Truffaux, N.; Butterfield, Y.; Morozova, O.; Philippe, C.; Castel, D.; Grasso, C.S.; Vinci, M.; Carvalho, D.; et al. Recurrent activating ACVR1 mutations in diffuse intrinsic pontine glioma. Nat. Genet. 2014, 46, 457–461. [Google Scholar] [CrossRef] [PubMed]
- Fontebasso, A.M.; Papillon-Cavanagh, S.; Schwartzentruber, J.; Nikbakht, H.; Gerges, N.; Fiset, P.O.; Bechet, D.; Faury, D.; De Jay, N.; Ramkissoon, L.A.; et al. Recurrent somatic mutations in ACVR1 in pediatric midline high-grade astrocytoma. Nat. Genet. 2014, 46, 462–466. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.; Zimmermann, M.T.; Wang, H.; Broski, S.M.; Sigafoos, A.N.; Macklin, S.K.; Urrutia, R.A.; Clark, K.J.; Atwal, P.S.; Pignolo, R.J.; et al. Molecular characterization of known and novel ACVR1 variants in phenotypes of aberrant ossification. Am. J. Med. Genet. A 2019, 179, 1764–1777. [Google Scholar] [CrossRef]
- Smith, K.A.; Joziasse, I.C.; Chocron, S.; van Dinther, M.; Guryev, V.; Verhoeven, M.C.; Rehmann, H.; van der Smagt, J.J.; Doevendans, P.A.; Cuppen, E.; et al. Dominant-negative ALK2 allele associates with congenital heart defects. Circulation 2009, 119, 3062–3069. [Google Scholar] [CrossRef] [PubMed]
- Joziasse, I.C.; Smith, K.A.; Chocron, S.; van Dinther, M.; Guryev, V.; van de Smagt, J.J.; Cuppen, E.; Ten Dijke, P.; Mulder, B.J.; Maslen, C.L.; et al. ALK2 mutation in a patient with Down’s syndrome and a congenital heart defect. Eur. J. Hum. Genet. 2011, 19, 389–393. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.B.; Chen, J.F.; Lai, F.C.; Li, X.; Xie, J.B.; Tu, Y.R.; Kang, M.Q. Involvement of activin a receptor type 1 (ACVR1) in the pathogenesis of primary focal hyperhidrosis. Biochem. Biophys. Res. Commun. 2020, 528, 299–304. [Google Scholar] [CrossRef] [PubMed]
- Salazar, V.S.; Gamer, L.W.; Rosen, V. BMP signaling in skeletal development, disease and repair. Nat. Rev. Endocrinol. 2016, 12, 203–221. [Google Scholar] [CrossRef]
- Urist, M.R. Bone: Formation by autoinduction. Science 1965, 150, 893–899. [Google Scholar] [CrossRef] [PubMed]
- Urist, M.R.; Strates, B.S. Bone morphogenetic protein. J. Dent. Res. 1971, 50, 1392–1406. [Google Scholar] [CrossRef] [PubMed]
- Mueller, T.D.; Nickel, J. Promiscuity and specificity in BMP receptor activation. FEBS Lett. 2012, 586, 1846–1859. [Google Scholar] [CrossRef]
- Katagiri, T.; Tsukamoto, S.; Nakachi, Y.; Kuratani, M. Discovery of Heterotopic Bone Inducing Activity in Hard Tissues and the TGF-β Superfamily. Int. J. Mol. Sci. 2018, 19, 3586. [Google Scholar] [CrossRef]
- Wrana, J.L.; Attisano, L.; Wieser, R.; Ventura, F.; Massagué, J. Mechanism of activation of the TGF-β receptor. Nature 1994, 370, 341–347. [Google Scholar] [CrossRef]
- Sampath, T.K.; Coughlin, J.E.; Whetstone, R.M.; Banach, D.; Corbett, C.; Ridge, R.J.; Ozkaynak, E.; Oppermann, H.; Rueger, D.C. Bovine osteogenic protein is composed of dimers of OP-1 and BMP-2A, two members of the transforming growth factor-beta superfamily. J. Biol. Chem. 1990, 265, 13198–13205. [Google Scholar] [CrossRef]
- Groppe, J.; Hinck, C.S.; Samavarchi-Tehrani, P.; Zubieta, C.; Schuermann, J.P.; Taylor, A.B.; Schwarz, P.M.; Wrana, J.L.; Hinck, A.P. Cooperative assembly of TGF-β superfamily signaling complexes is mediated by two disparate mechanisms and distinct modes of receptor binding. Mol. Cell 2008, 29, 157–168. [Google Scholar] [CrossRef]
- Martinez-Hackert, E.; Sundan, A.; Holien, T. Receptor binding competition: A paradigm for regulating TGF-β family action. Cytokine Growth Factor Rev. 2021, 57, 39–54. [Google Scholar] [CrossRef]
- Kang, Q.; Sun, M.H.; Cheng, H.; Peng, Y.; Montag, A.G.; Deyrup, A.T.; Jiang, W.; Luu, H.H.; Luo, J.; Szatkowski, J.P.; et al. Characterization of the distinct orthotopic bone-forming activity of 14 BMPs using recombinant adenovirus-mediated gene delivery. Gene Ther. 2004, 11, 1312–1320. [Google Scholar] [CrossRef]
- Katagiri, T.; Tsukamoto, S. The unique activity of bone morphogenetic proteins in bone: A critical role of the Smad signaling pathway. Biol. Chem. 2013, 394, 703–714. [Google Scholar] [CrossRef] [PubMed]
- Massagué, J.; Wotton, D. Transcriptional control by the TGF-beta/Smad signaling system. EMBO J. 2000, 19, 1745–1754. [Google Scholar] [CrossRef]
- Katagiri, T.; Tsukamoto, S.; Kuratani, M. Heterotopic bone induction via BMP signaling: Potential therapeutic targets for fibrodysplasia ossificans progressive. Bone 2018, 109, 241–250. [Google Scholar] [CrossRef]
- Katagiri, T. A door opens for fibrodysplasia ossificans progressiva. Trends Biochem. Sci. 2016, 41, 119–121. [Google Scholar] [CrossRef] [PubMed]
- Shafritz, A.B.; Shore, E.M.; Gannon, F.H.; Zasloff, M.A.; Taub, R.; Muenke, M.; Kaplan, F.S. Overexpression of an osteogenic morphogen in fibrodysplasia ossificans progressiva. N. Engl. J. Med. 1996, 335, 555–561. [Google Scholar] [CrossRef]
- Haga, N.; Nakashima, Y.; Kitoh, H.; Kamizono, J.; Katagiri, T.; Saijo, H.; Tsukamoto, S.; Shinoda, Y.; Sawada, R.; Nakahara, Y. Fibrodysplasia ossificans progressiva: Review and research activities in Japan. Pediatr. Int. 2020, 62, 3–13. [Google Scholar] [CrossRef]
- Kitterman, J.A.; Kantanie, S.; Rocke, D.M.; Kaplan, F.S. Iatrogenic harm caused by diagnostic errors in fibrodysplasia ossificans progressiva. Pediatrics 2005, 116, e654–e661. [Google Scholar] [CrossRef] [PubMed]
- Nakashima, Y.; Haga, N.; Kitoh, H.; Kamizono, J.; Tozawa, K.; Katagiri, T.; Susami, T.; Fukushi, J.; Iwamoto, Y. Deformity of the great toe in fibrodysplasia ossificans progressiva. J. Orthop. Sci. 2010, 15, 804–809. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, F.S.; Xu, M.; Glaser, D.L.; Collins, F.; Connor, M.; Kitterman, J.; Sillence, D.; Zackai, E.; Ravitsky, V.; Zasloff, M.; et al. Early diagnosis of fibrodysplasia ossificans progressiva. Pediatrics 2008, 121, e1295–e1300. [Google Scholar] [CrossRef] [PubMed]
- Fukuda, T.; Kanomata, K.; Nojima, J.; Kokabu, S.; Akita, M.; Ikebuchi, K.; Jimi, E.; Komori, T.; Maruki, Y.; Matsuoka, M.; et al. A unique mutation of ALK2, G356D, found in a patient with fibrodysplasia ossificans progressiva is a moderately activated BMP type I receptor. Biochem. Biophys. Res. Commun. 2008, 377, 905–909. [Google Scholar] [CrossRef] [PubMed]
- Shen, Q.; Little, S.C.; Xu, M.; Haupt, J.; Ast, C.; Katagiri, T.; Mundlos, S.; Seemann, P.; Kaplan, F.S.; Mullins, M.C.; et al. The fibrodysplasia ossificans progressiva R206H ACVR1 mutation activates BMP-independent chondrogenesis and zebrafish embryo ventralization. J. Clin. Investig. 2009, 119, 3462–3472. [Google Scholar] [CrossRef] [PubMed]
- Fukuda, T.; Kohda, M.; Kanomata, K.; Nojima, J.; Nakamura, A.; Kamizono, J.; Noguchi, Y.; Iwakiri, K.; Kondo, T.; Kurose, J.; et al. Constitutively activated ALK2 and increased SMAD1/5 cooperatively induce bone morphogenetic protein signaling in fibrodysplasia ossificans progressiva. J. Biol. Chem. 2009, 284, 7149–7156. [Google Scholar] [CrossRef]
- Gregson, C.L.; Hollingworth, P.; Williams, M.; Petrie, K.A.; Bullock, A.N.; Brown, M.A.; Tobias, J.H.; Triffitt, J.T. A novel ACVR1 mutation in the glycine/serine-rich domain found in the most benign case of a fibrodysplasia ossificans progressiva variant reported to date. Bone 2011, 48, 654–658. [Google Scholar] [CrossRef]
- Nakahara, Y.; Katagiri, T.; Ogata, N.; Haga, N. ACVR1 (587T>C) mutation in a variant form of fibrodysplasia ossificans progressiva: Second report. Am. J. Med. Genet. A 2014, 164A, 220–224. [Google Scholar] [CrossRef]
- Kaplan, F.S.; Xu, M.; Seemann, P.; Connor, J.M.; Glaser, D.L.; Carroll, L.; Delai, P.; Fastnacht Urban, E.; Forman, S.J.; Gillessen-Kaesbach, G.; et al. Classic and atypical fibrodysplasia ossificans progressiva (FOP) phenotypes are caused by mutations in the bone morphogenetic protein (BMP) type I receptor ACVR1. Hum. Mutat. 2009, 30, 379–390. [Google Scholar] [CrossRef]
- Petrie, K.A.; Lee, W.H.; Bullock, A.N.; Pointon, J.J.; Smith, R.; Russell, R.G.; Brown, M.A.; Wordsworth, B.P.; Triffitt, J.T. Novel mutations in ACVR1 result in atypical features in two fibrodysplasia ossificans progressiva patients. PLoS ONE 2009, 4, e5005. [Google Scholar] [CrossRef]
- Kaplan, F.S.; Kobori, J.A.; Orellana, C.; Calvo, I.; Rosello, M.; Martinez, F.; Lopez, B.; Xu, M.; Pignolo, R.J.; Shore, E.M.; et al. Multi-system involvement in a severe variant of fibrodysplasia ossificans progressiva (ACVR1 c.772G>A.; R258G): A report of two patients. Am. J. Med. Genet. A 2015, 167A, 2265–2271. [Google Scholar] [CrossRef]
- Bocciardi, R.; Bordo, D.; Di Duca, M.; Di Rocco, M.; Ravazzolo, R. Mutational analysis of the ACVR1 gene in Italian patients affected with fibrodysplasia ossificans progressiva: Confirmations and advancements. Eur. J. Hum. Genet 2009, 17, 311–318. [Google Scholar] [CrossRef] [PubMed]
- Ratbi, I.; Bocciardi, R.; Regragui, A.; Ravazzolo, R.; Sefiani, A. Rarely occurring mutation of ACVR1 gene in Moroccan patient with fibrodysplasia ossificans progressiva. Clin. Rheumatol. 2010, 29, 119–121. [Google Scholar] [CrossRef] [PubMed]
- Nakahara, Y.; Suzuki, R.; Katagiri, T.; Toguchida, J.; Haga, N. Phenotypic differences of patients with fibrodysplasia ossificans progressive due to p.Arg258Ser variants of ACVR1. Hum. Genome Var. 2015, 2, 15055. [Google Scholar] [CrossRef][Green Version]
- Whyte, M.P.; Wenkert, D.; Demertzis, J.L.; DiCarlo, E.F.; Westenberg, E.; Mumm, S. Fibrodysplasia ossificans progressiva: Middle-age onset of heterotopic ossification from a unique missense mutation (c.974G>C, p.G325A) in ACVR1. J. Bone Miner. Res. 2012, 27, 729–737. [Google Scholar] [CrossRef]
- Furuya, H.; Ikezoe, K.; Wang, L.; Ohyagi, Y.; Motomura, K.; Fujii, N.; Kira, J.; Fukumaki, Y. A unique case of fibrodysplasia ossificans progressiva with an ACVR1 mutation, G356D, other than the common mutation (R206H). Am. J. Med. Genet. A 2008, 146A, 459–463. [Google Scholar] [CrossRef]
- Katagiri, T.; Tsukamoto, S.; Nakachi, Y.; Kuratani, M. Recent topics in fibrodysplasia ossificans progressiva. Endocrinol. Metab. 2018, 33, 331–338. [Google Scholar] [CrossRef]
- Chaikuad, A.; Alfano, I.; Kerr, G.; Sanvitale, C.E.; Boergermann, J.H.; Triffitt, J.T.; von Delft, F.; Knapp, S.; Knaus, P.; Bullock, A.N. Structure of the bone morphogenetic protein receptor ALK2 and implications for fibrodysplasia ossificans progressiva. J. Biol. Chem. 2012, 287, 36990–36998. [Google Scholar] [CrossRef] [PubMed]
- Fujimoto, M.; Ohte, S.; Osawa, K.; Miyamoto, A.; Tsukamoto, S.; Mizuta, T.; Kokabu, S.; Suda, N.; Katagiri, T. Mutant activin-like kinase 2 in fibrodysplasia ossificans progressiva are activated via T203 by BMP type II receptors. Mol. Endocrinol. 2015, 29, 140–152. [Google Scholar] [CrossRef]
- Machiya, A.; Tsukamoto, S.; Ohte, S.; Kuratani, M.; Fujimoto, M.; Kumagai, K.; Osawa, K.; Suda, N.; Bullock, A.N.; Katagiri, T. Effects of FKBP12 and type II BMP receptors on signal transduction by ALK2 activating mutations associated with genetic disorders. Bone 2018, 111, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.W.; Li, B.Y.; Danielson, P.D.; Shah, P.C.; Rockwell, S.; Lechleider, R.J.; Donahoe, P.K. The immunophilin FKBP12 functions as a common inhibitor of the TGF β family type I receptors. Cell 1996, 86, 435–444. [Google Scholar] [CrossRef]
- Groppe, J.C.; Wu, J.; Shore, E.M.; Kaplan, F.S. In vitro analyses of the dysregulated R206H ALK2 kinase-FKBP12 interaction associated with heterotopic ossification in FOP. Cells Tissues Organs 2011, 194, 291–295. [Google Scholar] [CrossRef] [PubMed]
- Hino, K.; Ikeya, M.; Horigome, K.; Matsumoto, Y.; Ebise, H.; Nishio, M.; Sekiguchi, K.; Shibata, M.; Nagata, S.; Matsuda, S.; et al. Neofunction of ACVR1 in fibrodysplasia ossificans progressiva. Proc. Natl. Acad. Sci. USA 2015, 112, 15438–15443. [Google Scholar] [CrossRef] [PubMed]
- Hatsell, S.J.; Idone, V.; Wolken, D.M.A.; Huang, L.; Kim, H.J.; Wang, L.; Wen, X.; Nannuru, K.C.; Jimenez, J.; Xie, L.; et al. ACVR1R206H receptor mutation causes fibrodysplasia ossificans progressiva by imparting responsiveness to activin A. Sci. Transl. Med. 2015, 7, 303ra137. [Google Scholar] [CrossRef] [PubMed]
- Aykul, S.; Corpina, R.A.; Goebel, E.J.; Cunanan, C.J.; Dimitriou, A.; Kim, H.J.; Zhang, Q.; Rafique, A.; Leidich, R.; Wang, X.; et al. Activin A forms a non-signaling complex with ACVR1 and type II Activin/BMP receptors via its finger 2 tip loop. Elife 2020, 9, e54582. [Google Scholar] [CrossRef] [PubMed]
- Wosczyna, M.N.; Biswas, A.A.; Cogswell, C.A.; Goldhamer, D.J. Multipotent progenitors resident in the skeletal muscle interstitium exhibit robust BMP-dependent osteogenic activity and mediate heterotopic ossification. J. Bone Miner. Res. 2012, 27, 1004–1017. [Google Scholar] [CrossRef]
- Lees-Shepard, J.B.; Yamamoto, M.; Biswas, A.A.; Stoessel, S.J.; Nicholas, S.E.; Cogswell, C.A.; Devarakonda, P.M.; Schneider, M.J., Jr.; Cummins, S.M.; Legendre, N.P.; et al. Activin-dependent signaling in fibro/adipogenic progenitors causes fibrodysplasia ossificans progressiva. Nat. Commun. 2018, 9, 471. [Google Scholar] [CrossRef]
- Mundy, C.; Yao, L.; Sinha, S.; Chung, J.; Rux, D.; Catheline, S.E.; Koyama, E.; Qin, L.; Pacifici, M. Activin A promotes the development of acquired heterotopic ossification and is an effective target for disease attenuation in mice. Sci. Signal 2021, 14, eabd0536. [Google Scholar] [CrossRef] [PubMed]
- Dey, D.; Bagarova, J.; Hatsell, S.J.; Armstrong, K.A.; Huang, L.; Ermann, J.; Vonner, A.J.; Shen, Y.; Mohedas, A.H.; Lee, A.; et al. Two tissue-resident progenitor lineages drive distinct phenotypes of heterotopic ossification. Sci. Transl. Med. 2016, 8, 366ra163. [Google Scholar] [CrossRef]
- Mehler, M.F.; Mabie, P.C.; Zhang, D.; Kessler, J.A. Bone morphogenetic proteins in the nervous system. Trends Neurosci. 1997, 20, 309–317. [Google Scholar] [CrossRef]
- Tsukamoto, S.; Kuratani, M.; Katagiri, T. Functional characterization of a unique mutant of ALK2, p.K400E, that is associated with a skeletal disorder, diffuse idiopathic skeletal hyperostosis. Bone 2020, 137, 115410. [Google Scholar] [CrossRef]
- Matsuoka, M.; Tsukamoto, S.; Orihara, Y.; Kawamura, R.; Kuratani, M.; Haga, N.; Ikebuchi, K.; Katagiri, T. Design of primers for direct sequencing of nine coding exons in the human ACVR1 gene. Bone 2020, 138, 115469. [Google Scholar] [CrossRef] [PubMed]
- Mishina, Y.; Crombie, R.; Bradley, A.; Behringer, R.R. Multiple roles for activin-like kinase-2 signaling during mouse embryogenesis. Dev. Biol. 1999, 213, 314–326. [Google Scholar] [CrossRef]
- Kaartinen, V.; Nagy, A. Removal of the floxed neo gene from a conditional knockout allele by the adenoviral Cre recombinase in vivo. Genesis 2001, 31, 126–129. [Google Scholar] [CrossRef]
- Komatsu, Y.; Scott, G.; Nagy, A.; Kaartinen, V.; Mishina, Y. BMP type I receptor ALK2 is essential for proper patterning at late gastrulation during mouse embryogenesis. Dev. Dyn. 2007, 236, 512–517. [Google Scholar] [CrossRef] [PubMed]
- Yu, P.B.; Deng, D.Y.; Lai, C.S.; Hong, C.C.; Cuny, G.D.; Bouxsein, M.L.; Hong, D.W.; McManus, P.M.; Katagiri, T.; Sachidanandan, C.; et al. BMP type I receptor inhibition reduces heterotopic ossification. Nat. Med. 2008, 14, 1363–1369. [Google Scholar] [CrossRef]
- LaBonty, M.; Pray, N.; Yelick, P.C. A Zebrafish Model of Human Fibrodysplasia Ossificans Progressiva. Zebrafish 2017, 14, 293–304. [Google Scholar] [CrossRef]
- LaBonty, M.; Pray, N.; Yelick, P.C. Injury of Adult Zebrafish Expressing Acvr1l Q204D Does Not Result in Heterotopic Ossification. Zebrafish 2018, 15, 536–545. [Google Scholar] [CrossRef] [PubMed]
- Chakkalakal, S.A.; Zhang, D.; Culbert, A.L.; Convente, M.R.; Caron, R.J.; Wright, A.C.; Maidment, A.D.; Kaplan, F.S.; Shore, E.M. An Acvr1 R206H knock-in mouse has fibrodysplasia ossificans progressiva. J. Bone Miner. Res. 2012, 27, 1746–1756. [Google Scholar] [CrossRef] [PubMed]
- Fortin, J.; Tian, R.; Zarrabi, I.; Hill, G.; Williams, E.; Sanchez-Duffhues, G.; Thorikay, M.; Ramachandran, P.; Siddaway, R.; Wong, J.F.; et al. Mutant ACVR1 Arrests Glial Cell Differentiation to Drive Tumorigenesis in Pediatric Gliomas. Cancer Cell 2020, 37, 308–323. [Google Scholar] [CrossRef] [PubMed]
- Mohedas, A.H.; Xing, X.; Armstrong, K.A.; Bullock, A.N.; Cuny, G.D.; Yu, P.B. Development of an ALK2-biased BMP type I receptor kinase inhibitor. ACS Chem. Biol. 2013, 8, 1291–1302. [Google Scholar] [CrossRef]
- Williams, E.; Bagarova, J.; Kerr, G.; Xia, D.D.; Place, E.S.; Dey, D.; Shen, Y.; Bocobo, G.A.; Mohedas, A.H.; Huang, X.; et al. Saracatinib is an efficacious clinical candidate for fibrodysplasia ossificans progressiva. JCI Insight 2021, 6, e95042. [Google Scholar] [CrossRef]
- Shimono, K.; Tung, W.E.; Macolino, C.; Chi, A.H.; Didizian, J.H.; Mundy, C.; Chandraratna, R.A.; Mishina, Y.; Enomoto-Iwamoto, M.; Pacifici, M.; et al. Potent inhibition of heterotopic ossification by nuclear retinoic acid receptor-γ agonists. Nat. Med. 2011, 17, 454–460. [Google Scholar] [CrossRef] [PubMed]
- Chakkalakal, S.A.; Uchibe, K.; Convente, M.R.; Zhang, D.; Economides, A.N.; Kaplan, F.S.; Pacifici, M.; Iwamoto, M.; Shore, E.M. Palovarotene Inhibits Heterotopic Ossification and Maintains Limb Mobility and Growth in Mice with the Human ACVR1(R206H) Fibrodysplasia Ossificans Progressiva (FOP) Mutation. J. Bone Miner. Res. 2016, 31, 1666–1675. [Google Scholar] [CrossRef] [PubMed]
- Hino, K.; Horigome, K.; Nishio, M.; Komura, S.; Nagata, S.; Zhao, C.; Jin, Y.; Kawakami, K.; Yamada, Y.; Ohta, A.; et al. Activin-A enhances mTOR signaling to promote aberrant chondrogenesis in fibrodysplasia ossificans progressiva. J. Clin. Investig. 2017, 127, 3339–3352. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Katagiri, T.; Tsukamoto, S.; Kuratani, M. Accumulated Knowledge of Activin Receptor-Like Kinase 2 (ALK2)/Activin A Receptor, Type 1 (ACVR1) as a Target for Human Disorders. Biomedicines 2021, 9, 736. https://doi.org/10.3390/biomedicines9070736
Katagiri T, Tsukamoto S, Kuratani M. Accumulated Knowledge of Activin Receptor-Like Kinase 2 (ALK2)/Activin A Receptor, Type 1 (ACVR1) as a Target for Human Disorders. Biomedicines. 2021; 9(7):736. https://doi.org/10.3390/biomedicines9070736
Chicago/Turabian StyleKatagiri, Takenobu, Sho Tsukamoto, and Mai Kuratani. 2021. "Accumulated Knowledge of Activin Receptor-Like Kinase 2 (ALK2)/Activin A Receptor, Type 1 (ACVR1) as a Target for Human Disorders" Biomedicines 9, no. 7: 736. https://doi.org/10.3390/biomedicines9070736
APA StyleKatagiri, T., Tsukamoto, S., & Kuratani, M. (2021). Accumulated Knowledge of Activin Receptor-Like Kinase 2 (ALK2)/Activin A Receptor, Type 1 (ACVR1) as a Target for Human Disorders. Biomedicines, 9(7), 736. https://doi.org/10.3390/biomedicines9070736