
The NHance® Mutation-Equipped Anti-MET Antibody ARGX-111 Displays Increased Tissue Penetration and Anti-Tumor Activity in Advanced Cancer Patients


 , ,
, , 
Abstract
1. Introduction
2. Materials and Methods
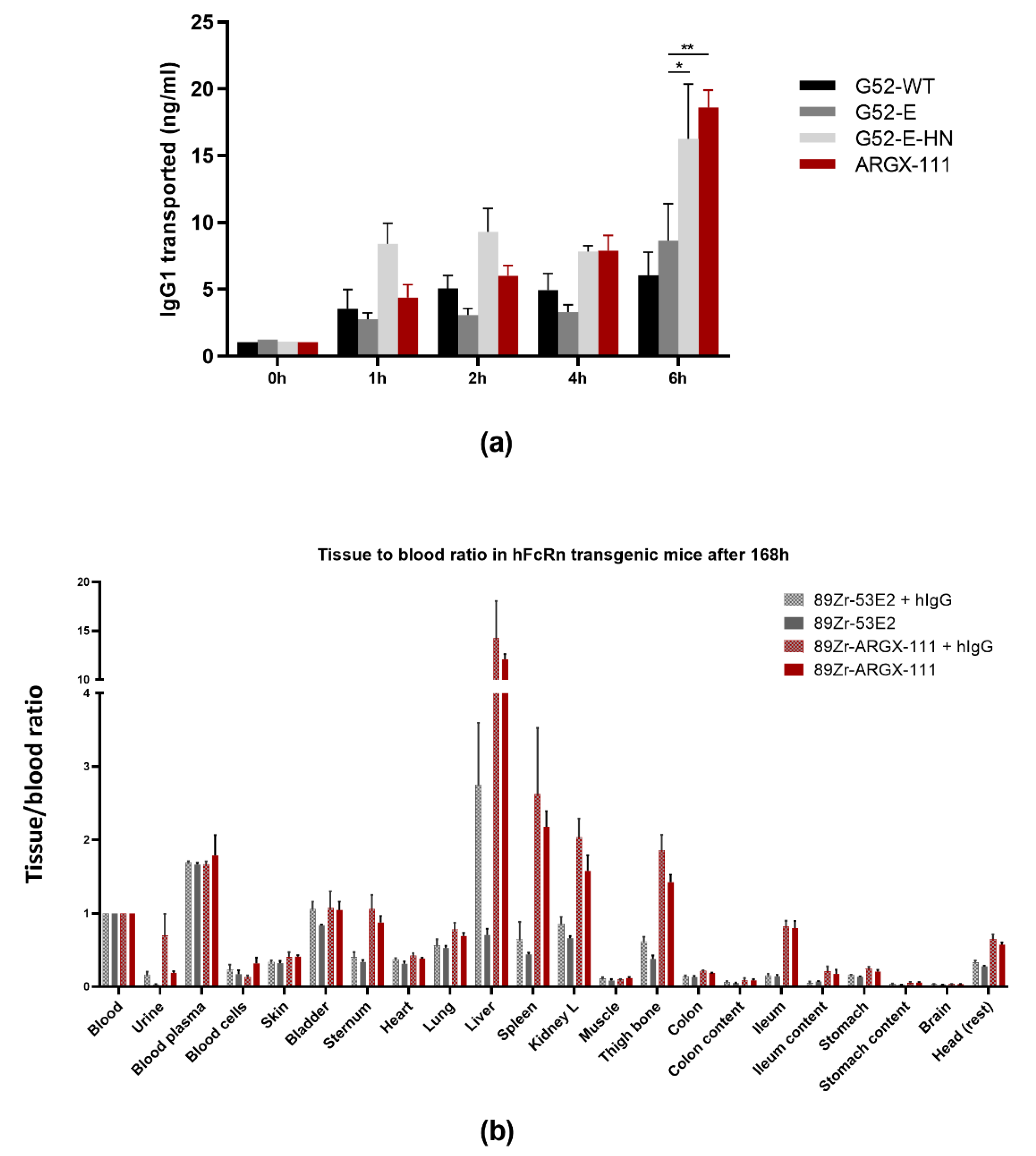
2.1. Transcytosis Assay with MDCKII Cells Expressing Human FcRn
2.2. Quantification of IgG Transport by ELISA
2.3. Preclinical In Vivo FcRn Experiments
2.4. Clinical Study Design
2.5. Patients and Eligibility Criteria
2.6. Rationale for Dose Selection and Treatment
2.7. Safety and Tolerability
2.8. Response Assessment
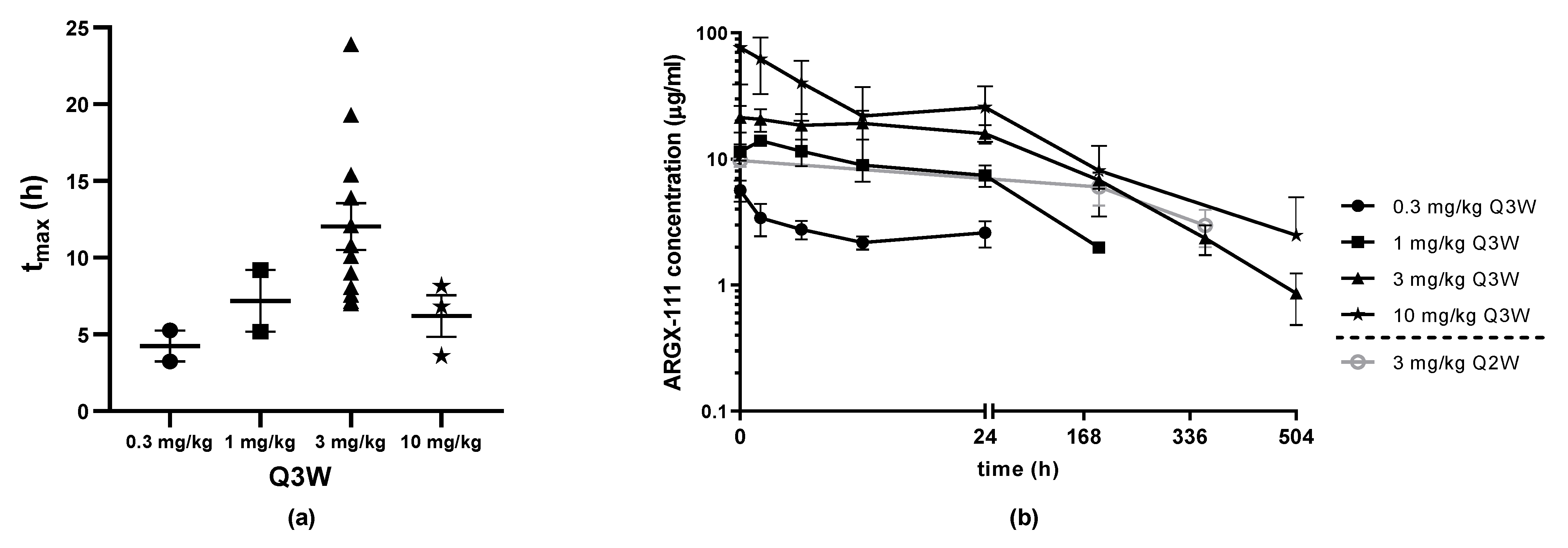
2.9. Pharmacokinetics and Additional Assessments
2.10. Statistical Methods
3. Results
3.1. Characterization of ARGX-111
3.2. Patient Demographics and Baseline Characteristics
3.3. Dose Escalation and Dose-Limiting Toxicity
3.4. Safety and Tolerability
3.5. Pharmacokinetics and Anti-Drug Antibodies
3.6. Exploratory Assessments of ADCC-Mediated Depletion of Circulation Tumor Cells (CTCs)
3.7. Duration of Study, Preliminary Anti-Tumor Activity and Best Response
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Trusolino, L.; Comoglio, P.M. Scatter-factor and semaphorin receptors: Cell signalling for invasive growth. Nat. Rev. Cancer 2002, 2, 289–300. [Google Scholar] [CrossRef]
- Lam, B.Q.; Dai, L.; Qin, Z. The role of HGF/c-MET signaling pathway in lymphoma. J. Hematol. Oncol. 2016, 9, 135. [Google Scholar] [CrossRef]
- Cappuzzo, F.; Marchetti, A.; Skokan, M.; Rossi, E.; Gajapathy, S.; Felicioni, L.; Del Grammastro, M.; Sciarrotta, M.G.; Buttitta, F.; Incarbone, M.; et al. Increased MET gene copy number negatively affects survival of surgically resected non-small-cell lung cancer patients. J. Clin. Oncol. 2009, 27, 1667–1674. [Google Scholar] [CrossRef] [PubMed]
- Heynen, G.J.; Fonfara, A.; Bernards, R. Resistance to targeted cancer drugs through hepatocyte growth factor signaling. Cell Cycle 2014, 13, 3808–3817. [Google Scholar] [CrossRef]
- Cecchi, F.; Rabe, D.C.; Bottaro, D.P. Targeting the HGF/Met signaling pathway in cancer therapy. Expert Opin. Ther. Targets 2012, 16, 553–572. [Google Scholar] [CrossRef]
- Cui, J.J. Targeting receptor tyrosine kinase MET in cancer: Small molecule inhibitors and clinical progress. J. Med. Chem. 2014, 57, 4427–4453. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.H.; Kim, H. Progress of antibody-based inhibitors of the HGF-cMET axis in cancer therapy. Exp. Mol. Med. 2017, 49, e307. [Google Scholar] [CrossRef]
- Bertotti, A.; Burbridge, M.F.; Gastaldi, S.; Galimi, F.; Torti, D.; Medico, E.; Giordano, S.; Corso, S.; Rolland-Valognes, G.; Lockhart, B.P.; et al. Only a subset of Met-activated pathways are required to sustain oncogene addiction. Sci. Signal. 2009, 2, er11. [Google Scholar] [CrossRef]
- Lai, A.Z.; Cory, S.; Zhao, H.; Gigoux, M.; Monast, A.; Guiot, M.C.; Huang, S.; Tofigh, A.; Thompson, C.; Naujokas, M.; et al. Dynamic reprogramming of signaling upon met inhibition reveals a mechanism of drug resistance in gastric cancer. Sci. Signal. 2014, 7, ra38. [Google Scholar] [CrossRef]
- Hultberg, A.; Morello, V.; Huyghe, L.; De Jonge, N.; Blanchetot, C.; Hanssens, V.; De Boeck, G.; Silence, K.; Festjens, E.; Heukers, R.; et al. Depleting MET-Expressing Tumor Cells by ADCC Provides a Therapeutic Advantage over Inhibiting HGF/MET Signaling. Cancer Res. 2015, 75, 3373–3383. [Google Scholar] [CrossRef]
- Vaccaro, C.; Bawdon, R.; Wanjie, S.; Ober, R.J.; Ward, E.S. Divergent activities of an engineered antibody in murine and human systems have implications for therapeutic antibodies. Proc. Natl. Acad. Sci. USA 2006, 103, 18709–18714. [Google Scholar] [CrossRef]
- Kuo, T.T.; Aveson, V.G. Neonatal Fc receptor and IgG-based therapeutics. MAbs 2011, 3, 422–430. [Google Scholar] [CrossRef] [PubMed]
- Ghetie, V.; Ward, E.S. Transcytosis and catabolism of antibody. Immunol. Res. 2002, 25, 97–113. [Google Scholar] [CrossRef]
- Morley, R.; Cardenas, A.; Hawkins, P.; Suzuki, Y.; Paton, V.; Phan, S.C.; Merchant, M.; Hsu, J.; Yu, W.; Xia, Q.; et al. Safety of Onartuzumab in Patients with Solid Tumors: Experience to Date from the Onartuzumab Clinical Trial Program. PLoS ONE 2015, 10, e0139679. [Google Scholar] [CrossRef] [PubMed]
- Petkova, S.B.; Akilesh, S.; Sproule, T.J.; Christianson, G.J.; Al Khabbaz, H.; Brown, A.C.; Presta, L.G.; Meng, Y.G.; Roopenian, D.C. Enhanced half-life of genetically engineered human IgG1 antibodies in a humanized FcRn mouse model: Potential application in humorally mediated autoimmune disease. Int. Immunol. 2006, 18, 1759–1769. [Google Scholar] [CrossRef]
- Latvala, S.; Jacobsen, B.; Otteneder, M.B.; Herrmann, A.; Kronenberg, S. Distribution of FcRn Across Species and Tissues. J. Histochem. Cytochem. 2017, 65, 321–333. [Google Scholar] [CrossRef] [PubMed]
- Simon, R.; Freidlin, B.; Rubinstein, L.; Arbuck, S.G.; Collins, J.; Christian, M.C. Accelerated titration designs for phase I clinical trials in oncology. J. Natl. Cancer Inst. 1997, 89, 1138–1147. [Google Scholar] [CrossRef] [PubMed]
- Noonan, S.A.; Berry, L.; Lu, X.; Gao, D.; Baron, A.E.; Chesnut, P.; Sheren, J.; Aisner, D.L.; Merrick, D.; Doebele, R.C.; et al. Identifying the Appropriate FISH Criteria for Defining MET Copy Number-Driven Lung Adenocarcinoma through Oncogene Overlap Analysis. J. Thorac. Oncol. 2016, 11, 1293–1304. [Google Scholar] [CrossRef]
- Basilico, C.; Hultberg, A.; Blanchetot, C.; de Jonge, N.; Festjens, E.; Hanssens, V.; Osepa, S.I.; De Boeck, G.; Mira, A.; Cazzanti, M.; et al. Four individually druggable MET hotspots mediate HGF-driven tumor progression. J. Clin. Investig. 2014, 124, 3172–3186. [Google Scholar] [CrossRef]
- Shibata-Koyama, M.; Iida, S.; Misaka, H.; Mori, K.; Yano, K.; Shitara, K.; Satoh, M. Nonfucosylated rituximab potentiates human neutrophil phagocytosis through its high binding for FcgammaRIIIb and MHC class II expression on the phagocytotic neutrophils. Exp. Hematol. 2009, 37, 309–321. [Google Scholar] [CrossRef]
- Foss, S.; Grevys, A.; Sand, K.M.K.; Bern, M.; Blundell, P.; Michaelsen, T.E.; Pleass, R.J.; Sandlie, I.; Andersen, J.T. Enhanced FcRn-dependent transepithelial delivery of IgG by Fc-engineering and polymerization. J. Control. Release 2016, 223, 42–52. [Google Scholar] [CrossRef]
- Shinkawa, T.; Nakamura, K.; Yamane, N.; Shoji-Hosaka, E.; Kanda, Y.; Sakurada, M.; Uchida, K.; Anazawa, H.; Satoh, M.; Yamasaki, M.; et al. The absence of fucose but not the presence of galactose or bisecting N-acetylglucosamine of human IgG1 complex-type oligosaccharides shows the critical role of enhancing antibody-dependent cellular cytotoxicity. J. Biol. Chem. 2003, 278, 3466–3473. [Google Scholar] [CrossRef]
- Golay, J.; Da Roit, F.; Bologna, L.; Ferrara, C.; Leusen, J.H.; Rambaldi, A.; Klein, C.; Introna, M. Glycoengineered CD20 antibody obinutuzumab activates neutrophils and mediates phagocytosis through CD16B more efficiently than rituximab. Blood 2013, 122, 3482–3491. [Google Scholar] [CrossRef]
- Aftimos, P.; Rolfo, C.; Rottey, S.; Offner, F.; Bron, D.; Maerevoet, M.; Soria, J.C.; Moshir, M.; Dreier, T.; Van Rompaey, L.; et al. Phase I Dose-Escalation Study of the Anti-CD70 Antibody ARGX-110 in Advanced Malignancies. Clin. Cancer Res. 2017, 23, 6411–6420. [Google Scholar] [CrossRef]
- Duvic, M.; Pinter-Brown, L.C.; Foss, F.M.; Sokol, L.; Jorgensen, J.L.; Challagundla, P.; Dwyer, K.M.; Zhang, X.; Kurman, M.R.; Ballerini, R.; et al. Phase 1/2 study of mogamulizumab, a defucosylated anti-CCR4 antibody, in previously treated patients with cutaneous T-cell lymphoma. Blood 2015, 125, 1883–1889. [Google Scholar] [CrossRef]
- Patel, K.; Parmar, S.; Shah, S.; Shore, T.; Gergis, U.; Mayer, S.; van Besien, K. Comparison of Subcutaneous versus Intravenous Alemtuzumab for Graft-versus-Host Disease Prophylaxis with Fludarabine/Melphalan-Based Conditioning in Matched Unrelated Donor Allogeneic Stem Cell Transplantation. Biol. Blood Marrow Transplant. 2016, 22, 456–461. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Wynne, C.; Harvey, V.; Schwabe, C.; Waaka, D.; McIntyre, C.; Bittner, B. Comparison of Subcutaneous and Intravenous Administration of Trastuzumab: A Phase I/Ib Trial in Healthy Male Volunteers and Patients with HER2-Positive Breast Cancer. J. Clin. Pharmacol. 2013, 53, 192–201. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Martin, C.; Plaza, J.C.; Pazo-Cid, R.; Salud, A.; Pons, F.; Fonseca, P.; Leon, A.; Alsina, M.; Visa, L.; Rivera, F.; et al. Level of HER2 gene amplification predicts response and overall survival in HER2-positive advanced gastric cancer treated with trastuzumab. J. Clin. Oncol. 2013, 31, 4445–4452. [Google Scholar] [CrossRef] [PubMed]
- Lennerz, J.K.; Kwak, E.L.; Ackerman, A.; Michael, M.; Fox, S.B.; Bergethon, K.; Lauwers, G.Y.; Christensen, J.G.; Wilner, K.D.; Haber, D.A.; et al. MET amplification identifies a small and aggressive subgroup of esophagogastric adenocarcinoma with evidence of responsiveness to crizotinib. J. Clin. Oncol. 2011, 29, 4803–4810. [Google Scholar] [CrossRef]
- Kwak, E.L.; LoRusso, P.; Hamid, O.; Janku, F.; Kittaneh, M.; Catenacci, D.V.T.; Chan, E.; Bekaii-Saab, T.S.; Amore, B.; Hwang, Y.C.; et al. Clinical activity of AMG 337, an oral MET kinase inhibitor, in adult patients (pts) with MET-amplified gastroesophageal junction (GEJ), gastric (G), or esophageal (E) cancer. J. Clin. Oncol. 2015, 33. [Google Scholar] [CrossRef]
- Angevin, E.; Spitaleri, G.; Rodon, J.; Dotti, K.; Isambert, N.; Salvagni, S.; Moreno, V.; Assadourian, S.; Gomez, C.; Harnois, M.; et al. A first-in-human phase I study of SAR125844, a selective MET tyrosine kinase inhibitor, in patients with advanced solid tumours with MET amplification. Eur. J. Cancer 2017, 87, 131–139. [Google Scholar] [CrossRef]
- Kang, Y.-K.; LoRusso, P.; Salgia, R.; Yen, C.-J.; Lin, C.-C.; Ramanathan, R.K.; Kaminker, P.; Sokolova, I.; Bhathena, A.; Wang, L.; et al. Phase I study of ABT-700, an anti-c-Met antibody, in patients (pts) with advanced gastric or esophageal cancer (GEC). J. Clin. Oncol. 2015, 33, 167. [Google Scholar] [CrossRef]
- Qiu, M.; Xu, R.; Zhou, Y.-x.; Huang, J.; Tang, E.-T.; Du, Z.; Zhang, F. Evaluation of MET and HER2 expression in primary and metastatic tumor in Chinese advanced gastric cancer patients. J. Clin. Oncol. 2016, 34, e15538. [Google Scholar] [CrossRef]
- Xu, C.W.; Wang, W.X.; Wu, M.J.; Zhu, Y.C.; Zhuang, W.; Lin, G.; Du, K.Q.; Huang, Y.J.; Chen, Y.P.; Chen, G.; et al. Comparison of the c-MET gene amplification between primary tumor and metastatic lymph nodes in non-small cell lung cancer. Thorac. Cancer 2017, 8, 417–422. [Google Scholar] [CrossRef] [PubMed]
- Bardelli, A.; Corso, S.; Bertotti, A.; Hobor, S.; Valtorta, E.; Siravegna, G.; Sartore-Bianchi, A.; Scala, E.; Cassingena, A.; Zecchin, D.; et al. Amplification of the MET receptor drives resistance to anti-EGFR therapies in colorectal cancer. Cancer Discov. 2013, 3, 658–673. [Google Scholar] [CrossRef]
- Bean, J.; Brennan, C.; Shih, J.Y.; Riely, G.; Viale, A.; Wang, L.; Chitale, D.; Motoi, N.; Szoke, J.; Broderick, S.; et al. MET amplification occurs with or without T790M mutations in EGFR mutant lung tumors with acquired resistance to gefitinib or erlotinib. Proc. Natl. Acad. Sci. USA 2007, 104, 20932–20937. [Google Scholar] [CrossRef]
- Tanizaki, J.; Okamoto, I.; Sakai, K.; Nakagawa, K. Differential roles of trans-phosphorylated EGFR, HER2, HER3, and RET as heterodimerisation partners of MET in lung cancer with MET amplification. Br. J. Cancer 2011, 105, 807–813. [Google Scholar] [CrossRef]
- Du, Y.; Yamaguchi, H.; Wei, Y.; Hsu, J.L.; Wang, H.L.; Hsu, Y.H.; Lin, W.C.; Yu, W.H.; Leonard, P.G.; Lee, G.R.t.; et al. Blocking c-Met-mediated PARP1 phosphorylation enhances anti-tumor effects of PARP inhibitors. Nat. Med. 2016, 22, 194–201. [Google Scholar] [CrossRef] [PubMed]
- Mondelo-Macia, P.; Rodriguez-Lopez, C.; Valina, L.; Aguin, S.; Leon-Mateos, L.; Garcia-Gonzalez, J.; Abalo, A.; Rapado-Gonzalez, O.; Suarez-Cunqueiro, M.; Diaz-Lagares, A.; et al. Detection of MET Alterations Using Cell Free DNA and Circulating Tumor Cells from Cancer Patients. Cells 2020, 9, 522. [Google Scholar] [CrossRef]
- Chen, D.; Xu, C.; Wu, J.; Zhang, Y.; Fang, M. A comparison of consistency of detecting c-MET gene amplification in peripheral blood and tumor tissue of nonsmall cell lung cancer patients. J. Cancer Res. Ther. 2015, 11, C63–C67. [Google Scholar] [CrossRef]
- Zhang, Y.; Tang, E.T.; Du, Z. Detection of MET Gene Copy Number in Cancer Samples Using the Droplet Digital PCR Method. PLoS ONE 2016, 11, e0146784. [Google Scholar] [CrossRef] [PubMed]
- He, H.J.; Das, B.; Cleveland, M.H.; Chen, L.; Camalier, C.E.; Liu, L.C.; Norman, K.L.; Fellowes, A.P.; McEvoy, C.R.; Lund, S.P.; et al. Development and interlaboratory evaluation of a NIST Reference Material RM 8366 for EGFR and MET gene copy number measurements. Clin. Chem. Lab. Med. 2019, 57, 1142–1152. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Boominathan, R.; Foulk, B.; Rao, C.; Kemeny, G.; Strickler, J.H.; Abbruzzese, J.L.; Harrison, M.R.; Hsu, D.S.; Healy, P.; et al. Development of a Novel c-MET-Based CTC Detection Platform. Mol. Cancer Res. 2016, 14, 539–547. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Anderson, M.G.; Oleksijew, A.; Vaidya, K.S.; Boghaert, E.R.; Tucker, L.; Zhang, Q.; Han, E.K.; Palma, J.P.; Naumovski, L.; et al. ABBV-399, a c-Met Antibody-Drug Conjugate that Targets Both MET-Amplified and c-Met-Overexpressing Tumors, Irrespective of MET Pathway Dependence. Clin. Cancer Res. 2017, 23, 992–1000. [Google Scholar] [CrossRef] [PubMed]
- Tseng, J.R.; Kang, K.W.; Dandekar, M.; Yaghoubi, S.; Lee, J.H.; Christensen, J.G.; Muir, S.; Vincent, P.W.; Michaud, N.R.; Gambhir, S.S. Preclinical efficacy of the c-Met inhibitor CE-355621 in a U87 MG mouse xenograft model evaluated by 18F-FDG small-animal PET. J. Nucl. Med. 2008, 49, 129–134. [Google Scholar] [CrossRef]
- Mira, A.; Morello, V.; Cespedes, M.V.; Perera, T.; Comoglio, P.M.; Mangues, R.; Michieli, P. Stroma-derived HGF drives metabolic adaptation of colorectal cancer to angiogenesis inhibitors. Oncotarget 2017, 8, 38193–38213. [Google Scholar] [CrossRef]
- Brauer, H.A.; Makowski, L.; Hoadley, K.A.; Casbas-Hernandez, P.; Lang, L.J.; Roman-Perez, E.; D’Arcy, M.; Freemerman, A.J.; Perou, C.M.; Troester, M.A. Impact of tumor microenvironment and epithelial phenotypes on metabolism in breast cancer. Clin. Cancer Res. 2013, 19, 571–585. [Google Scholar] [CrossRef] [PubMed]
- Akilesh, S.; Christianson, G.J.; Roopenian, D.C.; Shaw, A.S. Neonatal FcR expression in bone marrow-derived cells functions to protect serum IgG from catabolism. J. Immunol. 2007, 179, 4580–4588. [Google Scholar] [CrossRef] [PubMed]
- Yeung, Y.A.; Wu, X.; Reyes, A.E., 2nd; Vernes, J.M.; Lien, S.; Lowe, J.; Maia, M.; Forrest, W.F.; Meng, Y.G.; Damico, L.A.; et al. A therapeutic anti-VEGF antibody with increased potency independent of pharmacokinetic half-life. Cancer Res. 2010, 70, 3269–3277. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Q3W | Q2W | |||||
|---|---|---|---|---|---|---|
| 0.3 mg/kg | 1 mg/kg | 3 mg/kg | 10 mg/kg | Total | 3 mg/kg | |
| N | 2 | 2 | 12 | 3 | 19 | 5 |
| Age | ||||||
| Median | 60 | 52 | 60 | 47 | 60 | 57 |
| Range | 49–71 | 41–63 | 46–70 | 28–69 | 28–71 | 42–79 |
| Gender | ||||||
| Male | 1 | 2 | 7 | 1 | 11 | 4 |
| Female | 1 | 0 | 5 | 2 | 8 | 1 |
| Race | ||||||
| Caucasian | 2 | 2 | 12 | 3 | 19 | 5 |
| ECOG | ||||||
| Grade 0 | 0 | 2 | 4 | 0 | 6 | 1 |
| Grade 1 | 2 | 0 | 8 | 3 | 13 | 4 |
| Primary cancer type | ||||||
| Stomach | 1 | 0 | 0 | 0 | 1 | 2 |
| Esophagus | 0 | 2 | 1 | 0 | 3 | 0 |
| Cervix | 0 | 0 | 1 | 1 | 2 | 0 |
| Pancreas | 1 | 0 | 1 | 0 | 2 | 0 |
| Lung | 0 | 0 | 2 | 0 | 2 | 1 |
| Breast | 0 | 0 | 1 | 0 | 1 | 0 |
| Bile duct | 0 | 0 | 1 | 1 | 2 | 0 |
| Kidney | 0 | 0 | 2 | 1 | 3 | 2 |
| Vagina | 0 | 0 | 1 | 0 | 1 | 0 |
| Bladder | 0 | 0 | 1 | 0 | 1 | 0 |
| Bone | 0 | 0 | 1 | 0 | 1 | 0 |
| TEAE by PT | Any Graden, N (%) | Grade ≥ 3 n, N (%) |
|---|---|---|
| IRR | 58, 19 (79) | 2, 2 (8) |
| Fatigue | 20, 13 (54) | 1, 1 (4) |
| Constipation | 10, 10 (42) | 0, 0 (0) |
| Nausea | 11, 8 (33) | 0, 0 (0) |
| Decreased appetite | 11, 8 (33) | 0, 0 (0) |
| Myalgia | 8, 8 (33) | 0, 0 (0) |
| Vomiting | 7, 7 (29) | 0, 0 (0) |
| General physical health deterioration | 7, 7 (29) | 6, 6 (25) |
| Arthralgia | 5, 5 (21) | 1, 1 (4) |
| Q3W | |||||
|---|---|---|---|---|---|
| 0.3 mg/kg | 1 mg/kg | 3 mg/kg | 10 mg/kg | ||
| Cmax (µg/mg) | N | 2 | 2 | 12 | 3 |
| AM | 5.65 | 14.2 | 30.3 | 76.5 | |
| Sd | 1.51 | 0.07 | 17.4 | 64.6 | |
| median | 5.65 | 14.2 | 25.2 | 101 | |
| AUC0-t (µg·h/mL) | N | 2 | 2 | 12 | 3 |
| AM | 68.2 | 481 | 3020 | 4500 | |
| Sd | 0.81 | 309 | 1780 | 4420 | |
| median | 68.2 | 481 | 2620 | 4610 | |
| AUC0-∞ (µg·h/mL) | N | NA | 1 | 11 | 2 |
| AM | NA | 882 | 3580 | 7690 | |
| Sd | NA | NA | 1910 | 3320 | |
| median | NA | 882 | 8050 | ||
| CL (mL/h) | N | NA | 1 | 11 | 2 |
| AM | NA | 0.07 | 0.08 | 0.09 | |
| Sd | NA | NA | 0.04 | 0.02 | |
| median | NA | 0.07 | 0.08 | 0.09 | |
| Vd (L) | N | NA | 1 | 11 | 2 |
| AM | NA | 6.79 | 11.5 | 14.3 | |
| Sd | NA | NA | 4.33 | 7.22 | |
| median | NA | 6.79 | 10.8 | 14.3 | |
| t½ (h) | N | NA | 1 | 11 | 2 |
| AM | NA | 63.8 | 116 | 112 | |
| Sd | NA | NA | 54.6 | 72 | |
| median 0 | NA | 63.8 | 102 | 112 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aftimos, P.; Rolfo, C.; Rottey, S.; Barthélémy, P.; Borg, C.; Park, K.; Oh, D.-Y.; Kim, S.-W.; De Jonge, N.; Hanssens, V.; et al. The NHance® Mutation-Equipped Anti-MET Antibody ARGX-111 Displays Increased Tissue Penetration and Anti-Tumor Activity in Advanced Cancer Patients. Biomedicines 2021, 9, 665. https://doi.org/10.3390/biomedicines9060665
Aftimos P, Rolfo C, Rottey S, Barthélémy P, Borg C, Park K, Oh D-Y, Kim S-W, De Jonge N, Hanssens V, et al. The NHance® Mutation-Equipped Anti-MET Antibody ARGX-111 Displays Increased Tissue Penetration and Anti-Tumor Activity in Advanced Cancer Patients. Biomedicines. 2021; 9(6):665. https://doi.org/10.3390/biomedicines9060665
Chicago/Turabian StyleAftimos, Philippe, Christian Rolfo, Sylvie Rottey, Philippe Barthélémy, Christophe Borg, Keunchil Park, Do-Youn Oh, Sang-We Kim, Natalie De Jonge, Valérie Hanssens, and et al. 2021. "The NHance® Mutation-Equipped Anti-MET Antibody ARGX-111 Displays Increased Tissue Penetration and Anti-Tumor Activity in Advanced Cancer Patients" Biomedicines 9, no. 6: 665. https://doi.org/10.3390/biomedicines9060665
APA StyleAftimos, P., Rolfo, C., Rottey, S., Barthélémy, P., Borg, C., Park, K., Oh, D.-Y., Kim, S.-W., De Jonge, N., Hanssens, V., Zwanenpoel, K., Molthoff, C., Vugts, D., Dreier, T., Verheesen, P., van Dongen, G. A. M. S., Jacobs, J., Van Rompaey, L., Hultberg, A., ... Awada, A. (2021). The NHance® Mutation-Equipped Anti-MET Antibody ARGX-111 Displays Increased Tissue Penetration and Anti-Tumor Activity in Advanced Cancer Patients. Biomedicines, 9(6), 665. https://doi.org/10.3390/biomedicines9060665