
Multi-Omic Meta-Analysis of Transcriptomes and the Bibliome Uncovers Novel Hypoxia-Inducible Genes



Abstract
1. Introduction
2. Materials and Methods
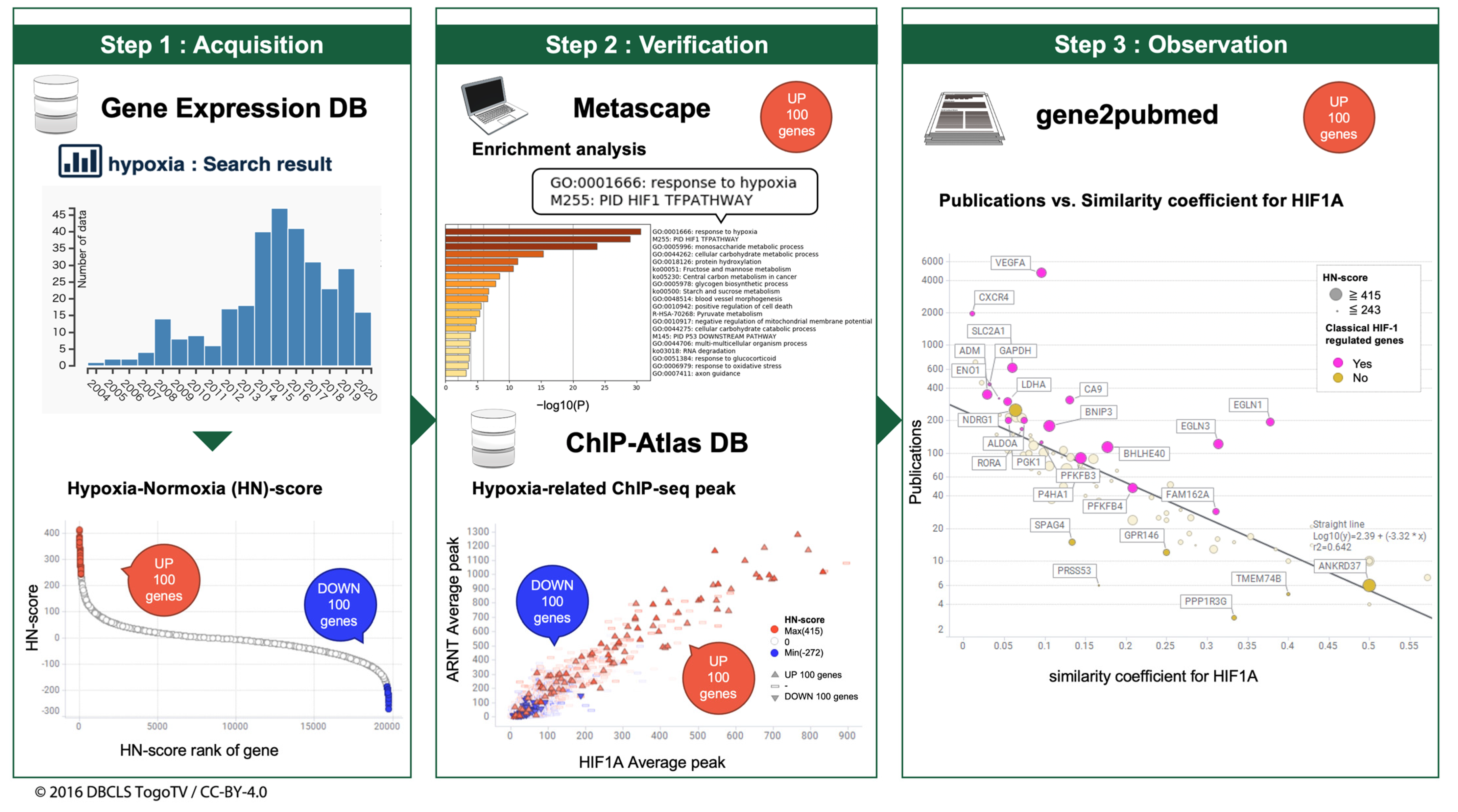
2.1. Curation of Public Gene Expression Data
2.2. Gene Expression Quantification
2.3. Calculation of HN-Ratio and HN-Score
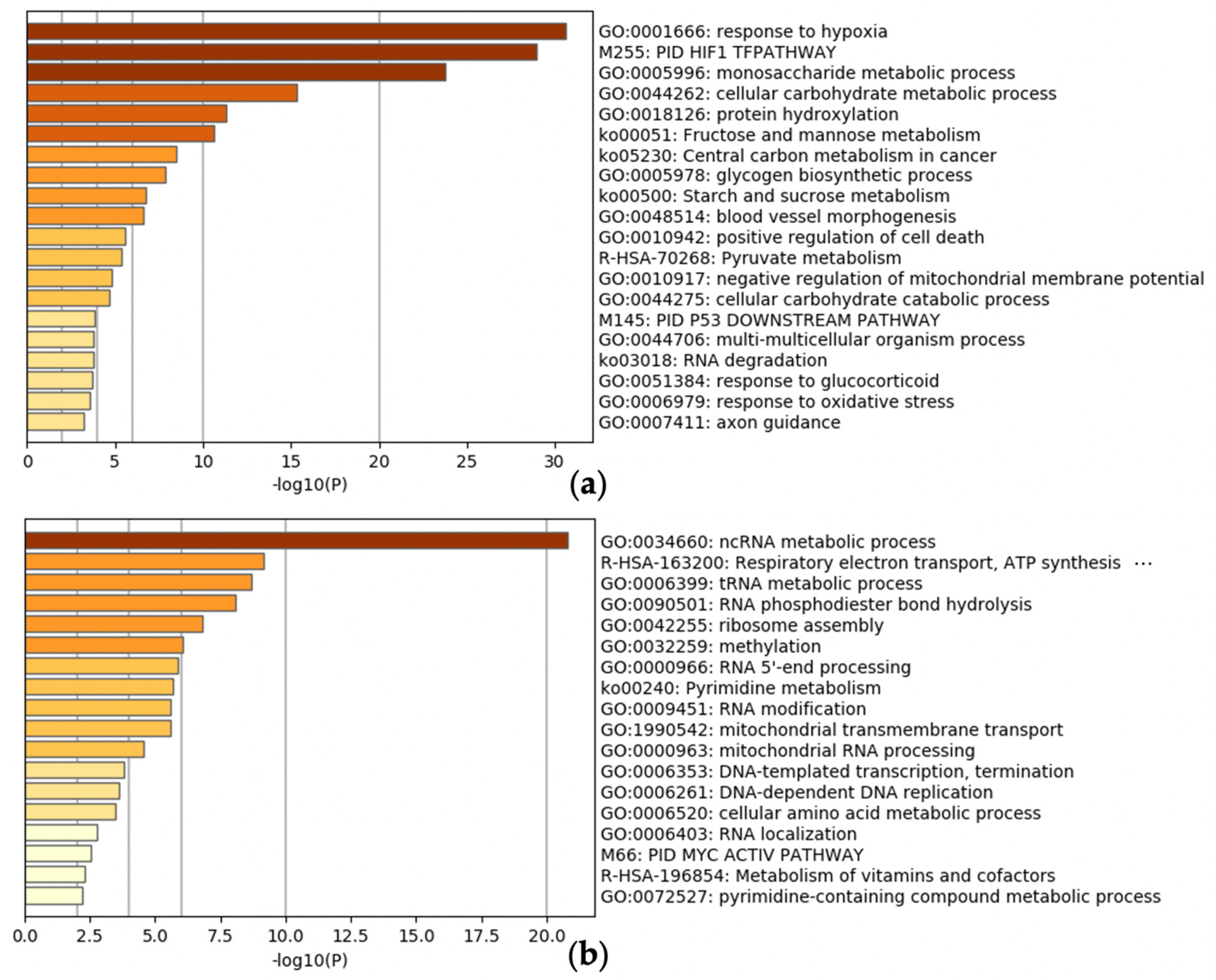
2.4. Enrichment Analysis
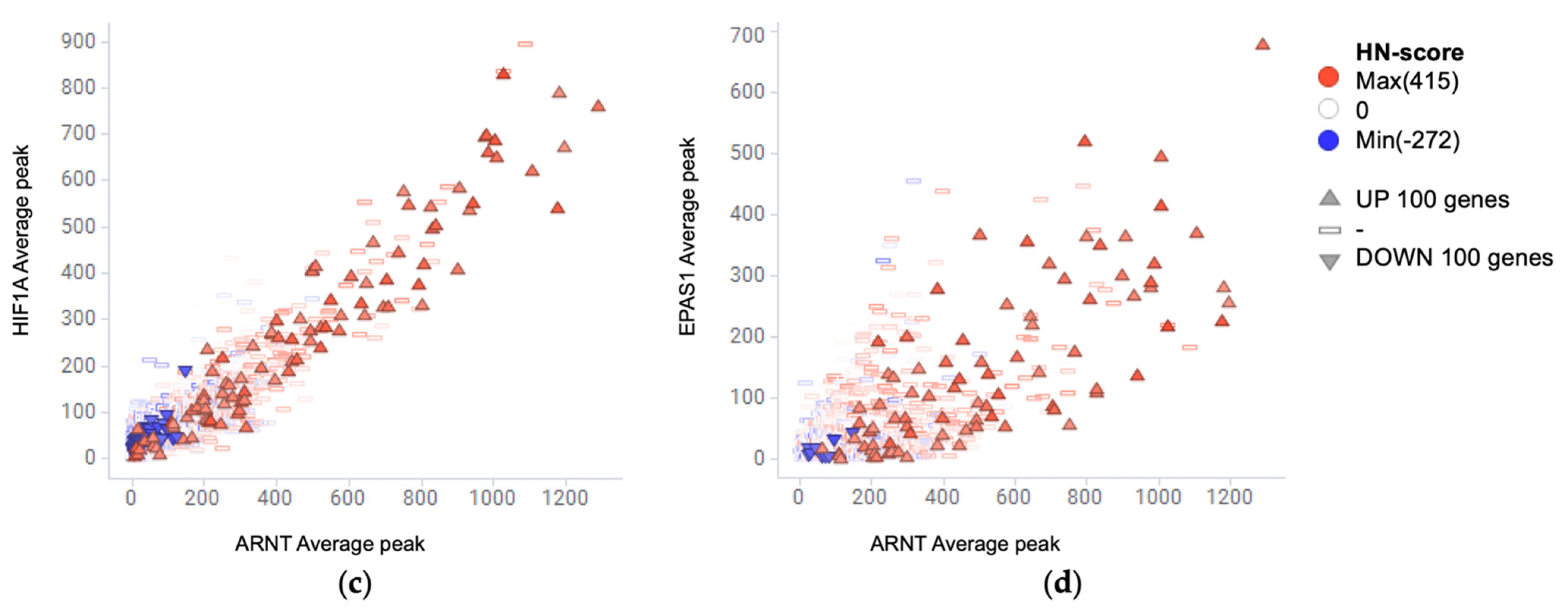
2.5. Meta-Analysis of ChIP-Seq Data
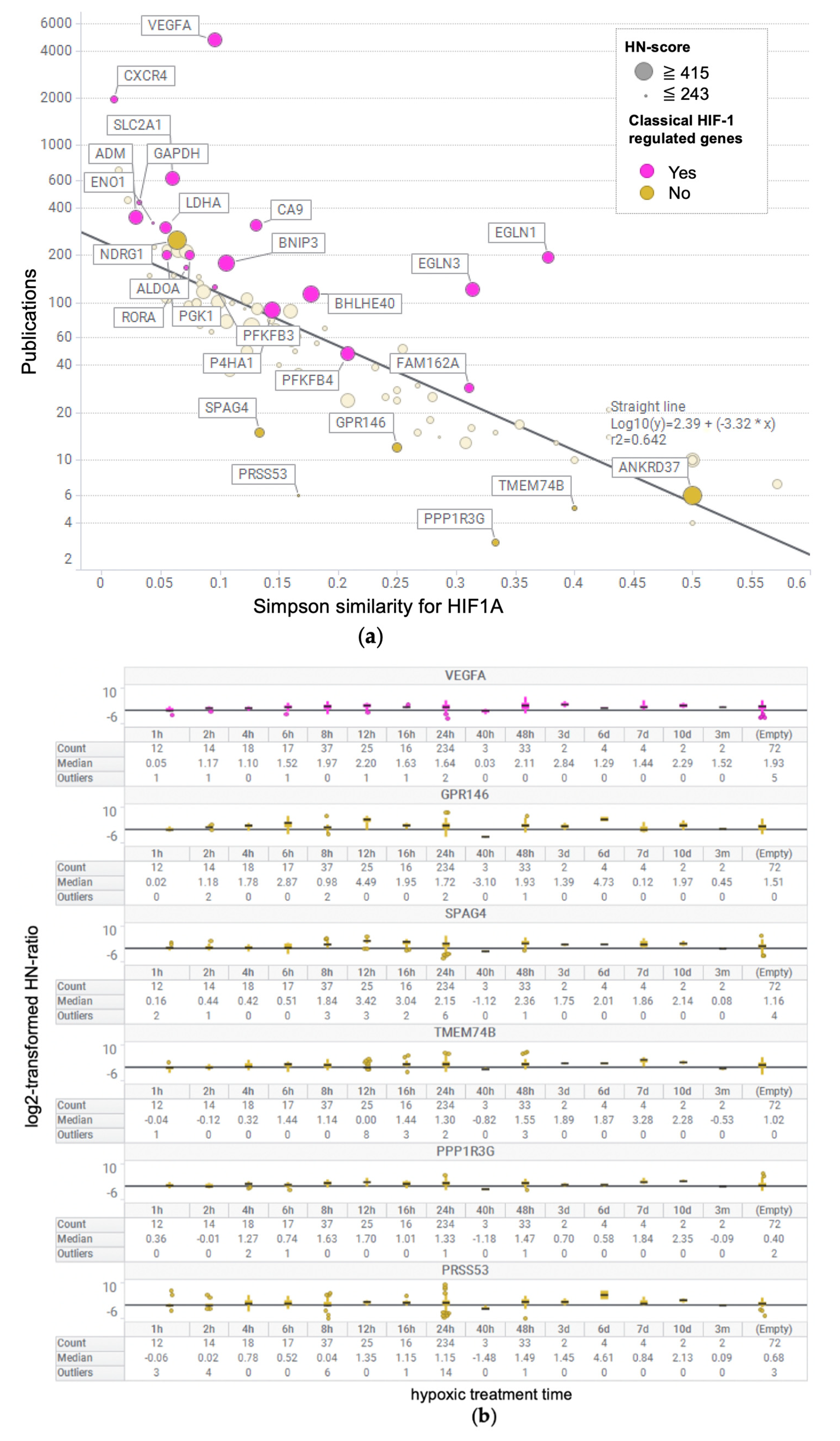
2.6. Calculation of the Number of Publications for Each Gene and Similarity Coefficient for HIF1A
2.7. Visualization and Integrated Functional Analysis of Genes
3. Results
3.1. Overview
3.2. Curation of Hypoxic Transcriptome Data in Public Databases
3.3. Meta-Analysis of Hypoxia-Inducible Genes
3.4. Evaluation of Hypoxia-Inducible Genes
3.5. Evaluation of Simpson Similarity between HIF1A and Genes
3.6. Box Plot of HN-Ratio by Treatment Time
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Carter, A.J.; Kraemer, O.; Zwick, M.; Mueller-Fahrnow, A.; Arrowsmith, C.H.; Edwards, A.M. Target 2035: Probing the Human Proteome. Drug Discov. Today 2019, 24, 2111–2115. [Google Scholar] [CrossRef]
- Barrett, T.; Wilhite, S.E.; Ledoux, P.; Evangelista, C.; Kim, I.F.; Tomashevsky, M.; Marshall, K.A.; Phillippy, K.H.; Sherman, P.M.; Holko, M.; et al. NCBI GEO: Archive for Functional Genomics Data Sets—Update. Nucleic Acids Res. 2013, 41, D991–D995. [Google Scholar] [CrossRef]
- Athar, A.; Füllgrabe, A.; George, N.; Iqbal, H.; Huerta, L.; Ali, A.; Snow, C.; Fonseca, N.A.; Petryszak, R.; Papatheodorou, I.; et al. ArrayExpress Update—From Bulk to Single-Cell Expression Data. Nucleic Acids Res. 2019, 47, D711–D715. [Google Scholar] [CrossRef]
- Kodama, Y.; Mashima, J.; Kosuge, T.; Ogasawara, O. DDBJ Update: The Genomic Expression Archive (GEA) for Functional Genomics Data. Nucleic Acids Res. 2019, 47, D69–D73. [Google Scholar] [CrossRef]
- Bono, H.; Hirota, K. Meta-Analysis of Hypoxic Transcriptomes from Public Databases. Biomedicines 2020, 8, 10. [Google Scholar] [CrossRef] [PubMed]
- Cheung, W.A.; Ouellette, B.F.; Wasserman, W.W. Inferring Novel Gene-Disease Associations Using Medical Subject Heading Over-Representation Profiles. Genome Med. 2012, 4, 75. [Google Scholar] [CrossRef] [PubMed]
- Fischer, I.; Barak, B. Molecular and Therapeutic Aspects of Hyperbaric Oxygen Therapy in Neurological Conditions. Biomolecules 2020, 10, 1247. [Google Scholar] [CrossRef] [PubMed]
- Mahon, P.C.; Hirota, K.; Semenza, G.L. FIH-1: A Novel Protein That Interacts with HIF-1alpha and VHL to Mediate Repression of HIF-1 Transcriptional Activity. Genes Dev. 2001, 15, 2675–2686. [Google Scholar] [CrossRef] [PubMed]
- Jaakkola, P.; Mole, D.R.; Tian, Y.M.; Wilson, M.I.; Gielbert, J.; Gaskell, S.J.; von Kriegsheim, A.; Hebestreit, H.F.; Mukherji, M.; Schofield, C.J.; et al. Targeting of HIF-Alpha to the von Hippel-Lindau Ubiquitylation Complex by O2-Regulated Prolyl Hydroxylation. Science 2001, 292, 468–472. [Google Scholar] [CrossRef] [PubMed]
- Ebert, B.L.; Bunn, H.F. Regulation of Transcription by Hypoxia Requires a Multiprotein Complex That Includes Hypoxia-Inducible Factor 1, an Adjacent Transcription Factor, and P300/CREB Binding Protein. Mol. Cell. Biol. 1998, 18, 4089–4096. [Google Scholar] [CrossRef]
- AOE. Available online: https://aoe.dbcls.jp/ (accessed on 27 February 2021).
- Bono, H. All of Gene Expression (AOE): An Integrated Index for Public Gene Expression Databases. PLoS ONE 2020, 15, e0227076. [Google Scholar] [CrossRef]
- Kodama, Y.; Shumway, M.; Leinonen, R.; on behalf of the International Nucleotide Sequence Database Collaboration. The Sequence Read Archive: Explosive Growth of Sequencing Data. Nucleic Acids Res. 2012, 40, D54–D56. [Google Scholar] [CrossRef]
- Ono, Y. Information on Hypoxic Conditions and Samples in the Dataset Used for the Meta-Analysis. figshare 2021. [Google Scholar] [CrossRef]
- Download SRA Sequences from Entrez Search Results. Available online: https://www.ncbi.nlm.nih.gov/sra/docs/sradownload/ (accessed on 27 February 2021).
- Yyoshiaki/Ikra: RNAseq Pipeline Centered on Salmon. Available online: https://github.com/yyoshiaki/ikra (accessed on 27 February 2021).
- Patro, R.; Duggal, G.; Love, M.I.; Irizarry, R.A.; Kingsford, C. Salmon Provides Fast and Bias-Aware Quantification of Transcript Expression. Nat. Methods 2017, 14, 417–419. [Google Scholar] [CrossRef]
- Ono, Y. Quantitative Gene Expression Data of Hypoxia Experimental Data Sets. figshare 2021. [Google Scholar] [CrossRef]
- Ono, Y. Ratio of Gene Expression under Hypoxic and Normoxic Conditions (HN-Ratio). figshare 2021. [Google Scholar] [CrossRef]
- Soneson, C.; Love, M.I.; Robinson, M.D. Differential Analyses for RNA-Seq: Transcript-Level Estimates Improve Gene-Level Inferences. F1000Res 2016, 4, 1521. [Google Scholar] [CrossRef] [PubMed]
- Metascape. Available online: https://metascape.org (accessed on 27 February 2021).
- Zhou, Y.; Zhou, B.; Pache, L.; Chang, M.; Khodabakhshi, A.H.; Tanaseichuk, O.; Benner, C.; Chanda, S.K. Metascape Provides a Biologist-Oriented Resource for the Analysis of Systems-Level Datasets. Nat. Commun. 2019, 10, 1523. [Google Scholar] [CrossRef]
- ChIP-Atlas. Available online: https://chip-atlas.org/ (accessed on 27 February 2021).
- Oki, S.; Ohta, T.; Shioi, G.; Hatanaka, H.; Ogasawara, O.; Okuda, Y.; Kawaji, H.; Nakaki, R.; Sese, J.; Meno, C. ChIP-Atlas: A Data-Mining Suite Powered by Full Integration of Public ChIP-Seq Data. EMBO Rep. 2018, 19, e46255. [Google Scholar] [CrossRef] [PubMed]
- No85j/Hypoxia_Code. Available online: https://github.com/no85j/hypoxia_code (accessed on 2 March 2021).
- Index of /Gene/DATA. Available online: https://ftp.ncbi.nlm.nih.gov/gene/DATA/ (accessed on 27 February 2021).
- Ono, Y. Score Based on the Ratio of Gene Expression between Hypoxic and Normoxic Conditions (HN-Score). figshare 2021. [Google Scholar] [CrossRef]
- Ono, Y. Genelist_Top 100 Human Genes Up-Regulated under Hypoxic Conditions. figshare 2021. [Google Scholar] [CrossRef]
- Ono, Y. Genelist_Top 100 Human Genes Down-Regulated under Hypoxic Conditions. figshare 2021. [Google Scholar] [CrossRef]
- Ono, Y. Number of Publications per Human Gene. figshare 2021. [Google Scholar] [CrossRef]
- Ono, Y. Simpson Similarity of Each Human Gene to HIF1A Based on Gene2pubmed. figshare 2021. [Google Scholar] [CrossRef]
- Hirota, K.; Semenza, G.L. Regulation of Angiogenesis by Hypoxia-Inducible Factor 1. Crit. Rev. Oncol./Hematol. 2006, 59, 15–26. [Google Scholar] [CrossRef]
- Ono, H.; Ogasawara, O.; Okubo, K.; Bono, H. RefEx, a Reference Gene Expression Dataset as a Web Tool for the Functional Analysis of Genes. Sci. Data 2017, 4, 170105. [Google Scholar] [CrossRef] [PubMed]
- Benita, Y.; Kikuchi, H.; Smith, A.D.; Zhang, M.Q.; Chung, D.C.; Xavier, R.J. An Integrative Genomics Approach Identifies Hypoxia Inducible Factor-1 (HIF-1)-Target Genes That Form the Core Response to Hypoxia. Nucleic Acids Res. 2009, 37, 4587–4602. [Google Scholar] [CrossRef] [PubMed]
- Shoji, K.; Murayama, T.; Mimura, I.; Wada, T.; Kume, H.; Goto, A.; Ohse, T.; Tanaka, T.; Inagi, R.; van der Hoorn, F.A.; et al. Sperm-Associated Antigen 4, a Novel Hypoxia-Inducible Factor 1 Target, Regulates Cytokinesis, and Its Expression Correlates with the Prognosis of Renal Cell Carcinoma. Am. J. Pathol. 2013, 182, 2191–2203. [Google Scholar] [CrossRef]
- Knaup, K.X.; Monti, J.; Hackenbeck, T.; Jobst-Schwan, T.; Klanke, B.; Schietke, R.E.; Wacker, I.; Behrens, J.; Amann, K.; Eckardt, K.-U.; et al. Hypoxia Regulates the Sperm Associated Antigen 4 (SPAG4) via HIF, Which Is Expressed in Renal Clear Cell Carcinoma and Promotes Migration and Invasion in Vitro. Mol. Carcinog. 2014, 53, 970–978. [Google Scholar] [CrossRef]
- Strausberg, R.L.; Feingold, E.A.; Grouse, L.H.; Derge, J.G.; Klausner, R.D.; Collins, F.S.; Wagner, L.; Shenmen, C.M.; Schuler, G.D.; Altschul, S.F.; et al. Generation and Initial Analysis of More than 15,000 Full-Length Human and Mouse CDNA Sequences. Proc. Natl. Acad. Sci. USA 2002, 99, 16899–16903. [Google Scholar] [CrossRef]
- Gerhard, D.S.; Wagner, L.; Feingold, E.A.; Shenmen, C.M.; Grouse, L.H.; Schuler, G.; Klein, S.L.; Old, S.; Rasooly, R.; Good, P.; et al. The Status, Quality, and Expansion of the NIH Full-Length CDNA Project: The Mammalian Gene Collection (MGC). Genome Res. 2004, 14, 2121–2127. [Google Scholar] [CrossRef]
- Kimura, K.; Wakamatsu, A.; Suzuki, Y.; Ota, T.; Nishikawa, T.; Yamashita, R.; Yamamoto, J.; Sekine, M.; Tsuritani, K.; Wakaguri, H.; et al. Diversification of Transcriptional Modulation: Large-Scale Identification and Characterization of Putative Alternative Promoters of Human Genes. Genome Res. 2006, 16, 55–65. [Google Scholar] [CrossRef]
- Labrecque, M.P.; Takhar, M.K.; Nason, R.; Santacruz, S.; Tam, K.J.; Massah, S.; Haegert, A.; Bell, R.H.; Altamirano-Dimas, M.; Collins, C.C.; et al. The Retinoblastoma Protein Regulates Hypoxia-Inducible Genetic Programs, Tumor Cell Invasiveness and Neuroendocrine Differentiation in Prostate Cancer Cells. Oncotarget 2016, 7, 24284–24302. [Google Scholar] [CrossRef]
- Qi, J.; Nakayama, K.; Cardiff, R.D.; Borowsky, A.D.; Kaul, K.; Williams, R.; Krajewski, S.; Mercola, D.; Carpenter, P.M.; Bowtell, D.; et al. Siah2-Dependent Concerted Activity of HIF and FoxA2 Regulates Formation of Neuroendocrine Phenotype and Neuroendocrine Prostate Tumors. Cancer Cell 2010, 18, 23–38. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Rimbert, A.; Palmer, A.E.; Toyohara, T.; Xia, Y.; Xia, F.; Ferreira, L.M.R.; Chen, Z.; Chen, T.; Loaiza, N.; et al. GPR146 Deficiency Protects Against Hypercholesterolemia and Atherosclerosis. Cell 2019, 179, 1276–1288.e14. [Google Scholar] [CrossRef] [PubMed]
- Richards, J.P.; Yosten, G.L.C.; Kolar, G.R.; Jones, C.W.; Stephenson, A.H.; Ellsworth, M.L.; Sprague, R.S. Low O2-Induced ATP Release from Erythrocytes of Humans with Type 2 Diabetes Is Restored by Physiological Ratios of C-Peptide and Insulin. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2014, 307, R862–R868. [Google Scholar] [CrossRef]
- Fang, S.; Zhang, L.; Guo, J.; Niu, Y.; Wu, Y.; Li, H.; Zhao, L.; Li, X.; Teng, X.; Sun, X.; et al. NONCODEV5: A Comprehensive Annotation Database for Long Non-Coding RNAs. Nucleic Acids Res. 2018, 46, D308–D314. [Google Scholar] [CrossRef]
- Ma, L.; Cao, J.; Liu, L.; Du, Q.; Li, Z.; Zou, D.; Bajic, V.B.; Zhang, Z. LncBook: A Curated Knowledgebase of Human Long Non-Coding RNAs. Nucleic Acids Res. 2019, 47, D128–D134. [Google Scholar] [CrossRef] [PubMed]
- Ono, Y. Multiomic Meta-Analysis of Hypoxic Stress from Public Databases. bioRxiv 2021. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Input List | Antigen | ID | Log P-val | Log Q-val | Fold Enrichment |
|---|---|---|---|---|---|
| UP 100 gene list | HIF1A | SRX4802348 | −88.8246 | −84.064 | 35.6249 |
| ARNT | SRX4802353 | −76.4303 | −72.3686 | 83.3136 | |
| EPAS1 | SRX3051209 | −73.1987 | −69.2831 | 34.9928 | |
| DOWN 100 gene list | SAP30 | SRX116447 | −34.1844 | −30.0149 | 4.96916 |
| MYC | SRX1497384 | −31.4158 | −27.5474 | 2.97453 | |
| HDAC1 | SRX186644 | −27.4205 | −24.1541 | 3.3231 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ono, Y.; Bono, H. Multi-Omic Meta-Analysis of Transcriptomes and the Bibliome Uncovers Novel Hypoxia-Inducible Genes. Biomedicines 2021, 9, 582. https://doi.org/10.3390/biomedicines9050582
Ono Y, Bono H. Multi-Omic Meta-Analysis of Transcriptomes and the Bibliome Uncovers Novel Hypoxia-Inducible Genes. Biomedicines. 2021; 9(5):582. https://doi.org/10.3390/biomedicines9050582
Chicago/Turabian StyleOno, Yoko, and Hidemasa Bono. 2021. "Multi-Omic Meta-Analysis of Transcriptomes and the Bibliome Uncovers Novel Hypoxia-Inducible Genes" Biomedicines 9, no. 5: 582. https://doi.org/10.3390/biomedicines9050582
APA StyleOno, Y., & Bono, H. (2021). Multi-Omic Meta-Analysis of Transcriptomes and the Bibliome Uncovers Novel Hypoxia-Inducible Genes. Biomedicines, 9(5), 582. https://doi.org/10.3390/biomedicines9050582