
Reduced SOCS1 Expression in Lung Fibroblasts from Patients with IPF Is Not Mediated by Promoter Methylation or Mir155

 , ,
, , 
Abstract
1. Introduction
2. Methods
2.1. Cell Cultures and Lung Tissue Samples
2.2. Isolation of RNA and DNA from FFPE Tissue Samples
2.3. Quantitative Real Time PCR Analysis
2.4. Western Blot Analysis of Activated STAT Expression
2.5. Methylation Analysis
2.6. Modulation of miRNA155 In Vitro
2.7. Statistical Analysis
3. Results
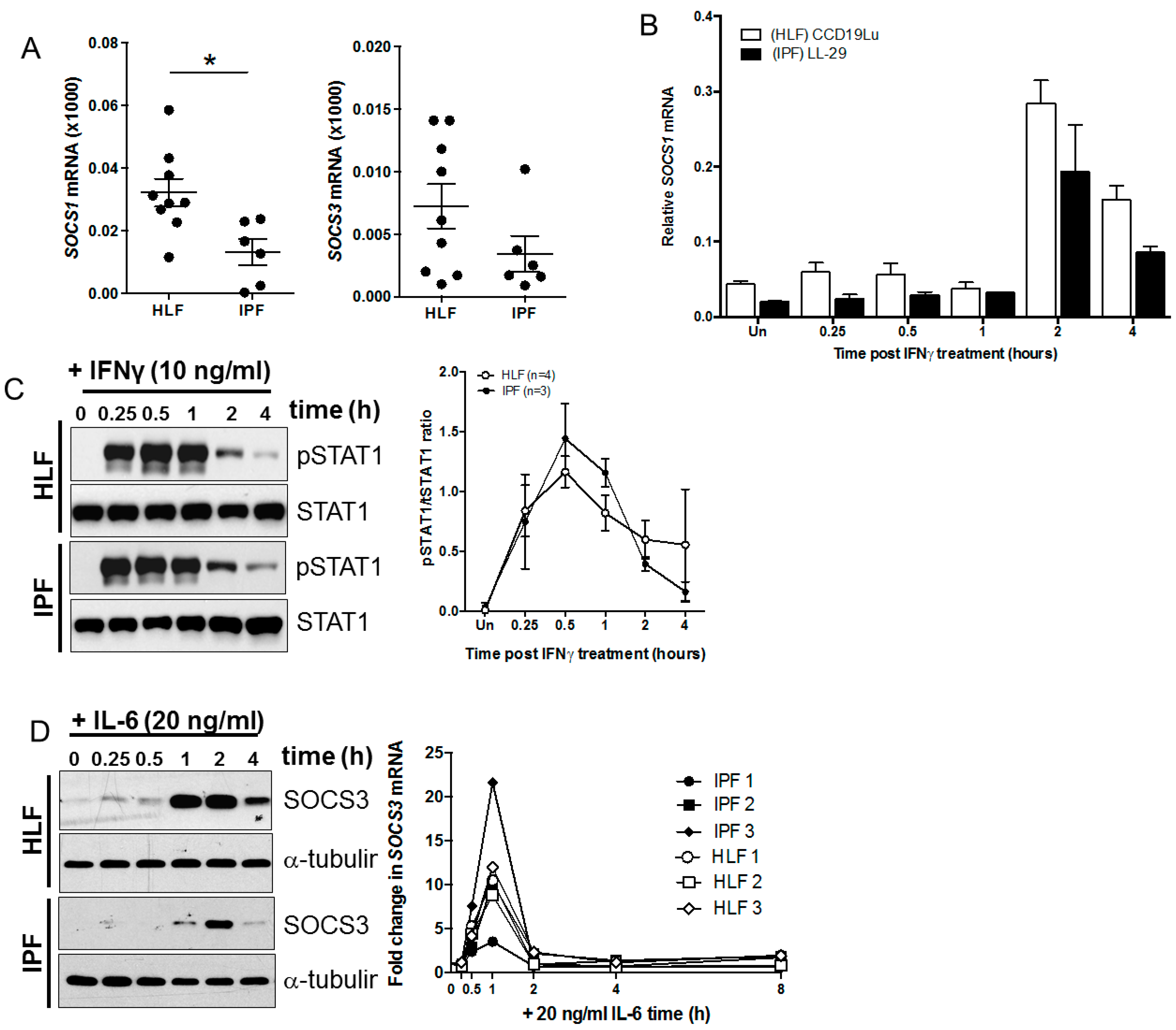
3.1. SOCS1 mRNA Levels Are Reduced in IPF Compared with Control Lung Fibroblasts
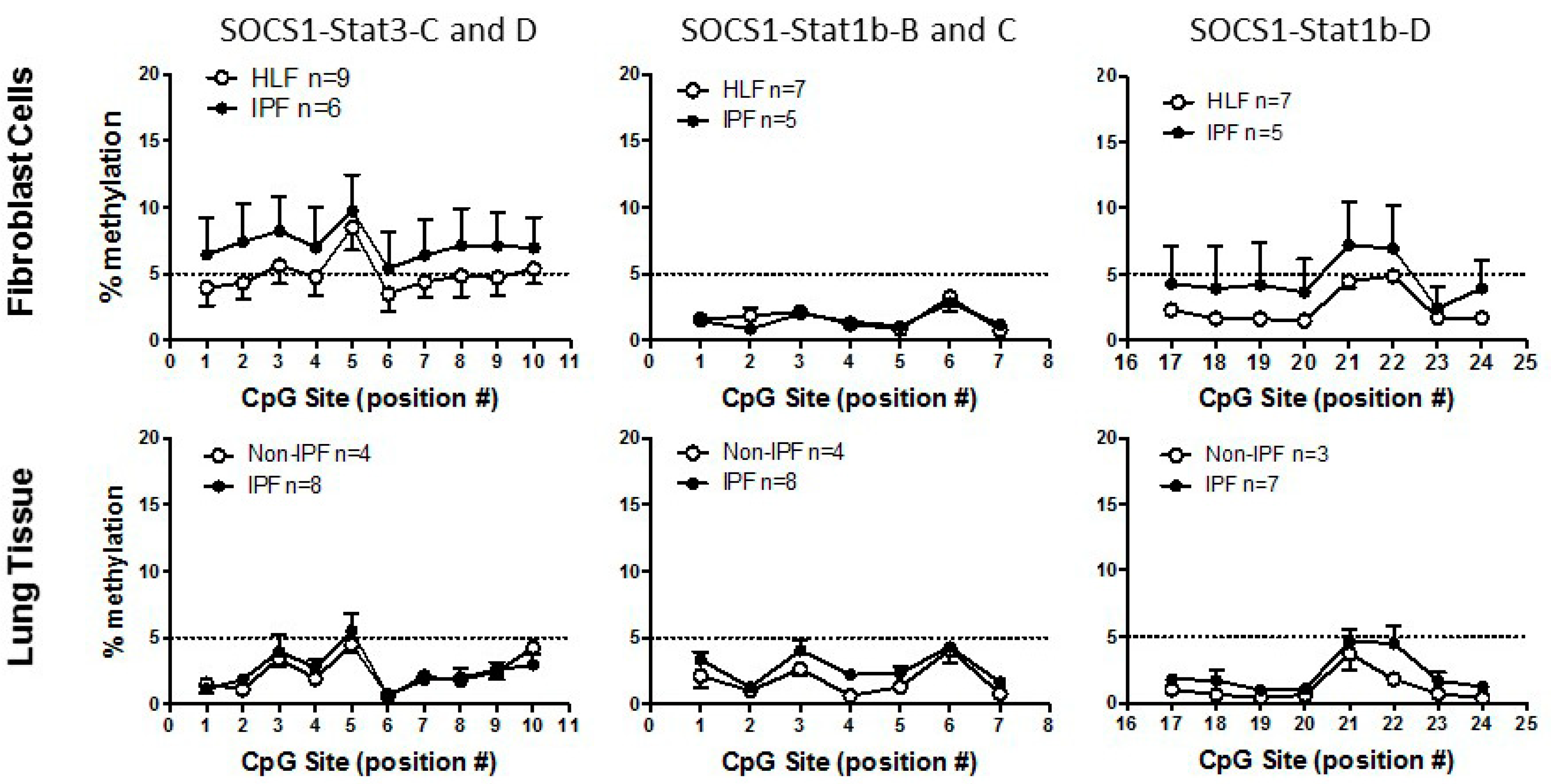
3.2. SOCS1 Methylation in IPF
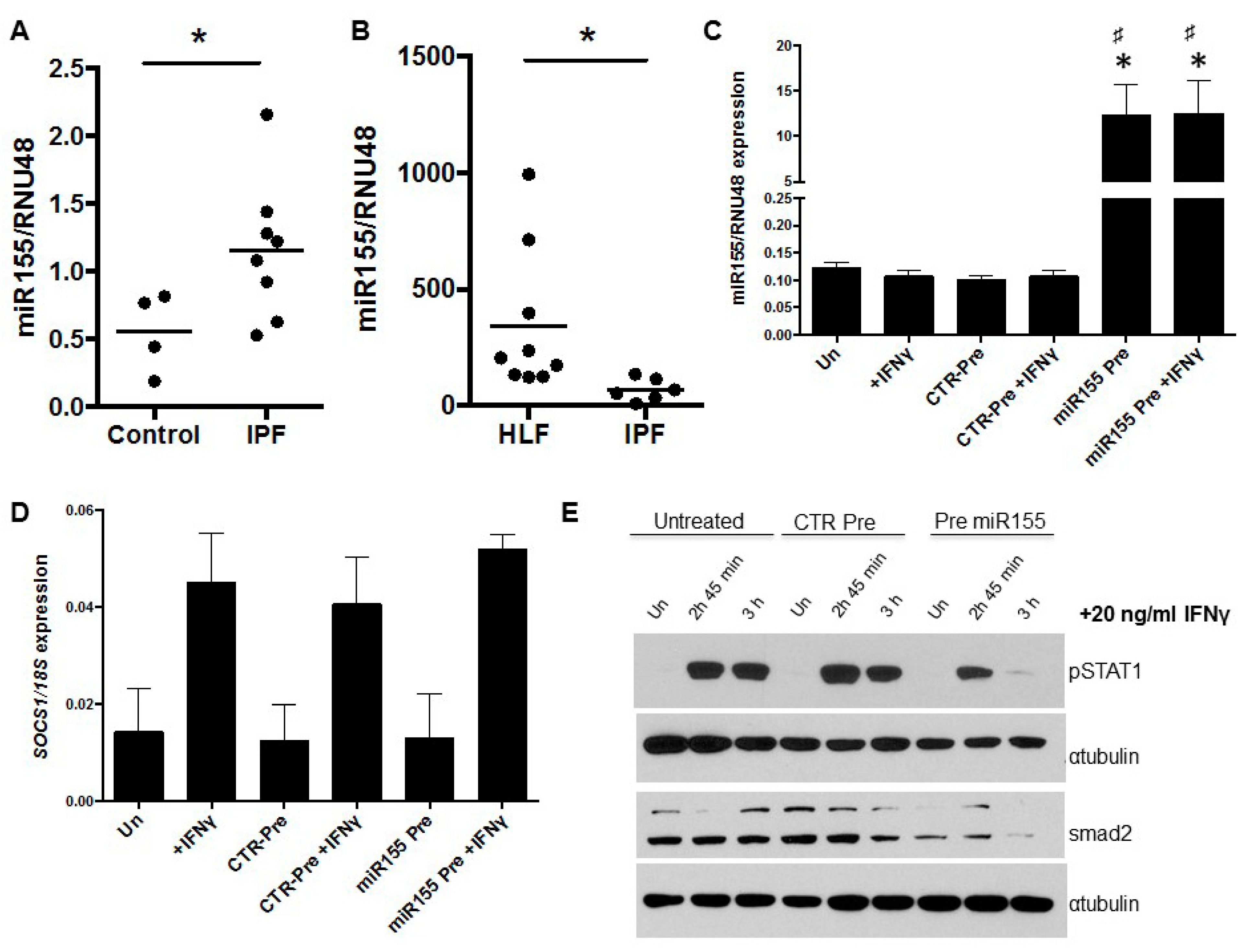
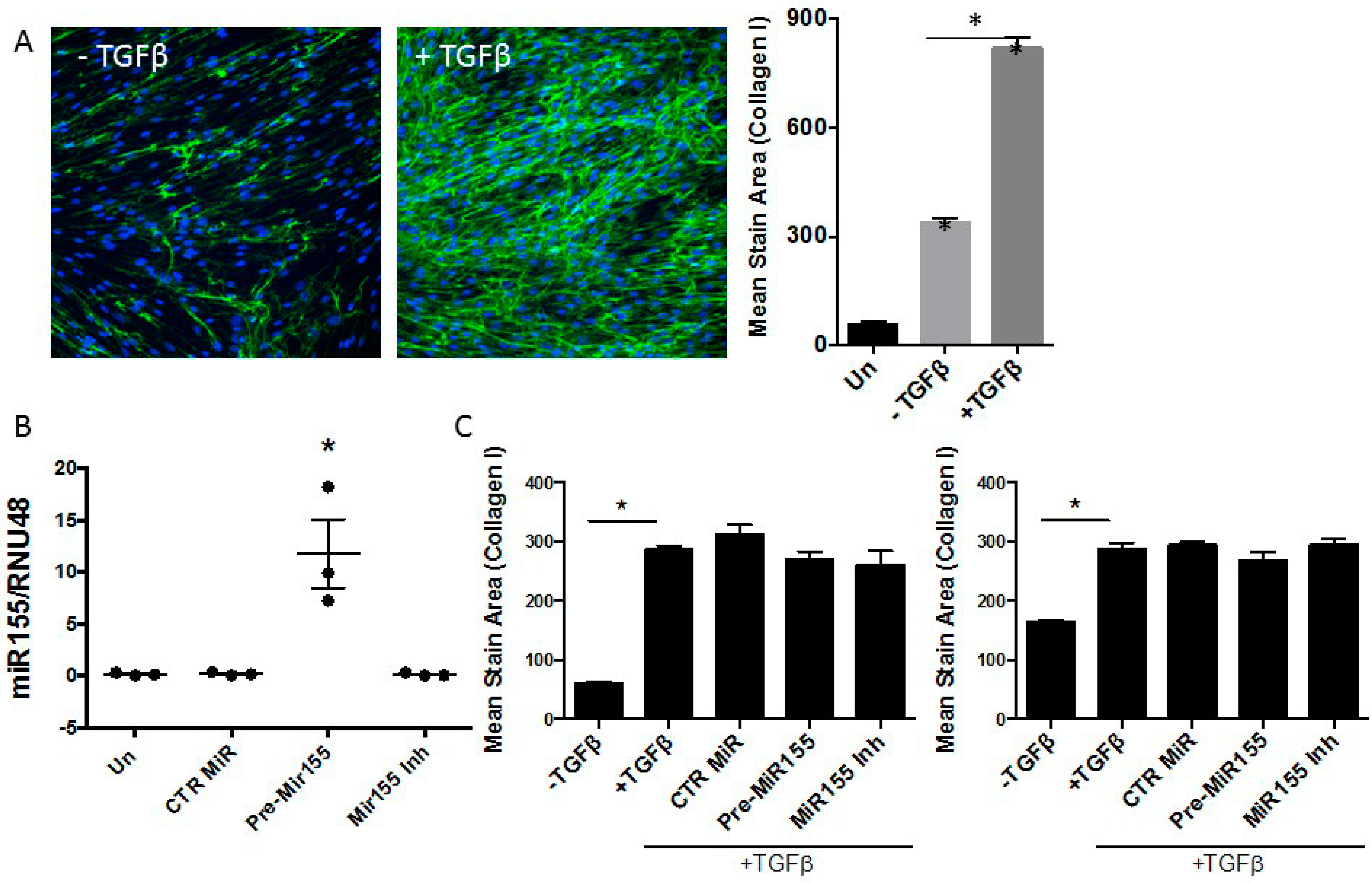
3.3. miR155 as a Potential Regulator of SOCS1 Expression in IPF Fibroblasts
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Khalil, N.; O’Connor, R. Idiopathic pulmonary fibrosis: Current understanding of the pathogenesis and the status of treatment. Can. Med. Assoc. J. 2004, 171, 153–160. [Google Scholar] [CrossRef]
- Klingsberg, R.C.; Mutsaers, S.E.; Lasky, J.A. Current clinical trials for the treatment of idiopathic pulmonary fibrosis. Respirology 2010, 15, 19–31. [Google Scholar] [CrossRef] [PubMed]
- King, T.E.; Schwarz, M.I.; Brown, K.; Tooze, J.A.; Colby, T.V.; Waldron, J.A.; Flint, A.; Thurlbeck, W.; Cherniack, R.M. Idiopathic Pulmonary Fibrosis: Relationship between Histopathologic Features and Mortality. Am. J. Respir. Crit. Care Med. 2001, 164, 1025–1032. [Google Scholar] [CrossRef]
- O’Donoghue, R.J.J.; Knight, D.A.; Richards, C.D.; Prele, C.M.; Lau, H.L.; Jarnicki, A.G.; Jones, J.; Bozinovski, S.; Vlahos, R.; Thiem, S.; et al. Genetic partitioning of interleukin-6 signalling in mice dissociates Stat3 from Smad3-mediated lung fibrosis. EMBO Mol. Med. 2012, 4, 939–951. [Google Scholar] [CrossRef]
- Pechkovsky, D.V.; Prêle, C.M.; Wong, J.; Hogaboam, C.M.; McAnulty, R.J.; Laurent, G.J.; Zhang, S.S.-M.; Selman, M.; Mutsaers, S.E.; Knight, D.A. STAT3-Mediated Signaling Dysregulates Lung Fibroblast-Myofibroblast Activation and Differentiation in UIP/IPF. Am. J. Pathol. 2012, 180, 1398–1412. [Google Scholar] [CrossRef]
- Prêle, C.M.; Yao, E.; O’Donoghue, R.J.J.; Mutsaers, S.E.; Knight, D.A. STAT3: A central mediator of pulmonary fibrosis? Proc. Am. Thorac. Soc. 2012, 9, 177–182. [Google Scholar] [CrossRef]
- Croker, B.A.; Krebs, D.L.; Zhang, J.-G.; Wormald, S.; Willson, T.A.; Stanley, E.G.; Robb, L.; Greenhalgh, C.J.; Förster, I.; Clausen, B.E.; et al. SOCS3 negatively regulates IL-6 signaling in vivo. Nat. Immunol. 2003, 4, 540–545. [Google Scholar] [CrossRef]
- Johnston, J.A.; O’Shea, J.J. Matching SOCS with function. Nat. Immunol. 2003, 4, 507–509. [Google Scholar] [CrossRef] [PubMed]
- Hanada, T.; Kinjyo, I.; Inagaki-Ohara, K.; Yoshimura, A. Negative regulation of cytokine signaling by CIS/SOCS family proteins and their roles in inflammatory diseases. Rev. Physiol. Biochem. Pharmacol. 2003, 149, 72–86. [Google Scholar] [PubMed]
- Naka, T.; Narazaki, M.; Hirata, M.; Matsumoto, T.; Minamoto, S.; Aono, A.; Nishimoto, N.; Kajita, T.; Taga, T.; Yoshizaki, K.; et al. Structure and function of a new STAT-induced STAT inhibitor. Nature 1997, 387, 924–929. [Google Scholar] [CrossRef] [PubMed]
- Wormald, S.; Hilton, D.J. The negative regulatory roles of suppressor of cytokine signaling proteins in myeloid signaling pathways. Curr. Opin. Hematol. 2007, 14, 9–15. [Google Scholar] [CrossRef]
- Alexander, W.S.; Hilton, D.J. The Role of Suppressors of Cytokine Signaling (SOCS) Proteins in Regulation of the Immune Response. Annu. Rev. Immunol. 2004, 22, 503–529. [Google Scholar] [CrossRef]
- Bagnyukova, T.V.; Tryndyak, V.P.; Muskhelishvili, L.; Ross, S.A.; Beland, F.A.; Pogribny, I.P. Epigenetic down-regulation of the suppressor of cytokine signaling 1 (Socs1) gene is associated with the STAT3 activation and development of hepatocellular carcinoma induced by methyl-deficiency in rats. Cell Cycle 2008, 7, 3202–3210. [Google Scholar] [CrossRef] [PubMed]
- Chim, C.S.; Fung, T.K.; Cheung, W.C.; Liang, R.; Kwong, Y.L. SOCS1 and SHP1 hypermethylation in multiple myeloma: Implications for epigenetic activation of the Jak/STAT pathway. Blood 2004, 103, 4630–4635. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, T.; Ogata, H.; Kamio, M.; Joo, A.; Shiraishi, H.; Tokunaga, Y.; Sata, M.; Nagai, H.; Yoshimura, A. SOCS1 Is a Suppressor of Liver Fibrosis and Hepatitis-induced Carcinogenesis. J. Exp. Med. 2004, 199, 1701–1707. [Google Scholar] [CrossRef] [PubMed]
- Flowers, L.O.; Subramaniam, P.S.; Johnson, H.M. A SOCS-1 peptide mimetic inhibits both constitutive and IL-6 induced activation of STAT3 in prostate cancer cells. Oncogene 2005, 24, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Nakashima, T.; Yokoyama, A.; Onari, Y.; Shoda, H.; Haruta, Y.; Hattori, N.; Naka, T.; Kohno, N. Suppressor of cytokine signaling 1 inhibits pulmonary inflammation and fibrosis. J. Allergy Clin. Immunol. 2008, 121, 1269–1276. [Google Scholar] [CrossRef] [PubMed]
- Bao, Z.; Zhang, Q.; Wan, H.; He, P.; Zhou, X.; Zhou, M. Expression of suppressor of cytokine signaling 1 in the peripheral blood of patients with idiopathic pulmonary fibrosis. Chin. Med. J. (Engl.). 2014, 127, 2117–2120. [Google Scholar]
- Shoda, H.; Yokoyama, A.; Nishino, R.; Nakashima, T.; Ishikawa, N.; Haruta, Y.; Hattori, N.; Naka, T.; Kohno, N. Overproduction of collagen and diminished SOCS1 expression are causally linked in fibroblasts from idiopathic pulmonary fibrosis. Biochem. Biophys. Res. Commun. 2007, 353, 1004–1010. [Google Scholar] [CrossRef] [PubMed]
- Keerthisingam, C.B.; Jenkins, R.G.; Harrison, N.K.; Hernandez-Rodriguez, N.A.; Booth, H.; Laurent, G.J.; Hart, S.L.; Foster, M.L.; McAnulty, R.J. Cyclooxygenase-2 Deficiency Results in a Loss of the Anti-Proliferative Response to Transforming Growth Factor-β in Human Fibrotic Lung Fibroblasts and Promotes Bleomycin-Induced Pulmonary Fibrosis in Mice. Am. J. Pathol. 2001, 158, 1411–1422. [Google Scholar] [CrossRef]
- Chen, C.Z.C.; Peng, Y.X.; Wang, Z.B.; Fish, P.V.; Kaar, J.L.; Koepsel, R.R.; Russell, A.J.; Lareu, R.R.; Raghunath, M. The Scar-in-a-Jar: Studying potential antifibrotic compounds from the epigenetic to extracellular level in a single well. Br. J. Pharmacol. 2009, 158, 1196–1209. [Google Scholar] [CrossRef]
- Kirschner, M.B.; Cheng, Y.Y.; Badrian, B.; Kao, S.C.; Creaney, J.; Edelman, J.J.B.; Armstrong, N.J.; Vallely, M.P.; Musk, A.W.; Robinson, B.W.; et al. Increased Circulating miR-625-3p: A Potential Biomarker for Patients with Malignant Pleural Mesothelioma. J. Thorac. Oncol. 2012, 7, 1184–1191. [Google Scholar] [CrossRef]
- Evans, I.C.; Barnes, J.L.; Garner, I.M.; Pearce, D.R.; Maher, T.M.; Shi-Wen, X.; Renzoni, E.A.; Wells, A.U.; Denton, C.P.; Laurent, G.J.; et al. Epigenetic regulation of cyclooxygenase-2 by methylation of c8orf4 in pulmonary fibrosis. Clin. Sci. 2016, 130, 575–586. [Google Scholar] [CrossRef]
- Nishida, N.; Kudo, M.; Nagasaka, T.; Ikai, I.; Goel, A. Characteristic patterns of altered DNA methylation predict emergence of human hepatocellular carcinoma. Hepatology 2012, 56, 994–1003. [Google Scholar] [CrossRef]
- Bibikova, M.; Le, J.; Barnes, B.; Saedinia-Melnyk, S.; Zhou, L.; Shen, R.; Gunderson, K.L. Genome-wide DNA methylation profiling using Infinium®assay. Epigenomics 2009, 1, 177–200. [Google Scholar] [CrossRef]
- Huang, C.; Li, H.; Wu, W.; Jiang, T.; Qiu, Z. Regulation of miR-155 affects pancreatic cancer cell invasiveness and migration by modulating the STAT3 signaling pathway through SOCS1. Oncol. Rep. 2013, 30, 1223–1230. [Google Scholar] [CrossRef]
- Jiang, S.; Zhang, H.-W.; Lu, M.-H.; He, X.-H.; Li, Y.; Gu, H.; Liu, M.-F.; Wang, E.-D. MicroRNA-155 Functions as an OncomiR in Breast Cancer by Targeting the Suppressor of Cytokine Signaling 1 Gene. Cancer Res. 2010, 70, 3119–3127. [Google Scholar] [CrossRef] [PubMed]
- Yao, R.; Ma, Y.-L.; Liang, W.; Li, H.-H.; Ma, Z.-J.; Yu, X.; Liao, Y.-H. MicroRNA-155 Modulates Treg and Th17 Cells Differentiation and Th17 Cell Function by Targeting SOCS1. PLoS ONE 2012, 7, e46082. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.D.; Zhang, W.; Liang, H.J.; Ji, W.Y. Overexpression of miR -155 promotes proliferation and invasion of human laryngeal squamous cell carcinoma via targeting SOCS1 and STAT3. PLoS ONE 2013, 8, e56395. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Li, X.; Tang, Y.; Chen, C.; Jing, R.; Liu, T. miR-155-5p Implicates in the Pathogenesis of Renal Fibrosis via Targeting SOCS1 and SOCS6. Oxid. Med. Cell. Longev. 2020, 2020, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Su, C.; Hou, Z.; Zhang, C.; Tian, Z.; Zhang, J. Ectopic expression of microRNA-155 enhances innate antiviral immunity against HBV infection in human hepatoma cells. Virol. J. 2011, 8, 354. [Google Scholar] [CrossRef]
- He, B.; You, L.; Uematsu, K.; Zang, K.; Xu, Z.; Lee, A.Y.; Costello, J.F.; McCormick, F.; Jablons, D.M. SOCS-3 is frequently silenced by hypermethylation and suppresses cell growth in human lung cancer. Proc. Natl. Acad. Sci. USA 2003, 100, 14133–14138. [Google Scholar] [CrossRef]
- Hodge, D.R.; Hurt, E.M.; Farrar, W.L. The role of IL-6 and STAT3 in inflammation and cancer. Eur. J. Cancer 2005, 41, 2502–2512. [Google Scholar] [CrossRef]
- Song, L.; Turkson, J.; Karras, J.G.; Jove, R.; Haura, E.B. Activation of Stat3 by receptor tyrosine kinases and cytokines regulates survival in human non-small cell carcinoma cells. Oncogene 2003, 22, 4150–4165. [Google Scholar] [CrossRef]
- Ortiz-Muñoz, G.; Lopez-Parra, V.; Lopez-Franco, O.; Fernandez-Vizarra, P.; Mallavia, B.; Flores, C.; Sanz, A.; Blanco, J.; Mezzano, S.; Ortiz, A.; et al. Suppressors of Cytokine Signaling Abrogate Diabetic Nephropathy. J. Am. Soc. Nephrol. 2010, 21, 763–772. [Google Scholar] [CrossRef] [PubMed]
- Souma, Y.; Nishida, T.; Serada, S.; Iwahori, K.; Takahashi, T.; Fujimoto, M.; Ripley, B.; Nakajima, K.; Miyazaki, Y.; Mori, M.; et al. Antiproliferative effect of SOCS-1 through the suppression of STAT3 and p38 MAPK activation in gastric cancer cells. Int. J. Cancer 2011, 131, 1287–1296. [Google Scholar] [CrossRef] [PubMed]
- Pandit, K.V.; Milosevic, J.; Kaminski, N. MicroRNAs in idiopathic pulmonary fibrosis. Transl. Res. 2011, 157, 191–199. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.-X.; Cai, Q.-X.; Peng, X.-M.; Chong, Y.-T.; Gao, Z.-L. Expression of SOCS-1 in the liver tissues of chronic hepatitis B and its clinical significance. World J. Gastroenterol. 2008, 14, 607–611. [Google Scholar] [CrossRef]
- Shimada, K.; Serada, S.; Fujimoto, M.; Nomura, S.; Nakatsuka, R.; Harada, E.; Iwahori, K.; Tachibana, I.; Takahashi, T.; Kumanogoh, A.; et al. Molecular mechanism underlying the antiproliferative effect of suppressor of cytokine signaling-1 in non-small-cell lung cancer cells. Cancer Sci. 2013, 104, 1483–1491. [Google Scholar] [CrossRef]
- Pottier, N.; Maurin, T.; Chevalier, B.; Puisségur, M.-P.; Lebrigand, K.; Robbe-Sermesant, K.; Bertero, T.; Cardenas, C.L.L.; Courcot, E.; Rios, G.; et al. Identification of Keratinocyte Growth Factor as a Target of microRNA-155 in Lung Fibroblasts: Implication in Epithelial-Mesenchymal Interactions. PLoS ONE 2009, 4, e6718. [Google Scholar] [CrossRef]
- Li, P.; Zhao, G.-Q.; Chen, T.-F.; Chang, J.-X.; Wang, H.-Q.; Chen, S.-S.; Zhang, G.-J. Serum miR-21 and miR-155 expression in idiopathic pulmonary fibrosis. J. Asthma 2013, 50, 960–964. [Google Scholar] [CrossRef]
- Waseda, Y.; Yasui, M.; Nishizawa, Y.; Inuzuka, K.; Takato, H.; Ichikawa, Y.; Tagami, A.; Fujimura, M.; Nakao, S. Angiotensin II type 2 receptor antagonist reduces bleomycin-induced pulmonary fibrosis in mice. Respir. Res. 2008, 9, 43. [Google Scholar] [CrossRef] [PubMed]
- Marshall, R.P.; Gohlke, P.; Chambers, R.C.; Howell, D.C.; Bottoms, S.E.; Unger, T.; McAnulty, R.J.; Laurent, G.J. Angiotensin II and the fibroproliferative response to acute lung injury. Am. J. Physiol. Cell. Mol. Physiol. 2004, 286, L156–L164. [Google Scholar] [CrossRef] [PubMed]
- Kurowska-Stolarska, M.; Hasoo, M.K.; Welsh, D.J.; Stewart, L.; McIntyre, D.; Morton, B.E.; Johnstone, S.; Miller, A.M.; Asquith, D.L.; Millar, N.L.; et al. The role of microRNA-155/liver X receptor pathway in experimental and idiopathic pulmonary fibrosis. J. Allergy Clin. Immunol. 2017, 139, 1946–1956. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Assay Name | Primer | Primer Sequence |
|---|---|---|
| SOCS1-Stat1b-B and C | Forward PCR primer | GAGGGTTTAGAAGAGAGGGAAATAG |
| Reverse PCR Primer | 5’Biotin-CCCCCAACTCCACTTTTATTT | |
| Sequencing primer | GAAGAGAGGGAAATAGG | |
| SOCS1-Stat1b-D | Forward PCR primer | GGTTTGATTATAGGTTTTAGAGGAATTT |
| Reverse PCR Primer | 5’Biotin-CCCCAACCTCAATTTCTC | |
| Sequencing primer | ATTATAGGTTTTAGAGGAATTTT | |
| SOCS1-Stat3-C and D | Forward PCR primer | GGGGTTTTTTTGAAGTTTGTGGTTAG |
| Reverse PCR Primer | 5’Biotin-CCCCTCCTAACCCCTACTC | |
| Sequencing primer | AGTTTGTGGTTAGGT |
| CpG ID | % Methylation | Change % Methylation | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CTR UT (n = 6) | CTR T (n = 6) | IPF UT (n = 5) | IPF T (n = 5) | CTR UT vs. CTR T | IPF UT vs. IPF T | CTR UT vs. IPF UT | CTR T vs. IPF T | |||||||||
| Mean | SD | Mean | SD | Mean | SD | Mean | SD | Change | p Value | Change | p Value | Change | p Value | Change | p Value | |
| cg00487159 | 11.713 | 1.195 | 14.184 | 2.885 | 11.790 | 1.135 | 14.947 | 4.414 | 2.472 | 0.811 | 3.157 | 0.863 | 0.077 | 0.983 | 0.762 | 0.969 |
| cg00674265 | 15.652 | 2.105 | 18.261 | 3.754 | 17.276 | 1.673 | 19.941 | 3.104 | 2.609 | 0.891 | 2.665 | 0.855 | 1.624 | 0.669 | 1.680 | 0.917 |
| cg01330880 | 5.561 | 1.180 | 5.060 | 1.770 | 5.159 | 0.956 | 4.460 | 1.141 | −0.501 | 1.000 | −0.699 | 1.000 | −0.402 | 0.887 | −0.600 | 0.948 |
| cg01717706 | 9.709 | 1.131 | 10.844 | 2.073 | 10.423 | 1.493 | 12.551 | 2.698 | 1.135 | 1.000 | 2.128 | 0.874 | 0.714 | 0.810 | 1.707 | 0.876 |
| cg03014241 | 32.513 | 13.765 | 31.706 | 11.822 | 19.027 | 5.034 | 23.322 | 6.284 | −0.807 | 1.000 | 4.295 | 0.913 | −13.486 | 0.469 | −8.384 | 0.816 |
| cg03195600 | 6.103 | 1.499 | 5.644 | 1.178 | 5.216 | 1.090 | 7.457 | 2.502 | −0.459 | 1.000 | 2.242 | 0.853 | −0.888 | 0.761 | 1.813 | 0.828 |
| cg03553358 | 14.261 | 3.334 | 17.253 | 5.228 | 17.211 | 4.445 | 18.966 | 6.256 | 2.992 | 0.979 | 1.755 | 1.000 | 2.950 | 0.684 | 1.713 | 0.952 |
| cg04004558 | 22.836 | 4.740 | 24.374 | 4.471 | 20.942 | 6.490 | 25.944 | 4.320 | 1.538 | 1.000 | 5.002 | 0.871 | −1.894 | 0.872 | 1.570 | 0.942 |
| cg04266460 | 2.923 | 0.626 | 3.668 | 1.114 | 3.271 | 0.444 | 3.116 | 0.981 | 0.746 | 1.000 | −0.155 | 1.000 | 0.348 | 0.858 | −0.553 | 0.940 |
| cg04289163 | 1.890 | 0.404 | 1.648 | 0.257 | 1.711 | 0.513 | 1.867 | 0.122 | −0.242 | 1.000 | 0.156 | 1.000 | −0.179 | 0.931 | 0.219 | 0.972 |
| cg04609482 | 11.691 | 1.423 | 13.629 | 3.087 | 11.725 | 1.424 | 13.837 | 2.642 | 1.938 | 0.926 | 2.112 | 0.871 | 0.034 | 0.994 | 0.208 | 0.991 |
| cg05181221 | 2.208 | 0.422 | 2.249 | 0.288 | 2.102 | 0.436 | 2.144 | 0.375 | 0.041 | 1.000 | 0.041 | 1.000 | −0.106 | 0.961 | −0.106 | 0.988 |
| cg05701125 | 3.816 | 1.301 | 3.615 | 0.475 | 7.013 | 2.437 | 6.152 | 2.259 | −0.201 | 1.000 | −0.861 | 1.000 | 3.197 | 0.367 | 2.537 | 0.648 |
| cg05730996 | 8.417 | 1.043 | 9.603 | 1.854 | 8.248 | 0.785 | 10.522 | 2.465 | 1.186 | 0.972 | 2.274 | 0.852 | −0.169 | 0.954 | 0.919 | 0.935 |
| cg05766667 | 6.259 | 0.312 | 5.939 | 1.122 | 4.897 | 1.079 | 4.925 | 1.198 | −0.320 | 1.000 | 0.028 | 1.000 | −1.362 | 0.464 | −1.014 | 0.883 |
| cg05902273 | 7.706 | 0.840 | 9.699 | 2.405 | 7.593 | 1.150 | 8.733 | 1.512 | 1.993 | 0.821 | 1.140 | 0.941 | −0.113 | 0.971 | −0.966 | 0.929 |
| cg06082432 | 6.052 | 1.483 | 5.389 | 1.249 | 6.880 | 0.831 | 5.944 | 1.423 | −0.663 | 1.000 | −0.936 | 0.972 | 0.828 | 0.765 | 0.556 | 0.948 |
| cg06220235 | 4.380 | 1.626 | 5.034 | 3.023 | 4.748 | 1.054 | 4.409 | 0.495 | 0.653 | 1.000 | −0.339 | 1.000 | 0.368 | 0.918 | −0.624 | 0.961 |
| cg06295404 | 3.795 | 0.746 | 3.422 | 0.912 | 3.135 | 0.501 | 3.817 | 1.184 | −0.374 | 1.000 | 0.682 | 1.000 | −0.660 | 0.726 | 0.396 | 0.959 |
| cg09567644 | 6.622 | 1.087 | 5.526 | 1.012 | 5.201 | 1.202 | 4.890 | 0.731 | −1.097 | 0.902 | −0.311 | 1.000 | −1.421 | 0.556 | −0.636 | 0.923 |
| cg10513253 | 1.958 | 0.331 | 1.875 | 0.411 | 1.897 | 0.349 | 1.727 | 0.498 | −0.083 | 1.000 | −0.170 | 1.000 | −0.061 | 0.978 | −0.148 | 0.983 |
| cg10784813 | 26.767 | 14.114 | 27.686 | 12.717 | 21.366 | 7.658 | 23.734 | 6.576 | 0.919 | 1.000 | 2.368 | 1.000 | −5.401 | 0.815 | −3.952 | 0.935 |
| cg11973052 | 1.450 | 0.288 | 1.675 | 0.309 | 1.181 | 0.198 | 1.321 | 0.225 | 0.225 | 1.000 | 0.140 | 1.000 | −0.269 | 0.874 | −0.354 | 0.951 |
| cg12642006 | 19.242 | 3.395 | 21.525 | 3.090 | 24.291 | 3.112 | 28.573 | 4.704 | 2.283 | 0.973 | 4.283 | 0.852 | 5.049 | 0.384 | 7.049 | 0.523 |
| cg26012103 | 7.832 | 1.312 | 8.621 | 1.213 | 8.350 | 1.031 | 9.473 | 2.089 | 0.789 | 1.000 | 1.123 | 0.981 | 0.518 | 0.859 | 0.852 | 0.926 |
| cg26301908 | 8.050 | 1.290 | 8.205 | 0.863 | 9.517 | 1.597 | 9.057 | 1.929 | 0.155 | 1.000 | −0.460 | 1.000 | 1.467 | 0.616 | 0.852 | 0.917 |
| cg27003951 | 11.410 | 2.728 | 14.376 | 2.487 | 11.229 | 0.894 | 16.208 | 5.881 | 2.966 | 0.805 | 4.979 | 0.852 | −0.181 | 0.975 | 1.833 | 0.929 |
| cg27540841 | 3.287 | 0.640 | 2.876 | 0.320 | 3.181 | 0.419 | 3.256 | 0.573 | −0.410 | 1.000 | 0.075 | 1.000 | −0.105 | 0.964 | 0.380 | 0.950 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Prêle, C.M.; Iosifidis, T.; McAnulty, R.J.; Pearce, D.R.; Badrian, B.; Miles, T.; Jamieson, S.E.; Ernst, M.; Thompson, P.J.; Laurent, G.J.; et al. Reduced SOCS1 Expression in Lung Fibroblasts from Patients with IPF Is Not Mediated by Promoter Methylation or Mir155. Biomedicines 2021, 9, 498. https://doi.org/10.3390/biomedicines9050498
Prêle CM, Iosifidis T, McAnulty RJ, Pearce DR, Badrian B, Miles T, Jamieson SE, Ernst M, Thompson PJ, Laurent GJ, et al. Reduced SOCS1 Expression in Lung Fibroblasts from Patients with IPF Is Not Mediated by Promoter Methylation or Mir155. Biomedicines. 2021; 9(5):498. https://doi.org/10.3390/biomedicines9050498
Chicago/Turabian StylePrêle, Cecilia M., Thomas Iosifidis, Robin J. McAnulty, David R. Pearce, Bahareh Badrian, Tylah Miles, Sarra E. Jamieson, Matthias Ernst, Philip J. Thompson, Geoffrey J. Laurent, and et al. 2021. "Reduced SOCS1 Expression in Lung Fibroblasts from Patients with IPF Is Not Mediated by Promoter Methylation or Mir155" Biomedicines 9, no. 5: 498. https://doi.org/10.3390/biomedicines9050498
APA StylePrêle, C. M., Iosifidis, T., McAnulty, R. J., Pearce, D. R., Badrian, B., Miles, T., Jamieson, S. E., Ernst, M., Thompson, P. J., Laurent, G. J., Knight, D. A., & Mutsaers, S. E. (2021). Reduced SOCS1 Expression in Lung Fibroblasts from Patients with IPF Is Not Mediated by Promoter Methylation or Mir155. Biomedicines, 9(5), 498. https://doi.org/10.3390/biomedicines9050498